
Abstract
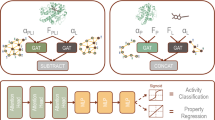
Protein kinases regulate various cellular functions and hold significant pharmacological promise in cancer and other diseases. Although kinase inhibitors are one of the largest groups of approved drugs, much of the human kinome remains unexplored but potentially druggable. Computational approaches, such as machine learning, offer efficient solutions for exploring kinase–compound interactions and uncovering novel binding activities. Despite the increasing availability of three-dimensional (3D) protein and compound structures, existing methods predominantly focus on exploiting local features from one-dimensional protein sequences and two-dimensional molecular graphs to predict binding affinities, overlooking the 3D nature of the binding process. Here we present KDBNet, a deep learning algorithm that incorporates 3D protein and molecule structure data to predict binding affinities. KDBNet uses graph neural networks to learn structure representations of protein binding pockets and drug molecules, capturing the geometric and spatial characteristics of binding activity. In addition, we introduce an algorithm to quantify and calibrate the uncertainties of KDBNet’s predictions, enhancing its utility in model-guided discovery in chemical or protein space. Experiments demonstrated that KDBNet outperforms existing deep learning models in predicting kinase–drug binding affinities. The uncertainties estimated by KDBNet are informative and well-calibrated with respect to prediction errors. When integrated with a Bayesian optimization framework, KDBNet enables data-efficient active learning and accelerates the exploration and exploitation of diverse high-binding kinase–drug pairs.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Data availability
The two kinase–drug binding-affinity datasets, Davis27 and KIBA28, were curated by and available in the Therapeutics Data Commons benchmark73. The PDBbind dataset (v.2020) was downloaded from http://www.pdbbind.org.cn/. The PDB codes of representative structures of kinases were obtained from the Kincore database50 (http://dunbrack.fccc.edu/kincore/home). The binding pocket structure of each kinase was downloaded from the KLIFS database20 (https://klifs.net/). Full AA sequences of kinases were obtained from UniProt56 (https://www.uniprot.org/). The 3D molecular structures were downloaded from PubChem57 (https://pubchem.ncbi.nlm.nih.gov/). Our processed version of the binding-affinity datasets and the identifier list of kinases and drugs are available on our GitHub repository (https://github.com/luoyunan/KDBNet).
Code availability
The source code of KDBNet is available at https://github.com/luoyunan/KDBNet and has been deposited to Zenodo74 at https://doi.org/10.5281/zenodo.7959829. KDBNet was developed using Python v.3.9, PyTorch v.1.16, PyTorch Geometric v.2.2, RDKit (v.2022.03.2), NumPy v.1.23.4 and SciPy v.1.9.3.
References
Oprea, T. I. et al. Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discov. 17, 317–332 (2018).
Attwood, M. M., Fabbro, D., Sokolov, A. V., Knapp, S. & Schiöth, H. B. Trends in kinase drug discovery: targets, indications and inhibitor design. Nat. Rev. Drug Discov. 20, 839–861 (2021).
Cohen, P., Cross, D. & Jänne, P. A. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat. Rev. Drug Discov. 20, 551–569 (2021).
Hanson, S. M. et al. What makes a kinase promiscuous for inhibitors? Cell Chem. Biol. 26, 390–399 (2019).
Arrowsmith, C. H. et al. The promise and peril of chemical probes. Nat. Chem. Biol. 11, 536–541 (2015).
Cichońska, A. et al. Crowdsourced mapping of unexplored target space of kinase inhibitors. Nat. Commun.12, 3307 (2021).
Bleakley, K. & Yamanishi, Y. Supervised prediction of drug–target interactions using bipartite local models. Bioinformatics 25, 2397–2403 (2009).
Cobanoglu, M. C., Liu, C., Hu, F., Oltvai, Z. N. & Bahar, I. Predicting drug–target interactions using probabilistic matrix factorization. J. Chem. Inf. Model. 53, 3399–3409 (2013).
Zheng, X., Ding, H., Mamitsuka, H. & Zhu, S. Collaborative matrix factorization with multiple similarities for predicting drug-target interactions. In Proc. 19th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining (eds Ghani, R. et al.) 1025–1033 (ACM, 2013).
Cichonska, A. et al. Computational-experimental approach to drug-target interaction mapping: a case study on kinase inhibitors. PLoS Comput. Biol. 13, e1005678 (2017).
Luo, Y. et al. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 8, 573 (2017).
Öztürk, H., Özgür, A. & Ozkirimli, E. Deepdta: deep drug–target binding affinity prediction. Bioinformatics 34, i821–i829 (2018).
Karimi, M., Wu, D., Wang, Z. & Shen, Y. Deepaffinity: interpretable deep learning of compound–protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 35, 3329–3338 (2019).
Tsubaki, M., Tomii, K. & Sese, J. Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics 35, 309–318 (2019).
Jiang, M. et al. Drug–target affinity prediction using graph neural network and contact maps. RSC Adv. 10, 20701–20712 (2020).
Nguyen, T. et al. Graphdta: predicting drug–target binding affinity with graph neural networks. Bioinformatics 37, 1140–1147 (2021).
Hie, B., Bryson, B. D. & Berger, B. Leveraging uncertainty in machine learning accelerates biological discovery and design. Cell Syst. 11, 461–477 (2020).
Rose, P. W. et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 45, gkw1000 (2016).
Van Linden, O. P., Kooistra, A. J., Leurs, R., De Esch, I. J. & De Graaf, C. KLIFS: a knowledge-based structural database to navigate kinase–ligand interaction space. J. Med. Chem. 57, 249–277 (2014).
Kanev, G. K., de Graaf, C., Westerman, B. A., de Esch, I. J. & Kooistra, A. J. KLIFS: an overhaul after the first 5 years of supporting kinase research. Nucleic Acids Res. 49, D562–D569 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with alphafold. Nature 596, 583–589 (2021).
Jing, B., Eismann, S., Suriana, P., Townshend, R. J. & Dror, R. Learning from protein structure with geometric vector perceptrons. Paper presented at the International Conference on Learning Representations (ICLR). (eds Oh, A., Murray, N. & Titov, I.) (2021); https://openreview.net/forum?id=1YLJDvSx6J4
Gainza, P. et al. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods 17, 184–192 (2020).
Lakshminarayanan, B., Pritzel, A. & Blundell, C. Simple and scalable predictive uncertainty estimation using deep ensembles. In Advances in Neural Information Processing Systems (NeurIPS) Vol 30 (eds Guyon, I. et al.) 6402–6413 (Curran Associates, Inc., 2017).
Zeng, H. & Gifford, D. K. Quantification of uncertainty in peptide-mhc binding prediction improves high-affinity peptide selection for therapeutic design. Cell Syst. 9, 159–166 (2019).
Soleimany, A. P. et al. Evidential deep learning for guided molecular property prediction and discovery. ACS Cent. Sci. 7, 1356–1367 (2021).
Davis, M. I. et al. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 (2011).
Tang, J. et al. Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis. J. Chem. Inf. Model. 54, 735–743 (2014).
Pahikkala, T. et al. Toward more realistic drug–target interaction predictions. Brief. Bioinform. 16, 325–337 (2015).
Goldman, S., Das, R., Yang, K. K. & Coley, C. W. Machine learning modeling of family wide enzyme-substrate specificity screens. PLoS Comput. Biol. 18, e1009853 (2022).
Singh, R., Sledzieski, S., Bryson, B., Cowen, L. & Berger, B. Contrastive learning in protein language space predicts interactions between drugs and protein targets. Proc. Natl Acad. Sci. 120, e2220778120 (2023).
Jiménez, J., Skalic, M., Martinez-Rosell, G. & De Fabritiis, G. K deep: protein–ligand absolute binding affinity prediction via 3D-convolutional neural networks. J. Chem. Inf. Model. 58, 287–296 (2018).
Townshend, R., Bedi, R., Suriana, P. & Dror, R. End-to-end learning on 3D protein structure for interface prediction. In Adv. Neural. Inf. Process. Syst. Vol 32 (eds Wallach, H. et al.) 15616–15625 (Curran Associate, Inc., 2019).
Townshend, R. J. et al. Atom3d: tasks on molecules in three dimensions. Preprint at https://arXiv.org/2012.04035 (2020).
Li, S. et al. Structure-aware interactive graph neural networks for the prediction of protein-ligand binding affinity. In Proc. 27th ACM SIGKDD Conference on Knowledge Discovery & Data Mining (eds Zhu, F., Ooi, B. C. & Miao, C.) 975–985 (ACM, 2021).
Liu, Z. et al. PDB-wide collection of binding data: current status of the PDBbind database. Bioinformatics 31, 405–412 (2015).
Lim, J. et al. Predicting drug–target interaction using a novel graph neural network with 3D structure-embedded graph representation. J. Chem. Inf. Model. 59, 3981–3988 (2019).
Zheng, L., Fan, J. & Mu, Y. Onionnet: a multiple-layer intermolecular-contact-based convolutional neural network for protein–ligand binding affinity prediction. ACS Omega 4, 15956–15965 (2019).
Zhou, J. et al. Distance-aware molecule graph attention network for drug-target binding affinity prediction. Preprint at https://arXiv.org/2012.09624 (2020).
Hassan-Harrirou, H., Zhang, C. & Lemmin, T. Rosenet: improving binding affinity prediction by leveraging molecular mechanics energies with an ensemble of 3D convolutional neural networks. J. Chem. Inf. Model. 60, 2791–2802 (2020).
Li, S. et al. Monn: a multi-objective neural network for predicting compound-protein interactions and affinities. Cell Syst. 10, 308–322 (2020).
Kuleshov, V., Fenner, N. & Ermon, S. Accurate uncertainties for deep learning using calibrated regression. In Proc. International Conference on Machine Learning (PMLR) (eds Dy, J. & Krause, A.) 2796–2804 (ACM, 2018).
Tran, K. et al. Methods for comparing uncertainty quantifications for material property predictions. Mach. Learn. Sci. Technol. 1, 025006 (2020).
Ali, K. et al. Inactivation of PI3K p110δ breaks regulatory t-cell-mediated immune tolerance to cancer. Nature 510, 407–411 (2014).
Angelopoulos, A. N. & Bates, S. Conformal prediction: a gentle introduction. Found. Trends Mach. Learn. 16, 494–591 (2023).
Bosc, N. et al. Large scale comparison of qsar and conformal prediction methods and their applications in drug discovery. J. Cheminform. 11, 4 (2019).
Levi, D., Gispan, L., Giladi, N. & Fetaya, E. Evaluating and calibrating uncertainty prediction in regression tasks. Sensors 22, 5540 (2023).
Song, H., Diethe, T., Kull, M. & Flach, P. Distribution calibration for regression. In Proc. International Conference on Machine Learning (PMLR) (eds Chaudhuri, K. & Salakhutdinov, R.) 5897–5906 (ACM, 2019).
PubChem3D release notes. PubChem https://pubchemdocs.ncbi.nlm.nih.gov/pubchem3d (2019).
Modi, V. & Dunbrack, R. Kincore: a web resource for structural classification of protein kinases and their inhibitors. Nucleic Acids Res. 50, D654–D664 (2022).
Zhou, G. et al. Uni-mol: a universal 3d molecular representation learning framework. In Proc. of the 11th International Conference on Learning Representations (eds Nickel, M. et al.) (OpenReview, 2023).
Lu, W. et al. Tankbind: trigonometry-aware neural networks for drug-protein binding structure prediction. In Advances in Neural Information Processing Systems Vol 35 (eds Koyejo, S. et al.) 7236–7249 (Curran Associates, Inc., 2022)
Luo, Y., Peng, J. & Ma, J. Next decade’s AI-based drug development features tight integration of data and computation. Health Data Sci. 2022, 9816939 (2022).
Burley, S. K. et al. RCSB protein data bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 49D437–D451 (2021).
Modi, V. & Dunbrack, R. L. Defining a new nomenclature for the structures of active and inactive kinases. Proc. Natl Acad. Sci. 116, 6818–6827 (2019).
Consortium, T. U. Uniprot: the universal protein knowledgebase in 2021. Nucleic Acids Res.49, D480–D489 (2021).
Kim, S. et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res.49, D1388–D1395 (2021).
Liu, Y., Palmedo, P., Ye, Q., Berger, B. & Peng, J. Enhancing evolutionary couplings with deep convolutional neural networks. Cell Syst.6, 65–74 (2018).
Ingraham, J., Garg, V., Barzilay, R. & Jaakkola, T. Generative models for graph-based protein design. In Proc. Advances in Neural Information Processing Systems 32 (eds Wallach, H. et al.) 15820–15831 (Curran, 2019).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. 118, e2016239118 (2021).
Luo, Y. et al. ECNet is an evolutionary context-integrated deep learning framework for protein engineering. Nat. Commun. 12, 5743 (2021).
Shaw, P., Uszkoreit, J. & Vaswani, A. Self-attention with relative position representations. In Proc. of the 2018 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 2 (Short Papers) (eds Walker, M., Ji, H. & Stent, A.) 464–468 (Association for Computational Linguistics, 2018).
Vaswani, A. et al. Attention is all you need. In Proc. Advances in Neural Information Processing Systems 30 (eds Guyon, I. et al.) 5998–6008 (Curran, 2017).
Shi, Y. et al. Masked label prediction: unified message passing model for semi-supervised classification. In Proceedings of the Thirtieth International Joint Conference on Artificial Intelligence (IJCAI) (2021).
Maas, A. L., Hannun, A. Y. & Ng, A. Y. Rectifier nonlinearities improve neural network acoustic models. In Proc. 30th International Conference on Machine Learning (ICML) (eds Dasgupta, S. & McAllester, D.) 3–8 (JMLR, 2013).
Ashukha, A., Lyzhov, A., Molchanov, D. & Vetrov, D. Pitfalls of in-domain uncertainty estimation and ensembling in deep learning. Paper presented at the 8th International Conference on Learning Representations (ICLR) (eds Song, D., Cho, K. & White, M.) (2020).
Eyke, N. S., Green, W. H. & Jensen, K. F. Iterative experimental design based on active machine learning reduces the experimental burden associated with reaction screening. React. Chem. Eng. 5, 1963–1972 (2020).
Roy, A. G. et al. Does your dermatology classifier know what it doesn’t know? Detecting the long-tail of unseen conditions. Med. Image Anal. 75, 102274 (2021).
Busk, J. et al. Calibrated uncertainty for molecular property prediction using ensembles of message passing neural networks. Mach. Learn. Sci. Technol. 3, 015012 (2021).
Chung, Y., Char, I., Guo, H., Schneider, J. & Neiswanger, W. Uncertainty toolbox: an open-source library for assessing, visualizing, and improving uncertainty quantification. Preprint at https://arXiv.org/2109.10254 (2021).
Brent, R. P. An algorithm with guaranteed convergence for finding a zero of a function. Comput. J. 14, 422–425 (1971).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Huang, K. et al. Therapeutics data commons: machine learning datasets and tasks for drug discovery and development. In Proc. Neural Information Processing Systems Track on Datasets and Benchmarks (eds Vanschoren, J. & Yeung, S.) (Conference on Neural Information Processing Systems, 2021).
Luo, Y. KDBNet: release v.0.1. Zenodo https://zenodo.org/record/7959829 (2023).
Acknowledgements
Y. Luo is supported in part by the National Institute of General Medical Sciences of the National Institutes of Health under award R35GM150890, the 2022 Amazon Research Award and the Seed Grant Program from the NSF AI Institute: Molecule Maker Lab Institute (grant no. 2019897) at the University of Illinois Urbana-Champaign. This work used the Delta GPU Supercomputer at NCSA of the University of Illinois Urbana-Champaign through allocation CIS230097 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) programme, which is supported by NSF grant nos. 2138259, 2138286, 2138307, 2137603 and 2138296. The authors acknowledge the computational resources provided by Microsoft Azure through the Cloud Hub program at GaTech IDEaS and the Microsoft Accelerate Foundation Models Research (AFMR) program.
Author information
Authors and Affiliations
Contributions
Y. Luo and J.P. conceived the research project. J.P. and Y. Luo supervised the project. Y. Luo developed the computational method and implemented the software. Y. Luo and Y. Liu performed the evaluation analyses. All authors analysed the results and participated in the interpretation. Y. Luo wrote the manuscript with support from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Tero Aittokallio and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Jacob Huth, in collaboration with the Nature Machine Intelligence team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Prediction performance evaluation on KIBA dataset.
(a) Four train-test split settings of evaluation, where the model is evaluated on data of unseen drugs (‘new-drug split’), unseen proteins (‘new-protein split’) or both (‘both-new split’), and unseen proteins with low (<50%) sequence identity (‘seq-id split’). (b) Comparisons of prediction performance of KDBNet with KronRLS, DeepDTA, GraphDTA, DGraphDTA, EnzPred, and ConPLex on the KIBA dataset using four train-test split settings. The performances of GP were not shown as it was not evaluated in the original study17 and it is computationally costly to run GP at the scale of KIBA dataset because of the high memory footprint of kernel computation. Performances were evaluated using three metrics, including Pearson correlation, Spearman correlation, and mean squared error (MSE) between predicted and true KIBA scores28. All bar plots represented the mean ± SD of evaluation results on five random train/test splits. Abbreviations: seq. id.: sequence identity.
Supplementary information
Supplementary Information
Supplementary Notes, Tables 1–3 and Figs. 1–5.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Luo, Y., Liu, Y. & Peng, J. Calibrated geometric deep learning improves kinase–drug binding predictions. Nat Mach Intell 5, 1390–1401 (2023). https://doi.org/10.1038/s42256-023-00751-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-023-00751-0
