
Abstract
We present a graph neural network approach that fully automates the prediction of defect formation enthalpies for any crystallographic site from the ideal crystal structure, without the need to create defected atomic structure models as input. Here we used density functional theory reference data for vacancy defects in oxides, to train a defect graph neural network (dGNN) model that replaces the density functional theory supercell relaxations otherwise required for each symmetrically unique crystal site. Interfaced with thermodynamic calculations of reduction entropies and associated free energies, the dGNN model is applied to the screening of oxides in the Materials Project database, connecting the zero-kelvin defect enthalpies to high-temperature process conditions relevant for solar thermochemical hydrogen production and other energy applications. The dGNN approach is applicable to arbitrary structures with an accuracy limited principally by the amount and diversity of the training data, and it is generalizable to other defect types and advanced graph convolution architectures. It will help to tackle future materials discovery problems in clean energy and beyond.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$99.00 per year
only $8.25 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others
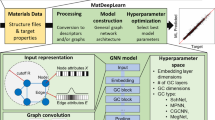
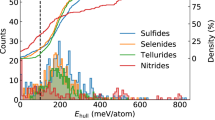

Data availability
The pre-trained models, datasets generated during and/or analyzed during the current study, and scripts to post-process them and reproduce the manuscript figures, are available in the Zenodo repository: ‘A database of vacancy formation enthalpies for materials discovery’ at https://doi.org/10.5281/zenodo.8087871 (ref. 65). Source data are provided with this paper.
Code availability
The open-source dGNN code for training models the models presented in this article has been distributed on the Paper1 branch at the following GitHub repository (https://github.com/mwitman1/cgcnndefect/tree/Paper1)66, which is a modified fork of the original open-source CGCNN code (https://github.com/txie-93/cgcnn).
References
Muhich, C. L. et al. A review and perspective of efficient hydrogen generation via solar thermal water splitting. WIREs Energy Environ. 5, 261–287 (2016).
Komsa, H. P. et al. Two-dimensional transition metal dichalcogenides under electron irradiation: defect production and doping. Phys. Rev. Lett. 109, 1–5 (2012).
Jeen, H. et al. Reversible redox reactions in an epitaxially stabilized SrCoOx oxygen sponge. Nat. Mater. 12, 1057–1063 (2013).
Menéndez, C., Chu, D. & Cazorla, C. Oxygen-vacancy induced magnetic phase transitions in multiferroic thin films. npj Comput. Mater. 6, 1–9 (2020).
Ong, S. P. et al. Python Materials Genomics (pymatgen): a robust, open-source Python library for materials analysis. Comput. Mater. Sci. 68, 314–319 (2013).
Jain, A. et al. The Materials Project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Stevanović, V., Lany, S., Zhang, X. & Zunger, A. Correcting density functional theory for accurate predictions of compound enthalpies of formation: fitted elemental-phase reference energies. Phys. Rev. B 85, 115104 (2012).
Choudhary, K. et al. The joint automated repository for various integrated simulations (JARVIS) for data-driven materials design. npj Comput. Mater. 6, 173 (2020).
Bertoldo, F., Ali, S., Manti, S. & Thygesen, K. S. Quantum point defects in 2D materials - the QPOD database. npj Comput. Mater. 8, 56 (2022).
Saal, J. E., Kirklin, S., Aykol, M., Meredig, B. & Wolverton, C. Materials design and discovery with high-throughput density functional theory: the Open Quantum Materials Database (OQMD). JOM 65, 1501–1509 (2013).
Deml, A. M., Holder, A. M., O’Hayre, R. P., Musgrave, C. B. & Stevanović, V. Intrinsic material properties dictating oxygen vacancy formation energetics in metal oxides. J. Phys. Chem. Lett. 6, 1948–1953 (2015).
Varley, J. B., Samanta, A. & Lordi, V. Descriptor-based approach for the prediction of cation vacancy formation energies and transition levels. J. Phys. Chem. Lett. 8, 5059–5063 (2017).
Goyal, A. et al. On the dopability of semiconductors and governing material properties. Chem. Mater. 32, 4467–4480 (2020).
Frey, N. C., Akinwande, D., Jariwala, D. & Shenoy, V. B. Machine learning-enabled design of point defects in 2D materials for quantum and neuromorphic information processing. ACS Nano 14, 13406–13417 (2020).
Wan, Z., Wang, Q. D., Liu, D. & Liang, J. Data-driven machine learning model for the prediction of oxygen vacancy formation energy of metal oxide materials. Phys. Chem. Chem. Phys. 23, 15675–15684 (2021).
Wexler, R. B., Gautam, G. S., Stechel, E. B. & Carter, E. A. Factors governing oxygen vacancy formation in oxide perovskites. J. Am. Chem. Soc. 143, 13212–13227 (2021).
Mannodi-Kanakkithodi, A. et al. Universal machine learning framework for defect predictions in zinc blende semiconductors. Patterns 3, 100450 (2022).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Park, C. W. & Wolverton, C. Developing an improved crystal graph convolutional neural network framework for accelerated materials discovery. Phys. Rev. Mater. 4, 063801 (2020).
Schütt, K., Unke, O. & Gastegger, M. Equivariant message passing for the prediction of tensorial properties and molecular spectra. In Proc. 38th International Conference on Machine Learning Vol. 139, 9377–9388 (2021).
Unke, O. T. et al. SpookyNet: learning force fields with electronic degrees of freedom and nonlocal effects. Nat. Commun. 12, 7273 (2021).
Fung, V., Zhang, J., Juarez, E. & Sumpter, B. G. Benchmarking graph neural networks for materials chemistry. npj Comput. Mater. 7, 1–8 (2021).
McDaniel, A. H. et al. Sr- and Mn-doped LaAlO3−δ for solar thermochemical H2 and CO production. Energy Environ. Sci. 6, 2424 (2013).
Gorman, B. T., Lanzarini-Lopes, M., Johnson, N. G., Miller, J. E. & Stechel, E. B. Techno-economic analysis of a concentrating solar power plant using redox-active metal oxides as heat transfer fluid and storage media. Front. Energy Res. https://doi.org/10.3389/fenrg.2021.734288 (2021).
Lany, S. The electronic entropy of charged defect formation and its impact on thermochemical redox cycles. J. Chem. Phys. 148, 071101 (2018).
Park, J. E. et al. Computationally accelerated discovery and experimental demonstration of Gd0.5La0.5Co0.5Fe0.5O3 for solar thermochemical hydrogen production. Front. Energy Res. https://doi.org/10.3389/fenrg.2021.750600 (2021).
Sai Gautam, G., Stechel, E. B. & Carter, E. A. A first-principles-based sub-lattice formalism for predicting off-stoichiometry in materials for solar thermochemical applications: the example of ceria. Adv. Theory Simul. 3, 2000112 (2020).
Millican, S. L., Clary, J. M., Musgrave, C. B. & Lany, S. Redox defect thermochemistry of FeAl2O4 hercynite in water splitting from first-principles methods. Chem. Mater. 34, 519–528 (2022).
Vieten, J. et al. Materials design of perovskite solid solutions for thermochemical applications. Energy Environ. Sci. 12, 1369–1384 (2019).
Maiti, D. et al. Earth abundant perovskite oxides for low temperature CO2 conversion. Energy Environ. Sci. 11, 648–659 (2018).
Tezsevin, I., van de Sanden, M. C. M. & Er, S. High-throughput computational screening of cubic perovskites for solid oxide fuel cell cathodes. J. Phys. Chem. Lett. 12, 4160–4165 (2021).
Belsky, A., Hellenbrandt, M., Karen, V. L. & Luksch, P. New developments in the Inorganic Crystal Structure Database (ICSD): accessibility in support of materials research and design. Acta Crystallogr. Sect. B 58, 364–369 (2002).
Goyal, A., Gorai, P., Peng, H., Lany, S. & Stevanović, V. A computational framework for automation of point defect calculations. Comput. Mater. Sci. 130, 1–9 (2017).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Sharan, A. & Lany, S. Computational discovery of stable and metastable ternary oxynitrides. J. Chem. Phys. 154, 234706 (2021).
Pavone, M., Munoz-Garcia, A. B., Ritzmann, A. M. & Carter, E. A. First-principles study of lanthanum strontium manganite: Insights into electronic structure and oxygen vacancy formation. J. Phys. Chem. C 118, 13346–13356 (2014).
Santana, J. A., Krogel, J. T., Kent, P. R. & Reboredo, F. A. Diffusion quantum Monte Carlo calculations of SrFeO3 and LaFeO3. J. Chem. Phys. 147, 034701 (2017).
Peng, H. et al. Convergence of density and hybrid functional defect calculations for compound semiconductors. Phys. Rev. B 88, 115201 (2013).
Lany, S. Semiconductor thermochemistry in density functional calculations. Phys. Rev. B 78, 245207 (2008).
Emery, A. A., Saal, J. E., Kirklin, S., Hegde, V. I. & Wolverton, C. High-throughput computational screening of perovskites for thermochemical water splitting applications. Chem. Mater. 28, 5621–5634 (2016).
Cheng, Y. et al. Vacancy formation energy and its connection with bonding environment in solid: a high-throughput calculation and machine learning study. Comput. Mater. Sci. 183, 109803 (2020).
Choudhary, K. & Sumpter, B. G. A deep-learning model for fast prediction of vacancy formation in diverse materials. Preprint at arXiv https://doi.org/10.48550/arXiv.2205.08366 (2022).
Witman, M. D., Goyal, A., Ogitsu, T., McDaniel, A. H. & Lany, S. Graph neural network modeling of vacancy formation enthalpy for materials discovery and its application in solar thermochemical water splitting. Preprint at ChemRxiv https://doi.org/10.26434/chemrxiv-2022-frcns (2022).
Meredig, B. & Wolverton, C. First-principles thermodynamic framework for the evaluation of thermochemical H2O- or CO2-splitting materials. Phys. Rev. B 80, 245119 (2009).
Barcellos, D. R., Sanders, M. D., Tong, J., McDaniel, A. H. & O’Hayre, R. P. BaCe0.25Mn0.75O3−δ - a promising perovskite-type oxide for solar thermochemical hydrogen production. Energy Environ. Sci. 11, 3256–3265 (2018).
Bergeson-Keller, A. M., Sanders, M. D. & O’Hayre, R. P. Reduction thermodynamics of Sr1−xCexMnO3 and CexSr2−xMnO4 perovskites for solar thermochemical hydrogen production. Energy Technol. 10, 2100515 (2022).
Zunger, A., Wei, S.-H., Ferreira, L. G. & Bernard, J. E. Special quasirandom structures. Phys. Rev. Lett. 65, 353–356 (1990).
Titov, Y. et al. Effect of isovalent substitution on the crystal structure and properties of two-slab indates BaLa2−xSmxIn2O7. Open Chem. 18, 1294–1303 (2020).
Tirmali, P. M. et al. Structural, magnetic and dielectric relaxation behaviour study of La2MnCoO6 and fully substituted b-site La2FeCoO6. J. Chin. Adv. Mater. Soc. 6, 207–221 (2018).
Zhang, C. et al. Ferromagnetic Y2CoMnO6: spin-glass-like behavior and dielectric relaxation. J. Electron. Mater. 43, 1071–1075 (2014).
Mevs, H. & Müller-Buschbaum, H. Neue verbindungen mit Ba6Ln2M43+O15-TYP: Ba6Nd2Fe4O15, Ba5SrLa2Fe4O15 und Ba5SrNd2Fe4O15. J. Less Common Met. 158, 147–152 (1990).
Suescun, L., Chmaissem, O., Mais, J., Dabrowski, B. & Jorgensen, J. D. Crystal structures, charge and oxygen-vacancy ordering in oxygen deficient perovskites SrMnOx (x < 2.7). J. Solid State Chem. 180, 1698–1707 (2007).
Nair, M. M. & Abanades, S. Experimental screening of perovskite oxides as efficient redox materials for solar thermochemical CO2 conversion. Sustain. Energy Fuels 2, 843–854 (2018).
Fuks, D., Mastrikov, Y., Kotomin, E. & Maier, J. Ab initio thermodynamic study of (Ba,Sr)(Co,Fe)O3 perovskite solid solutions for fuel cell applications. J. Mater. Chem. A 1, 14320 (2013).
Kim, Y.-M. et al. Probing oxygen vacancy concentration and homogeneity in solid-oxide fuel-cell cathode materials on the subunit-cell level. Nat. Mater. 11, 888–894 (2012).
Lee, Y.-L., Kleis, J., Rossmeisl, J., Shao-Horn, Y. & Morgan, D. Prediction of solid oxide fuel cell cathode activity with first-principles descriptors. Energy Environ. Sci. 4, 3966–3970 (2011).
Gokon, N., Yawata, T., Bellan, S., Kodama, T. & Cho, H.-S. Thermochemical behavior of perovskite oxides based on LaxSr1−x-(Mn, Fe, Co)-O3−δ and BaySr1−yCoO3−δ redox system for thermochemical energy storage at high temperatures. Energy 171, 971–980 (2019).
Xiang, D., Gu, C., Xu, H. & Xiao, G. Self-assembled structure evolution of Mn-Fe oxides for high temperature thermochemical energy storage. Small 17, 2101524 (2021).
Naghavi, S. S. et al. Giant onsite electronic entropy enhances the performance of ceria for water splitting. Nat. Commun. 8, 285 (2017).
Chueh, W. C. et al. High-flux solar-driven thermochemical dissociation of CO2 and H2O using nonstoichiometric ceria. Science 330, 1797–1801 (2010).
McDaniel, A. H. Renewable energy carriers derived from concentrating solar power and nonstoichiometric oxides. Curr. Opin. Green Sustainable Chem. 4, 37–43 (2017).
Witman, M., Goyal, A., Ogitsu, T., McDaniel, A. H. & Lany, S. A database of vacancy formation enthalpies for materials discovery (0.0.1) [data set]. Zenodo https://doi.org/10.5281/zenodo.8087871 (2023).
Witman, M. mwitman1/cgcnndefect: release for paper1 (v0.0.0). Zenodo https://doi.org/10.5281/zenodo.8051401 (2023).
Paszke, A. et al. PyTorch: an imperative style, high-performance deep learning library. In Adv. Neural Information Processing Systems Vol. 32, 8024–8035 (2019).
Zhang, C., Bengio, S., Hardt, M., Recht, B. & Vinyals, O. Understanding deep learning (still) requires rethinking generalization. Commun. ACM 64, 107–115 (2021).
Bengio, Y. & Grandvalet, Y. No unbiased estimator of the variance of K-fold cross-validation. J. Mach. Learn. Res. 5, 1089–1105 (2004).
Aykol, M., Dwaraknath, S. S., Sun, W. & Persson, K. A. Thermodynamic limit for synthesis of metastable inorganic materials. Sci. Adv. 4, eaaq0148 (2018).
Acknowledgements
This material is based on work supported by the US Department of Energy (DOE), Office of Energy Efficiency and Renewable Energy (EERE), specifically the Hydrogen and Fuel Cell Technologies Office. Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC (NTESS), a wholly owned subsidiary of Honeywell International Inc., for the US Department of Energy’s National Nuclear Security Administration (DOE/NNSA) under contract DE-NA0003525. This written work is authored by an employee of NTESS. The employee, not NTESS, owns the right, title and interest in and to the written work and is responsible for its contents. Part of this work was supported by the Sandia Laboratory Directed Research and Development program. Part of the work was performed under the auspices of the US Department of Energy by Lawrence Livermore National Laboratory under contract DE-AC52-07NA27344. The National Renewable Energy Laboratory (NREL) is operated by the Alliance for Sustainable Energy, LLC, for the DOE under contract DE-AC36-08GO28308. This work used high-performance computing resources at NREL, sponsored by DOE-EERE. Any subjective views or opinions that might be expressed in the written work do not necessarily represent the views of the US government. The publisher acknowledges that the US government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this written work or allow others to do so, for US government purposes. The DOE will provide public access to results of federally sponsored research in accordance with the DOE Public Access Plan.
Author information
Authors and Affiliations
Contributions
M.D.W., A.G., T.O., A.H.M. and S.L. conceptualized the study. Methodology, software and validation were the responsibility of M.D.W., A.G. and S.L.; M.D.W., A.G. and S.L. completed formal analyses and investigations. M.D.W., A.G. and S.L. curated the data. A.H.M. and S.L. acquired the funding. M.W., A.G. and S.L. wrote the original draft. All authors reviewed and edited the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Computational Science thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Kaitlin McCardle, in collaboration with the Nature Computational Science team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Sections 1–10, Tables 1–5 and Figs. 1–7.
Supplementary Data 1
Summary of training compounds. Additional summary information of the compounds investigated with DFT and their supercell properties used to compute vacancy formation energies, as summarized in Supplementary Section 1.
Supplementary Data 2
Final summary data for MP screened oxides. Promising STCH candidates with summary information as described in Supplementary Section 10.
Source data
Source data for Fig. 2
(x, y) data of the various subplots.
Source data for Fig. 3
(x, y) data of the various subplots.
Source data for Fig. 4
(x, y) data of the various subplots.
Source data for Fig. 5
(x, y) data of the various subplots.
Source data for Fig. 6
(x, y) data of the various subplots.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Witman, M.D., Goyal, A., Ogitsu, T. et al. Defect graph neural networks for materials discovery in high-temperature clean-energy applications. Nat Comput Sci 3, 675–686 (2023). https://doi.org/10.1038/s43588-023-00495-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43588-023-00495-2
