
Abstract
Benzene, toluene, ethylbenzene and xylenes can contribute to hydroxyl reactivity and secondary aerosol formation in the atmosphere. These aromatic hydrocarbons are typically classified as anthropogenic air pollutants, but there is growing evidence of biogenic sources, such as emissions from plants and phytoplankton. Here we use a series of shipborne measurements of the remote marine atmosphere, seawater mesocosm incubation experiments and phytoplankton laboratory cultures to investigate potential marine biogenic sources of these compounds in the oceanic atmosphere. Laboratory culture experiments confirmed marine phytoplankton are a source of benzene, toluene, ethylbenzene, xylenes and in mesocosm experiments their sea-air fluxes varied between seawater samples containing differing phytoplankton communities. These fluxes were of a similar magnitude or greater than the fluxes of dimethyl sulfide, which is considered to be the key reactive organic species in the marine atmosphere. Benzene, toluene, ethylbenzene, xylenes fluxes were observed to increase under elevated headspace ozone concentration in the mesocosm incubation experiments, indicating that phytoplankton produce these compounds in response to oxidative stress. Our findings suggest that biogenic sources of these gases may be sufficiently strong to influence atmospheric chemistry in some remote ocean regions.
Similar content being viewed by others

Introduction
Current global and regional trace gas emission inventories and chemistry-climate models generally include a limited number of marine volatile organic compounds (VOCs) emissions that are known to be important to particle formation, remote OH reactivity and halogen-ozone chemistry. These include dimethyl sulfide (DMS)1,2,3,4, isoprene5,6, C2-C3 hydrocarbons7, amines8, organohalogens, and monoterpenes6,9. However, many known VOCs emissions from phytoplankton are not taken into account, either to economize computational resources, because quantitative information on fluxes from the ocean are lacking or because of a perceived lack of impact.
For instance, Colomb et al.10 observed phytoplankton emissions of organochlorines including chlorobenzene, typically associated with anthropogenic emissions such as shipping and offshore gas exploitation. More recently, previously unexplored biological sources of benzenoid compounds were reported the emission of which have previously been almost solely attributed to anthropogenic and biomass burning emissions. Indeed, in addition to emissions from terrestrial plants, Misztal et al.11 reported benzenoids emissions from marine phytoplankton from a series of laboratory, mesocosm, and ship-borne experiments.
Importantly for their relevance to climate, marine emissions of such aromatic compounds in remote ocean regions, may contribute to local ozone photochemistry, OH reactivity, and secondary organic aerosol formation12. For instance, yields of SOA from benzenoid compounds have been observed to be higher in cleaner (low NOx) air masses, typical of remote marine atmospheres, compared to polluted environments.
The composition and sources of marine organic compounds, the processes that mediate their air-sea exchange, and their atmospheric chemical transformations are complex and not currently well understood13,14. To unravel some of these complex systems requires multiscale studies from the laboratory to the open ocean in order to understand their biological production pathways and the drivers of their seasonal and spatial variations for use in regional and global ocean-atmosphere models.
Here we report on a series of experiments measuring the fluxes of the volatile monoaromatic compounds benzene, toluene, ethylbenzene, and xylenes (BTEX) across scales, from a series of controlled laboratory studies of marine phytoplankton monocultures to ship-borne mesocosm experiments, as well as measurements of the marine atmosphere of the South Pacific, South Atlantic, and Southern Indian Oceans.
Results
Air-sea fluxes of BTEX in mesocosm studies of natural seawaters from the Southwest Pacific Ocean
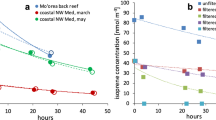
As part of the 2020 Sea2Cloud campaign aboard the R/V Tangaroa15, experiments were undertaken in which ~1 m3 samples of surface seawaters were collected in three distinct locations in the Southwest Pacific east of New Zealand and studied in situ in two Air-Sea Interaction Tanks (ASIT). Key elements of seawater biogeochemistry and the headspace composition in the ASITs were continuously monitored (Figure S.1) over the proceeding 1.5–3.5 days. Here we report on measurements of the net sea-to-air fluxes of BTEX. determined from the headspace concentrations using the method described by Sinha et al.16—see Methods. Times series of BTEX and DMS fluxes are shown in Fig. 1, the absence of bars indicates a gap in the data due to instrument calibrations, instrument failures, and data removed due to ship exhaust contamination.

Times series of fluxes in ng m−2 s−1 of benzene, toluene, and the sum of ethylbenzene plus xylenes (yellow bars) and Photosynthetically Active Radiation (PAR) in µmol m−2 s−1 in dark green. Gray areas are plotted to indicate when the ASITs are opened. The absence of bars indicates gaps in the measurements.
In the open ocean, multiple complex physical parameters influence the air-sea exchange of a given chemical species including, light, temperature, wind speed, and turbulence. Due to atmospheric transport, mixing, and chemistry, the mixing ratios of trace gases at a given location in the atmosphere over the open ocean are typically dislocated from their marine sources, which limits our ability to directly observe air-sea interactions via in situ shipborne observations of ambient air and seawater.
Thus while there are enclosure artefacts, which are not representative of open ocean conditions (i.e., very low wind and turbulence)17, the ASIT conditions were well suited to monitor the changes in seawater biogeochemistry and plankton community dynamics under natural light and temperature conditions, and to explore their link with the air-sea fluxes of trace gases and subsequent role in new particle formation16.
Air was continuously flushed through the ASITs (23 L min−1) and the reported fluxes can be considered representative of emission fluxes at very low winds (<1 m s−1).
The estimated lifetimes of benzene, toluene, and xylenes in ambient air were 9 days, 2 days and 6 h, respectively (OH ~2.0 × 106 molecules cm−3)18, and it was assumed the net fluxes of BTEX were not substantially affected by photochemistry within the ~40 min residence time within the ASIT headspace but instead were dominated by their production in seawater and sea-air gas transfer rates.
The median fluxes [interquartile range] of benzene, toluene, and the sum of ethylbenzene + xylenes over the three experiments were 0.09 [0.06–0.26], 0.19 [0.13–0.83], and 0.18 [0.09–0.47] ng m−2 s−1, respectively, with a median total BTEX flux of 0.46 [0.28–1.56] ng m−2 s−1 (Table S.1). These BTEX fluxes were of a similar magnitude or greater than the fluxes of DMS considered the key reactive organic species in the marine atmosphere (0.36 [0.26–1.02] ng m−2 s−1). Analysis of water samples from the ASITs with gas chromatography (see Methods) confirms the presence of BTEX compounds in the sampled seawater, that were in close agreement to levels measured via workboat sampling in the surrounding ocean.
Although relatively few data points (18.5% of data points) were obtained during daytime, there were no significant difference between daytime (0.18 ± 0.13 ng m−2 s−1 e.g., benzene flux) and nighttime (0.17 ± 0.14 ng m−2 s−1 for benzene flux) emission fluxes.
There was a consistent, strong linear relationship between the fluxes of B and the sum of E + X across the three experiments (Slope = 0.67, r2 = 0.94) and both B and E + X were correlated with the fluxes of the known biogenic compounds DMS (r² = 0.86 and 0.81, n = 444, p < 0.001) and monoterpenes (r² = 0.84 and 0.73, n = 444, p < 0.001), strongly indicating a common biological origin for these VOCs (Table S.2). Conversely, the T/B ratio varied up to a factor three within the course of an experiment and by a factor of 10 from one experiment to the other, indicating a different emission pathway for T than those of B and E + X (Fig S.2).
Toluene fluxes as a function of phytoplankton abundance
The ASIT’s seawater wase analysed daily for Chlorophyll-a (Chl-a), and small phytoplanktonic class cell abundances. None of the BTEX species correlated with Chl-a, which is a broad nonspecific indicator of phytoplankton biomass. Moderate linear correlations were observed between toluene fluxes and Synechococcus (picophytoplankton, <2 µm) cell abundance (R² =0.57), whereas benzene and xylenes did not correlate with any of the planktonic classes analysed (Table S.3). The toluene flux could be expressed as a function of the Synechococcus cell abundance as:
The toluene fluxes reported here (average 1.16 ng m−2 s−1 during Exp. B) were within the range of those reported by Misztal et al.11 (mean = 0.14 ng m−2 s−1; max ~1.38 ng m−2 s−1) from a Norwegian fjord mesocosm experiment in which fluxes also showed significant relationships with the abundance of picophytoplankton, as well as the globally dominant coccolithophore E. huxleyi. Further laboratory experiments are necessary to directly verify BTEX emission from Synechococcus.
Laboratory studies of BTEX emissions from marine phytoplankton cultures
A series of laboratory studies were re-examined in order to investigate whether direct biosynthesis of BTEX by marine phytoplankton could have contributed to the fluxes observed in the Sea2Cloud mesocosm studies. Laboratory cultures of 5 marine phytoplankton species (two coccolithophores, two diatoms, and a chlorophyte) were analysed for BTEX by GC-MS. The initial concentration of each compound in the culture was determined and normalized against the concentration of Chlorophyll-a (Chl-a), used here as a proxy for phytoplankton biomass; see Colomb et al.10 and Yassaa et al.19 and Figure S.3 for the Method and details).
The normalized biomass concentrations of BTEX for each phytoplankton species are shown in Fig. 2. E. huxleyi cultures exhibited high levels of benzene, toluene, and ethylbenzene concentrations, whereas toluene and xylenes were the dominant BTEX compounds detected in C. neogracilis cultures and were higher than its DMS emissions (0.53 ± 0.02 pmol/L/Chl-a). Only minor amounts of BTEX were observed from other phytoplankton cultures (C. leptoporous, P. tricornutum, and D. tertiolecta), suggesting BTEX emissions are species specific, as has been found for other biogenic VOC emitted from phytoplankton10 and terrestrial plants11.

Concentration of benzene, toluene, ethylbenzene, and xylenes after subtraction of the medium concentration, normalized by the chlorophyll-a concentration (pmol L−1 /Chl-a) observed in the headspace of monocultures of coccolithophore species (Emiliania huxleyi (4–5 µm), Calcidiscus leptoporus (5–8 µm)), diatoms (Phaeodactylum tricornutum (5–12 µm), Chaetoceros neogracilis (10–50 µm)), and chlorophyte (Dunaliella tertiolecta (5–25 µm)). All cultures were kept at room temperature between 20– 25 °C and adapted to a 12–12 h light–dark cycle (light intensity of ~250 µE s−1 m−2). The samplings of VOC emissions were performed in the middle of the light cycle (between 10:00 and 14:00) where all algae were in the transition from the exponential to the stationary phase of growth.
While the mechanisms responsible for biosynthesis of BTEX in phytoplankton are not clear, studies of BTEX production pathways in terrestrial plants implicate both the shikimate pathway and the nonmevalonate pathway11,20, which is supported in this study by the observed correlations of BTEX compounds with monoterpenes and isoprene in the ASITs experiments (Table S.2).
Shipborne observations of BTEX in the atmosphere over the remote Southern Oceans
Ambient mixing ratios of benzene, toluene, and the sum of ethylbenzene + xylenes were determined for remote open ocean air masses East of New Zealand measured during the Sea2Cloud campaign, and carefully filtered for ship stack contamination and/or terrestrial influence (see Methods). BTEX concentrations were found above the detection limit of the instrument for 26.1, 47.6, and 41.5% of the time, with average concentrations (and standard deviation) of 0.026 ± 0.023 ppb, 0.038 ± 0.042 ppb, and 0.026 ± 0.041 ppb for benzene, toluene, and xylenes, respectively.
Ambient fluxes of BTEX were calculated from the Sea2Cloud data via the nocturnal accumulation method21,22, which assumes negligible nighttime photochemical losses and overnight accumulation of sea surface emission within the well-mixed marine boundary, with horizontally homogeneous fluxes and steady surface layer height (see Methods). Benzenoid ambient fluxes during Sea2Cloud were 1.2 ± 1.8, 3.7 ± 4.9, and 1.0 ± 8.3 ng m−2 s−1 for benzene, toluene, and xylenes respectively. These fluxes were higher than the BTEX fluxes measured in the ASITs (Fig. 1). Higher seawater turbulence, as well as higher wind stress, are likely responsible for higher fluxes than those measured in the ASITs. Calculated ambient fluxes showed a higher variability, and were occasionally negative, which might also be due to the processing and dilution of these species within the marine boundary layer and loss processes at the sea surface.
In situ observation of BTEX from four other voyages was re-examined in order to explore the potential spatial and temporally heterogeneity of atmospheric BTEX over the Southern Oceans. The highest ambient atmospheric concentrations of benzene and toluene were observed over a chlorophyll enriched upwelling zone IIa (30–40 °S) in the Southern Indian Ocean during the MANCHOT campaign23 with averaged concentrations of toluene and benzene of 0.278 ± 0.075 and 0.085 ± 0.036 ppb. Besides levels of BTEX observed in ambient air during the Sea2Cloud campaign were of the same order of magnitude as those reported from previous ship campaigns in the Southern Ocean for which the datasets were revisited in the framework of the present study.
The MANCHOT campaign took place during southern hemispheric summer (December), in contrast to the other four voyages (Drake Passage in 2009;24 SOAP in 2012;25 CAPRICORN in 2016 and Sea2Cloud in 2020; Fig. 3 and S.4, Table S.4) reported here which took place during the autumn season (March-April), and the higher concentrations observed during MANCHOT might point to a seasonal variation in phytoplankton type and abundance and subsequent BTEX emissions. Indeed, ambient BTEX levels were enhanced in zone II and III of the MANCHOT voyage23 (Fig. 3) coinciding with seawaters dominated by diatoms, such as C. neogracilis and haptophytes coccolithophores E. huxleyi, which laboratory studies presented here demonstrate were capable of producing BTEX.

Toluene ambient concentrations (ppb) measured during five voyages around the world over the Southern Ocean (2004–2020). Top inset for MANCHOT voyage shows enhanced toluene concentrations coinciding with seawaters dominated by diatoms (e.g., C. neogracilis) and haptophyte coccolithophores (e.g., E. huxleyi), which laboratory studies demonstrated are capable of producing BTEX.
Sensitivity of BTEX emissions to ozone in ambient air
In order to investigate the sensitivity of VOC fluxes to ambient ozone concentrations during the Sea2Cloud voyage, one ASIT was simply flushed with ambient air containing on average 6.7 ± 1.5 ppb of ozone, while the second tank (ASIT-O3) was flushed with ambient air enriched with an ozone concentration of 14.5 ± 2.9 ppb during the experiments. In comparison, ambient concentrations of ozone during the voyage were 14.6 ± 1.8 ppb. The ASIT-control headspace contained lower levels of ozone compared to ambient air, due to losses in lines and ASITs walls, and reactions with other trace species in the headspace. For the three experiments, the emission fluxes of benzene and xylenes were 166 ± 155 and 185 ± 155% higher in the ozone-enriched ASIT (Fig. 4; Fig S.1). The toluene emission flux also increased under a high ozone scenario but only in experiment B (39 ± 34% increase). The effect of ozone on benzene and xylenes fluxes was greater than that for DMS which showed a 39 ± 45% difference between the ASIT-Control and the ASIT-O3 over the three experiments.

Comparison of toluene, benzene, and xylenes fluxes (ng m−2 s−1) over the three experiments with boxplots. The bottom and top of each box are the 25th and 75th percentile, line in the middle of each box is the median and whiskers are the maximum and minimum values.
Only minor differences in phytoplankton community cell abundances were observed between the ASIT-Control and the ASIT-Ozone during the course of each experiment (Fig. S.5), with an increase of the cell abundances of Synechococcus (8.31% in EXP C) and picoeukaryotic phytoplankton (4.47% in EXP B) suggesting ozone did not influence phytoplankton community composition.
Many VOC emissions are metabolically linked to photosynthesis26,27 although also occur during nighttime28. The observed changes in fluxes in response to ozone may be explained by changes in biological production due to abiotic stressors, such as oxidative stress which has been shown to influence the metabolism of microorganisms and the regulation of photosynthetic activity of seagrasses29,30 and alter VOCs production, for terpenoids in plants in particular31,32,33,34 which are emitted in reaction to ozone stress to prevent oxidative damage, protect membrane integrity and quench ozone products31,35,36. In addition, Ciuraru et al.37 showed that illumination initiated degradation of organic surfactant in dissolved organic matter (DOM) initiates degradation of organic surfactant at the sea-air interface leading to the formation of benzene and toluene. During the nighttime, changes in VOCs emission could also be due to the heterogeneous reaction of ozone with organic compounds in and on the seawater although ozonolysis is unlikely to be primarily responsible for a change in emissions of primary compounds such as BTEX13,38.
By analogy, BTEX emissions could be enhanced in ASIT-ozone experiments as a response to the oxidative stress produced by ozone to prevent damage to the microorganisms. Overall, even for more conventional BVOCs, such as isoprene, the physiological production and function of VOC in phytoplankton is not yet fully understood, and further research is needed to elucidate the underlying mechanisms.
Tropospheric ozone is strongly seasonal (±10 ppb) with maxima in summer and is generally exhibiting a positive long-term global trend39,40. The observed sensitivity of marine BTEX fluxes to ambient ozone concentrations reported here may have potentially important implications for understanding seasonal and longer-term trends in the spatial and temporal variability of marine BTEX emissions to the atmosphere.
Ambient ozone concentrations during Sea2Cloud were 14.6 ± 1.8 ppb compared to 14.5 ± 2.7 ppb during MANCHOT and 24.1 ± 7.0 ppb during the Drake Passage voyage. Indicating the higher BTEX fluxes observed during MANCHOT cannot be directly linked to higher ambient ozone levels, and are more likely to be dominated by phytoplankton abundance and productivity which can be influenced by seasonal variation in sunlight and temperature41.
Conclusion
Here we present several different lines of evidence for potentially marine sources of BTEX that are not currently included in model inventories and global budgets. The combined results of this and other recent studies are revealing increasing diversity in biogenic marine VOC emission profiles7,11,16,42,43, and complexity in ocean-atmosphere interactions that mediate their production and air-sea exchange13,38 as evidenced in the sensitivity of BTEX fluxes to changes in ambient levels of ozone explored here.
Given the extent of the global oceans, the magnitude of the marine BTEX fluxes reported in this study and their potential contribution to the reactive chemistry of the remote marine atmosphere (in particular OH chemistry and SOA formation)12,42,44, further observation and modeling studies of marine benzenoids are clearly warranted.
Methods
The Sea2Cloud voyage on the RV Tangaroa traversed a region in the Pacific Ocean off the coast of New Zealand, next to Chatham Rise (44°S, 174–181°E). The campaign took place from 17 to 27 March 2020. The regional context has been developed in Law et al.25 from the Surface Ocean Aerosol Production (SOAP) voyage 2012. Briefly, the Southern Pacific Ocean is an ideal area to study the impact of marine emissions, such as and aerosols and the inorganic and organic and precursors (e.g., VOCs) due to the presence of diverse and abundant phytoplankton populations in this region.
Air was sampled from four separate sampling points on the ship:
-
Ambient air was sampled via a ~5 m stainless steel inlet (OD 100 mm) inlet over the starboard-side of the boat.
-
Two Air-Sea Interface Tanks (ASITs) located on the rear deck of the ship.
-
ASITS headspace flush air was sampled upstream of the tanks (ASIT bypass).
The sample streams converged in a four-way valve system, located in the rear deck laboratory, which housed the trace gas-phase and aerosol monitoring instruments. The valve switched between the two ASITs, ambient air, and bypass every 20 min. After the valve, the sampled air was split between aerosol measurements and gas-phase instruments using ¼ inch Teflon tubing.
The sampling sites and instrumental descriptions are given briefly in the following sections, and detailed in Sellegri et al.14 Relevant to this study, ambient, ASIT, and ASIT bypass gas-phase species were continuously analysed by Proton-Transfer-Reaction-Mass-Spectrometry for volatile organic compounds (II-1-2), and for ozone and sulfur dioxide using a Thermo Fisher gas analysers (see II-1-4)
1 Instrumental description
1-1 Air-sea interface tanks (ASITs)
Two cylindrical 1.82 m3 chambers were built to capture and monitor aerosol and gases emissions of natural seawaters sampled during the voyage. The walls of the ASITs were covered with a Teflon film, while the lid was made out of PMMA that is chemically inert and UV transparent. The Air-Sea Interface Tanks (ASITs) were half filled with seawater from the ocean surface via an overside inlet and pump system. The ASITS headspace was continuously flushed with ambient air supplied from a common inlet located on the “monkey island” at the top of the boat’ consisting of 400 mm ECOLO Polyurethane Antistatic hose with a flow rate of 1000 L min−1). The ASITS was flushed with a sub-sample of this flow (23 L min−1), which was then passed through a filter to remove particles before entering the ASITs. Air was sampled from the ASITs via a 2.14 m, 3/8 inch OD stainless steel tube.
A four-way valve system was used to switch measurement between the two ASITs, ambient air, and bypass every 20 min. After the valve, the sampled air was split between aerosol measurements and gas-phase instruments using ¼ inch Teflon tubing.
A concentration of about 100 ppb of ozone was generated (MGC101, Environmental S.A., Poissy, France) and added continuously in one of the two tanks.
From 17 to 27 March, four experiments were performed. Each experiment lasted almost 2 days. Between experiments, the valve was switched to continuous ambient measurement, the ASITs were emptied, flushed for several hours, and re-filled and a steady state was established before sampling.
As an additional check, BTEX concentrations were also measured in three samples of the seawater ASITs using an extraction method (see II-1-3) (Figure S.6). The seawater concentrations observed in the ASITs agreed with the seawater concentrations measured from workboat samples taken off the ship, indicating that the ASIT’s seawater was similar to the surrounding natural seawater and was not contaminated by the ASITs and seawater sampling systems themselves.
1-2 Sea2Cloud VOCs measurements via PTR-MS
A dedicated PTFE/PFA line of about 5 m and ¼ inch linked the fou-way valve to the PTR-MS and other gas measurements (ozone and sulfur dioxide) with a sample flow of 1.5 L min−1. A Proton-Transfer Reaction-Mass Spectrometer (PTR-MS, Ionicon Analytik, Innsbruck, Austria) was used to measure VOCs during the voyage. This on-line technique is described in detail elsewhere45,46. Briefly, VOCs are chemically ionized via proton transfer from H3O + and the charged products are separated according to mass/charge (m/z) and detected with a quadrupole mass spectrometer. The raw ion signals in count per second (cps) were normalized to 106 H3O + ions and expressed as normalized count per second (ncps)47. The PTR-MS continuously scanned 16 selected m/z with a 1 sec dwell time, which equates to about 6 mass scans per 20 min valve switching cycle. The ion signals at m/z 79, m/z 93, and m/z 107 were selected to measure the protonated ion signals of benzene, toluene, and the sum of ethylbenzene + xylenes, respectively.
The instrument operating parameters were checked daily with an inlet temperature of 60 °C, a 600 V drift tube voltage, and a 2.0 mbar drift tube pressure. The ratio O2 + /H3O + was always <3%. Zero measurements were performed daily whereby the VOCS were removed from the sample air by passing it through a platinum wool catalyst at 300 °C. The zero measurements were performed for each ASITs, averaged, and subtracted from the ASITs sample ion signals. Daily calibrations were made with a ~1 ppm gravimetrically prepared mixture of VOC in nitrogen (acetaldehyde, methanol, acetonitrile, acetone, methacrolein (MACR), methyl ethyl ketone (MEK), benzene, toluene, m-xylene, and α-pinene) and a ~1 ppm mixture in nitrogen of isoprene and dimethyl sulfide (DMS) (Apel-Riemer Environmental, USA). For each cylinder the stated accuracy was ± 5%. Multipoint calibrations were achieved by stepwise addition (5–20 mL min−1) of the calibration standard to a flow 1500 mL min−1 of ambient air that had been passed through the zero furnace.
The empirically derived calibration factors for benzene, toluene, and m-xylenes were 7.6 ± 0.6, 7.1 ± 0.6, and 5.7 ± 0.4, ncps ppb−1, respectively. The minimal detection limit (MDL) was calculated as the 95th percentile of deviations from the mean zero. The detection limits for ASIT with and without ozone are summarized in the Table 1.
Minimum detection limits of benzene, toluene, and xylenes concentrations in ASITs were on average 0.060, 0.100, and 0.080 ppb, respectively. Table 1 provides the fraction of observations detected above the MDL in the ASIT-control, ASIT-O3, and ambient air data.
Identification and filtering of data impacted by ship exhaust
BTEX compounds are enhanced in ship exhausts from diesel combustion, waste incineration, and kitchens and must be carefully screened to ensure the quality of atmospheric data collected from marine research vessels. PTR-MS data from the four voyages reported in this study employed different exhaust screening methods as follows:
Sea2Cloud—Air masses impacted by ship and kitchen exhausts were identified using SO2 and the PTR-MS signal at m/z 57. When a spike of these compounds was observed in ambient and bypass air, data in the ASITs were excluded during the peak period and the following 2 h (3 x residence time in the ASIT’s headspace). In the ambient air, only spikes of high SO2 data were excluded, after it was checked that the BTEX concentrations decreased at the same rate as SO2, showing that no slow inlet desorption was occurring after ship stack contamination.
SOAP—a “baseline wind switch” was employed in the SOAP campaign which switched the PTR-MS sample to zero air when wind conditions made ingestion of ship exhaust emission likely. The data were further screened using CO2, aerosol, and VOC exhaust tracers to ensure complete removal of exhaust impacted air masses21.
MANCHOT—a CO filter correction was applied to the ambient PTR-MS data with time periods flagged for removal when a peak > 65 ppb of CO appeared.
CAPRICORN—the exhaust data filter described in Humphries et al.48 was applied to the PTR-MS data from CAPRICORN. The data were further screened using VOC exhaust tracers to ensure complete removal of exhaust impacted air masses.
1-3 VOCs measurements via GC-MS
Sorbent tubes (TENAX TA, 250 mg) were used to sample VOCs during the voyage principally in the ASIT without ozone. After collection of 4 L ambient air, they were desorbed via an auto-sampler Thermal Desorber (ATD TurboMatrix) and analysed with a gas chromatograph-mass spectrometer instrument (Clarus 600, Perkin Elmer, Waltham, MA). Benzene, Toluene, Ethylbenzene, (m + p)-xylene, and o-xylenes have been calibrated via a reference solution: MegaMix standard (200 µg/mL in methanol provided by Restek, Bellefonte, PA). The method of analysis was as follows: the column temperature was held at 35 °C for 5 min, increased at 5 °C/min to 160 °C, and then increased at 45 °C/min to 300 °C where it was held for 5 min.
The limit of detection (LOD) for the compounds were 20, 47, 5, 15, and 436 ppt for ethylbenzene, (m + p)-xylenes, o-xylenes, toluene, and benzene respectively. The precision is 5% and the accuracy 15%.
A good correlation was observed between PTR-MS and GC-MS BTEX measurements (Fig. S.7). The coefficient of correlation (R²) for benzene, toluene, and xylenes is 0.76, 0.98, 0.86 with a slope of 1.68, 1.69, and 0.81 (intercept = 441 ppt, −38 ppt and 18 ppt, respectively). Blank cartridge measurement (orange dot in the Fig. S.7) shows that residuals of benzene are present in the cartridges and explain the difference of benzene concentration measurement by PTR-MS and GC-MS.
A Stir Bar Sorptive Extraction (SBSE) technique was used to characterize VOC concentrations in seawater samples. This technique has previously been used to determine VOCs concentration in clouds water49 and is described therein. Briefly, the stir bar coated with polydimethylsiloxane (PDMS) was placed in 5 mL of seawater sample and stirred for 1 h. Then, the stir bar was analysed by GC-MS under the same conditions as mentioned for TENAX cartridges. Efficiencies of extraction of 15, 22, 30, 31, and 32% were calculated for benzene, toluene, ethylbenzene, (m + p)-xylenes, and o-xylenes, respectively. Seawater concentrations measured from the ASIT seawater were slightly lower than the seawater concentration measured in open water from the workboat on the same day, indicating that no contamination of these compounds occurred during the ASIT filling procedure or within the course of the experiment from the ASITs themselves.
The concentrations were on average 2.33 µg L−1(water) (B), 2.73 µg L−1 (water) (T), 0.36 µg L−1 (water) (E) and 0.46 µg L−1 (water) (X) (Figure S.6), confirming that measured fluxes originate from BTEX in the seawater. These seawater concentrations agreed with the seawater concentrations measured from workboat samples taken outside the ship, indicating that the ASIT’s seawater was not contaminated by the ASITs themselves or the headspace.
1-4 Additional measurements
Ozone and sulfur dioxide (SO2) have been continuously measured with a UV photometric analyser (TEI49i, Thermo Fisher Scientific, Waltham, MA USA) and a SO2 analyser using pulsed fluorescence (TEI43i, Thermo Fisher Scientific, Waltham, MA USA).
Meteorological parameters were recorded on the boat by an automatic weather station (AWS) mounted on top of the crow’s nest above the bridge.
2 Quantification of VOC concentrations and fluxes
2-1 PTR-MS quantification of VOCs
In the PTR-MS, the raw ion signals (counts per second, cps) are normalized to 106 cps (ncps) of the primary reagent ion (H3O + ) at measured at m/z 19. The concentrations of VOCs were calculated using the calibrated compound sensitivity (S) and the zero-corrected calibration signals (ncps). The sensitivity is derived from measurements of certified calibration gas mixtures diluted with VOC-free ambient air and have been calculated as follows:
Where I(X)zero.corr.cal is the zero-corrected ion signal in normalized count per second (ncps) at m/z = X during the calibration measurement, calculated as:
The concentration is obtained with the Eq. 3:
Where I(x)zero.corr.samp is the zero-corrected sample signal (in ncps) at m/z = X. Zero correction is performed by analogous procedure to Eq. 2 above.
2-2 ASIT Fluxes calculations
Calculation of the net sea-air flux in the ASITs experiments were performed as per Sinha et al.15 with the following equation:
Where FVOC is the flux of VOCs in the ASITs in µg m−2 s−1, Q is the flow rate of the bypass air into the mesocosm, A is the surface area of the seawater enclosed in the ASITs in m2, MVOC is the molecular weight of X compound in g mol−1, Vm is the molar gas volume in m3 kmol−1 (= 23.233 at 1015.25 hPa and 283 K) and [X]ASITS (ppb) = [X]ASITS (ppb) - [X]bypass (ppb), where [X]ASITS (ppb) is the concentration in the ASITs and [X]bypass (ppb) the concentration in the bypass.
2-3 Ambient air fluxes calculations using nocturnal boundary layer height
Ambient air fluxes calculation were performed via the following equation, described by Marandino et al. and employed in a previous cruise in this region in Lawson et al:21,22.
Where C is the concentration in ng/m3, dt difference of time between the measurement of the highest and the lowest concentration of DMS and hMBL the nocturnal Mixed Boundary layer (MBL) in m deduced from radiosonde measurements (range between 670 m and 1450 m for the whole campaign). Fambient VOC is the flux of VOCs in ambient air in ng m−2 s−1 deduced from nocturnal VOC measurements. These fluxes can be estimated based on the reasonable assumption of minimal oxidation of VOCs during the nighttime, which favors the nocturnal accumulation of primary VOCs. The highest level of VOC concentrations were observed at ~06h00 LT and the lowest at ~17h00 LT22.
Three nights without terrestrial influence were selected for the calculation: from 21 March 21 h LT to 22 March 06 h LT, from 22 March 20 h LT to 23 March 0 h LT and from 23 March 20 h LT to 6 h LT. For each night, the MBL was 1200 m, 670 m and 770 m, respectively.
These nights were selected as linear increase in BTEX concentration were observed over several hours.
Data availability
Datasets reported in this manuscript are available at the Sea2Cloud project data repository at https://sea2cloud.data-terra.org/en/catalogue/ or https://en.aeris-data.fr/metadata/?8d5b9e0e-1b0d-5208-afc1-fd0212ea97cd.
References
Simó, R. & Dachs, J. Global ocean emission of dimethylsulfide predicted from biogeophysical data. Global Biogeochem. Cycles. 16, 26-1–26–10 (2002).
Boucher, O. et al. DMS atmospheric concentrations and sulphate aerosol indirect radiative forcing: a sensitivity study to the DMS source representation and oxidation. Atmos. Chem. Phys. 3, 49–65 (2003).
Asher, E. C., Merzouk, A. & Tortell, P. D. Fine-scale spatial and temporal variability of surface water dimethylsufide (DMS) concentrations and sea-air fluxes in the NE Subarctic Pacific. Mar. Chem. 126, 63–75 (2011).
Lana, A., Simó, R., Vallina, S. M. & Dachs, J. Re-examination of global emerging patterns of ocean DMS concentration. Biogeochemistry. 110, 173–182 (2012).
Gantt, B. et al. A new physically-based quantification of marine isoprene and primary organic aerosol emissions Linking aerosol ‘types’ to their chemical composition View project GESAMP WG 38 ‘Changing Atmospheric Acidity and the Oceanic Solubility of Nutrients’ View project Atmospheric Chemistry and Physics A new physically-based quantification of marine isoprene and primary organic aerosol emissions. Atmos. Chem. Phys. 9, 4915–4927 (2009).
Myriokefalitakis, S. et al. Global Modeling of the Oceanic Source of Organic Aerosols. Adv. Meteorol. 2010, 16 (2010).
McKay, W. A., Turner, M. F., Jones, B. M. R. & Halliwell, C. M. Emissions of hydrocarbons from marine phytoplankton - Some results from controlled laboratory experiments. Atmos. Environ. 30, 2583–2593 (1996).
Brean, J. et al. Open ocean and coastal new particle formation from sulfuric acid and amines around the Antarctic Peninsula. Nat. Geosci. 14, 383–388 (2021).
Meskhidze, N. et al. Global distribution and climate forcing of marine organic aerosol: 1. Model improvements and evaluation. Atmos. Chem. Phys. 11, 11689–11705 (2011).
Colomb, A., Yassaa, N., Williams, J., Peeken, I. & Lochte, K. Screening volatile organic compounds (VOCs) emissions from five marine phytoplankton species by head space gas chromatography/mass spectrometry (HS-GC/MS). J. Environ. Monit. 10, 325–330 (2008).
Misztal, P. K. et al. Atmospheric benzenoid emissions from plants rival those from fossil fuels. Sci. Rep. 5, 1–10 (2015).
Ng, N. L. et al. Secondary organic aerosol formation from m-xylene, toluene, and benzene. Atmos. Chem. Phys. 7, 3909–3922 (2007).
Carpenter, L. J. & Nightingale, P. D. Chemistry and release of gases from the surface ocean. Chem. Rev. 115, 4015–4034 (2015).
Engel, A. et al. The ocean’s vital skin: toward an integrated understanding of the sea surface microlayer. Front. Mari. Sci. 4, 165 (2017).
Sellegri, K. et al. Sea2Cloud R/V Tangaroa voyage: from biogenic emission fluxes to Cloud properties in the South Western Pacific (Prep. BAMS, 2021).
Sinha, V. et al. Air-sea fluxes of methanol, acetone, acetaldehyde, isoprene and DMS from a Norwegian fjord following a phytoplankton bloom in a mesocosm experiment. Atmos. Chem. Phys. 7, 739–755 (2007).
Watts, M. C. & Bigg, G. R. Modelling and the monitoring of mesocosm experiments: two case studies. J. Plankt. Reasearch. 23, 1081–1093 (2001).
Atkinson, R. et al. Evaluated kinetic and photochemical data for atmospheric chemistry: volume II-gas phase reactions of organic species. Atmos. Chem. Phys 6, 3625–4055 (2006).
Yassaa, N., Colomb, A., Lochte, K., Peeken, I. & Williams, J. Development and application of a headspace solid-phase microextraction and gas chromatography/mass spectrometry method for the determination of dimethylsulfide emitted by eight marine phytoplankton species. Limnol. Oceanogr. Methods 4, 374–381 (2006).
Fasbender, L., Yáñez-Serrano, A. M., Kreuzwieser, J., Dubbert, D. & Werner, C. Real-time carbon allocation into biogenic volatile organic compounds (BVOCs) and respiratory carbon dioxide (CO 2) traced by PTR-TOF-MS, 13 CO 2 laser spectroscopy and 13 C-pyruvate labelling. PLoS ONE. 13, e0204398 (2018).
Lawson, S. J. et al. Methanethiol, dimethyl sulfide and acetone over biologically productive waters in the southwest Pacific Ocean. Atmos. Chem. Phys. 20, 3061–3078 (2020).
Marandino, C. A., De Bruyn, W. J., Miller, S. D. & Saltzman, E. S. Eddy correlation measurement of the air/sea flux of dimethylsulfide over the North Pacific Ocean. J. Geophys. Res. Atmos. 112, D03301 (2007).
Colomb, A. et al. Variation of atmospheric volatile organic compounds over the Southern Indian Ocean (3049°S). Environ. Chem. 6, 70–82 (2009).
Colomb, A., Paris, R., Losno, R., Desboeufs, K. & Provost, C. Continental background in oceanic air masses and marine emission of Volatile Organic Compounds in Drake Passage. Geophys. Res. Abstr. 12, 13988 (2010).
Law, C. S. et al. Overview and preliminary results of the Surface Ocean Aerosol Production (SOAP) campaign. Atmos. Chem. Phys. 17, 13645–13667 (2017).
Srikanta Dani, K. G. et al. Relationship between isoprene emission and photosynthesis in diatoms, and its implications for global marine isoprene estimates. Mar. Chem. 189, 17–24 (2017).
Achyuthan, K. E., Harper, J. C., Manginell, R. P. & Moorman, M. W. Volatile metabolites emission by in vivo microalgae—an overlooked opportunity? Metabolites. 7, 39 (2017).
Dani, K. G. S., Torzillo, G., Michelozzi, M. & Baraldi, R. & Loreto, F. Isoprene emission in darkness by a facultative heterotrophic green aAlga. Front. Plant Sci. 11, 598786 (2020).
Wirgot, N. et al. Metabolic modulations of Pseudomonas graminis in response to H2O2 in cloud water. Sci. Rep. 9, 1–14 (2019).
Phandee, S. & Buapet, P. Photosynthetic and antioxidant responses of the tropical intertidal seagrasses Halophila ovalis and Thalassia hemprichii to moderate and high irradiances. Bot. Mar. 61, 247–256 (2018).
Loreto, F. & Velikova, V. Isoprene produced by leaves protects the photosynthetic apparatus against ozone damage, quenches ozone products, and reduces lipid peroxidation of cellular membranes. Plant Physiol. 127, 1781–1787 (2001).
Behnke, K. et al. RNAi-mediated suppression of isoprene biosynthesis in hybrid poplar impacts ozone tolerance. Tree Physiol. 29, 725–736 (2009).
Ryan, A., Cojocariu, C., Possell, M., Davies, W. J. & Hewitt, C. N. Defining hybrid poplar (Populus deltoides x Populus trichocarpa) tolerance to ozone: identifying key parameters. Plant, Cell Environ. 32, 31–45 (2009).
Loreto, F. & Schnitzler, J. P. Abiotic stresses and induced BVOCs. Trends Plant Sci. 15, 154–166 (2010).
Sharkey, T. D., Wiberley, A. E. & Donohue, A. R. Isoprene emission from plants: Why and how. Ann. Bot. 101, 5–18 (2008).
Hewitt, C. N., Kok, G. L. & Fall, R. Hydroperoxides in plants exposed to ozone mediate air pollution damage to alkene emitters. Nature. 344, 56–58 (1990).
Ciuraru, R. et al. Photosensitized production of functionalized and unsaturated organic compounds at the air-sea interface. Sci. Rep. 5, 12741 (2015).
Zhou, S. et al. Formation of gas-phase carbonyls from heterogeneous oxidation of polyunsaturated fatty acids at the air-water interface and of the sea surface microlayer. Atmos. Chem. Phys. 14, 1371–1384 (2014).
Lelieved, J. et al. Increasing ozone over the Atlantic Ocean. Science. 304, 1483–1487 (2004).
Cooper, O. R. et al. Multi-decadal surface ozone trends at globally distributed remote locations. Elementa. 8, 23 (2020).
Rasconi, S., Winter, K. & Kainz, M. J. Temperature increase and fluctuation induce phytoplankton biodiversity loss – Evidence from a multi-seasonal mesocosm experiment. Ecol. Evol. 7, 2936–2946 (2017).
Taraborrelli, D. et al. Influence of aromatics on tropospheric gas-phase composition. Atmos. Chem. Phys. 21, 1–26 (2020).
Shaw, S. L., Gantt, B. & Meskhidze, N. Production and emissions of marine isoprene and monoterpenes: a eview. Adv. Meteorol. 2010, 1–24 (2010).
Lewis, A. C. et al. A larger pool of ozone-forming carbon compounds in urban atmospheres. Nature. 405, 778–781 (2000).
Blake, R. S., Monks, P. S. & Ellis, A. M. Proton-transfer reaction mass spectrometry. Chem. Rev. 109, 861–896 (2009).
Lindinger, W., Hansel, A. & Jordan, A. On-line monitoring of volatile organic compounds at pptv levels by means of Proton-Transfer-Reaction Mass Spectrometry (PTR-MS) Medical applications, food control and environmental research. Int. J. Mass Spectrom. Ion Process. 173, 191–241 (1998).
De Gouw, J. A. et al. Validation of proton transfer reaction-mass spectrometry (PTR-MS) measurements of gas-phase organic compounds in the atmosphere during the New England Air Quality Study (NEAQS) in 2002. J. Geophys. Res. 108, 4682 (2003).
Humphries, R. S. et al. Identification of platform exhaust on the RV Investigator. Atmos. Meas. Tech. 12, 3019–3038 (2019).
Wang, M. et al. Anthropogenic and biogenic hydrophobic VOCs detected in clouds at the puy de Dôme station using Stir Bar Sorptive Extraction: deviation from the Henry’s law prediction. Atmos. Res. 237, 104844 (2020).
Acknowledgements
This research received funding from the European Research Council (ERC) under the Horizon 2020 research and innovation programme (Grant agreement number − 771369). We would also like to acknowledge Māori iwi, hapū, whānau and Moriori who are manawhenua of the lands and waters across Aotearoa on which Sea2Cloud voyage was conducted.
Author information
Authors and Affiliations
Contributions
K.Se. and C.L designed the experiment. N.B., C.L., K.Se and M.P. conceived the ASITs. N.B, E.D, J.H, M.P., K.Se and C.L. set-up the ASITs experiments. M.R and M.P. collected atmospheric ASITs data. M.R., E.D. and A.C. analyzed BTX data. K.Sa., A. S-M., S. D. and A.M. provided Sea2Cloud phytoplankton data, A.C. and J.W. provide laboratory phytoplankton culture and MANCHOT data and E.D., J.H., SOAP and CAPRICORN data. M.R., K.Se. and A.C. wrote the original draft. Further writing of the manuscript were performed by M.R., E.D., J.W, C.L., A.C. and K.Se. All co-authors contributed to the revision of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Communications Earth & Environment thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Clare Davis.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rocco, M., Dunne, E., Peltola, M. et al. Oceanic phytoplankton are a potentially important source of benzenoids to the remote marine atmosphere. Commun Earth Environ 2, 175 (2021). https://doi.org/10.1038/s43247-021-00253-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-021-00253-0
This article is cited by
-
Atmospheric isoprene measurements reveal larger-than-expected Southern Ocean emissions
Nature Communications (2024)
-
Aerosolization flux, bio-products, and dispersal capacities in the freshwater microalga Limnomonas gaiensis (Chlorophyceae)
Communications Biology (2023)
-
Molecular rearrangement of bicyclic peroxy radicals is a key route to aerosol from aromatics
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
