
Abstract
Photocatalytic splitting of seawater for hydrogen evolution has attracted a great deal of attention in recent years. However, the poor energy conversion efficiency and stability of photocatalysts in a salty environment have greatly hindered further applications of this technology. Moreover, the effects of electrolytes in seawater remain controversial. Here we present electrolyte-assisted charge polarization over an N-doped TiO2 photocatalyst, which demonstrates the stoichiometric evolution of H2 and O2 from the thermo-assisted photocatalytic splitting of seawater. Our extensive characterizations and computational studies show that ionic species in seawater can selectively adsorb on photo-polarized facets of the opposite charge, which can prolong the charge-carrier lifetime by a factor of five, leading to an overall energy conversion efficiency of 15.9 ± 0.4% at 270 °C. Using a light-concentrated furnace, a steady hydrogen evolution rate of 40 mmol g−1 h−1 is demonstrated, which is of the same order of magnitude as laboratory-scale electrolysers.

Similar content being viewed by others


Main
The photocatalytic overall water splitting (POWS) reaction using particulate catalysts is widely recognized as a promising approach for solar hydrogen production, but its performance is greatly limited by the rapid recombination of photo-generated charge carriers, generally limiting the achieved solar-to-hydrogen conversion efficiencies (ηSTH) to lower than 5% (refs. 1,2,3). By alleviating the rate-limiting step of oxygen-vacancy regeneration at elevated temperatures, we have previously demonstrated that a local electric field (LEF) or a local magnetic field can promote the POWS reaction by facilitating charge separation, leading to a remarkable enhancement in performance4,5,6. Recently, increasing attention has been directed to the water splitting reaction using seawater7,8, as more than 95% of water resources on the Earth’s surface are in seas and oceans, not to mention that freshwater supplies are already depleted worldwide9, and the desalination of seawater greatly leads to increases in overall capital costs. Although the direct electrolysis of seawater has made great progress in recent years, there are major challenges that are yet to be overcome10,11,12. For example, the use of seawater compromises both the efficiency and stability of electrolysers, and the consumption of electricity further increases the cost of the H2 obtained from seawater electrolysis13,14,15,16,17. In sharp contrast, such challenges could be overcome by the photocatalytic splitting of seawater using robust photocatalysts with rationally designed band positions where the energy is provided by free solar light. However, in the limited number of photocatalytic seawater splitting systems reported so far, the use of sacrificial reagents is indispensable, which is non-sustainable and leads to the generation of carbon-containing by-products18,19,20. Also, the ionic species in seawater complicate the photocatalytic mechanism. Some early attempts at photocatalytic seawater splitting have demonstrated the negative effects of seawater on photocatalytic performance21,22. For example, Tian and colleagues showed that black phosphorus-based nanosheets split seawater at a much lower rate than with pure water due to passivation of the nanosheets by the ionic species23. However, on the contrary, it has been reported recently that the presence of Cl− ions could facilitate the reaction kinetics, resulting in improved photocatalytic activity24. Obviously, contradictory views regarding the effect of seawater are prevalent in this research field, so the role of these ionic species in seawater and their interactions with the catalyst particles need to be unravelled unambiguously.
In this Article we show that water with a high salt content can substantially promote the thermo-assisted POWS performance over a robust facet-charge-polarized N-doped titanium dioxide photocatalyst (N–TiO2) at 270 °C via electrolyte-assisted polarization, exhibiting steady evolution of H2 and O2 in a stoichiometric ratio without producing any by-products (such as Cl2, H2O2 and H2S). The photocatalytic activity is improved by a factor of four, and the internal quantum efficiency (IQE) from 575 to 850 nm is enhanced by six times in artificial Dead Sea water, as compared with pure water. Consequently, a high ηSTH of 20.2 ± 0.5% and an overall energy conversion efficiency of 15.9 ± 0.4% are achieved in this sacrificial reagent-free thermo-assisted photocatalytic seawater splitting system. A high H2 evolution rate of 246.9 l h−1 m−2 (∼40 mmol g−1 h−1) is demonstrated in a light-concentrated furnace, which is of the same order of magnitude as laboratory-scale electrolysers. The effects of the ionic species in seawater have also been elucidated: photo-generated electrons and holes migrate selectively to the (101) and (001) crystal facets upon photoexcitation. This then results in the selective adsorption of ions with the opposite charge. The adsorbed ions, in turn, generate a strong LEF without the use of any local-field promoters, prolonging the charge-carrier lifetime. A wide range of ionic species have been studied, showing that, apart from Cl− anions, cations like Na+ that are widely considered as inert in photocatalytic seawater splitting could also exert a strong polarization effect. Direct evidence of this electrolyte-assisted charge polarization has been provided with the help of various characterization techniques, including time-resolved photoluminescence (TRPL) spectroscopy, high-angle annular dark-field scanning transmission electron microscopy (HAADF–STEM), ambient pressure X-ray photoelectron spectroscopy (AP-XPS), density functional theory (DFT) calculations, and so on. Consequently, linear relationships between photocatalytic activity, charge-carrier lifetime and the electrolyte-assisted charge polarization energy have been identified. This electrolyte-assisted charge polarization can be a versatile technique that is not limited to seawater, but is also applicable in artificial salty solutions or waste water.
Results
Effects of ionic species at elevated temperature
Facet-controlled N–TiO2 nanostructures were fabricated and comprehensively characterized (Supplementary Fig. 1 and Supplementary Note 1). We have demonstrated that the POWS reaction can be promoted substantially at elevated temperatures, so other effects such as surface polarization effects can be better investigated under such conditions. More background information about high-temperature photocatalysis is available in refs. 4,5,6,25 and Supplementary Note 2. The POWS activity in this work was evaluated at 270 °C, which has been shown to be the optimal temperature for this system5,6,25. Comprehensive control experiments without light irradiation or heating were carried out, and non-photoactive materials such as Pt/Al2O3 were tested as well. All of these control experiments showed that this POWS reaction at elevated temperatures is indeed photocatalytic rather than thermally driven (Supplementary Table 1). In addition, the photocatalytic performance exhibits a linear response to the light-irradiation intensity (Supplementary Fig. 2). Pt nanoparticles (NPs, 1 wt%) were loaded onto the N–TiO2 as a co-catalyst (optimizations of metal loading and catalyst quantities are presented in Supplementary Figs. 3 and 4).
The role of elevated temperature in this photocatalytic system and its relationship with electrolyte effects were unravelled by isotopic experiments (Supplementary Note 2 and Supplementary Fig. 5). It has been recognized widely that the oxygen evolution reaction (OER) over metal-oxide catalysts can trigger the surface adsorbate evolution mechanism (AEM) and/or the lattice oxygen-mediated mechanism (LOM)26,27. Obviously, in the AEM, liquid H2O (OH−) is the only oxygen source, whereas in the LOM, both liquid H2O and lattice oxygen provide the oxygen source for the OER. In this study we used ∼50 wt% H218O in H216O as the reaction medium instead of normal deionized H216O (the exact amounts of H218O and H216O used are given in Supplementary Fig. 5), and the oxygen in the N–TiO2 catalyst was predominantly 16O. The natural 18O isotopic abundance of ∼0.2% has been considered27. The POWS reaction was performed at different temperatures (ranging from 150 to 270 °C). The evolved dioxygen (including 18O18O, 18O16O and 16O16O) was measured accurately using MS during the POWS reaction, to obtain the 18O/16O ratio of the evolved O2. This ratio was then compared with that of the original water used for the reaction. Obviously, if the OER triggers the AEM, the two ratios will be similar, whereas the LOM leads to a decrease in the 18O/16O ratio because the 16O in the N–TiO2 is involved in the OER. As shown in Supplementary Fig. 5, at each temperature from 180 to 270 °C, the 18O/16O ratio drops sharply within the first 1 h, during which period the LOM becomes increasingly important. Subsequently, the 18O/16O ratio increases slightly because the surface oxygen of the N–TiO2 is partly substituted by 18O from the water. Finally, a steady state is reached at ∼2 h, after which the 18O/16O ratio remains stable (Supplementary Fig. 5b). The study of the effect of different temperatures has shown that, as the reaction temperature increases, the steady-state 18O/16O ratio drops sharply, indicative of the increasing contribution of LOM to oxygen evolution (Supplementary Fig. 5c). In contrast, at the lower temperature of 150 °C, the 18O/16O ratio does not change obviously throughout the POWS reaction. All the above results indicate that the AEM is dominant for the OER at lower temperatures, and O2 is mainly generated from the adsorbed H2O/OH− species in this case. The contribution of LOM increases substantially as the reaction temperature increases over this redox material, to give a higher rate of water decomposition at elevated temperatures, as discussed already. If the POWS reaction is performed at a low temperature (for example, at room temperature), it will be difficult for the OER, the generation of surface oxygen vacancies and oxygen-vacancy regeneration by the lattice oxygen via the LOM process to take place. Accordingly, the slow AEM process for OER is dominant and becomes the rate-limiting step. As a consequence, the dramatic effect of electrolytes in promoting the charge-carrier lifetime will not be visible. Thus, to observe the electrolyte effects, it is essential to perform such photocatalysis at elevated temperatures.
To understand the effect of ionic species, NaCl aqueous solutions were initially studied, and a wide range of concentrations up to 6.0 mol l−1 were investigated (Fig. 1a). The POWS activity increases proportionally with NaCl concentration up to 3.0 mol l−1, showing a remarkable enhancement from 6.43 to 24.75 mmol g−1 h−1, then the enhancement becomes less notable when the concentration is further increased beyond 3.0 mol l−1. We detected no gaseous Cl2 formation after the POWS reaction (as determined by gas chromatography), presumably because the required potential for oxidizing Cl− ions is higher than what the N–TiO2 valence band (VB) could provide28. Also, if Cl− is oxidized to Cl2, this will inevitably lead to a pH increase of the resulting solution (equation (3)). For this reason, the pH was measured before and after the reaction, and showed no obvious change. Using titration, we also carefully analysed the chlorine balance before and after the reaction. The concentrations of Cl− before and after the POWS reaction agree with each other within experimental uncertainty, indicating that Cl− is not consumed during the reaction. To detect any potentially dissolved Cl2 and HClO, we further scrutinized the post-reaction solution. A potassium iodide solution was added to the post-reaction solution, together with starch as the indicator (details of the procedure are provided in the Methods). No colour change was observed, suggesting that there were no detectable oxidative Cl2 or HClO species in the solution. In addition, the continuous O2 evolution was monitored carefully during each reaction, showing a H2-to-O2 ratio of ∼2:1 throughout (Supplementary Fig. 6). The H2 and O2 evolution rates and the exact H2-to-O2 ratio, with errors, are presented in Supplementary Table 2. Such a stoichiometric ratio between H2 and O2 indicates the overall water-splitting nature of the reaction, and oxidation of Cl− or other side reactions can thus be neglected in the system. It should be emphasized that N2 was not detected in the gaseous phase, which suggests that the detected O2 is generated from the POWS reaction rather than as contamination from air. The absence of gaseous N2 also indicates that the stable doped N in the catalyst is not oxidized by holes during the reaction. Post-reaction XPS measurements of the catalyst showed a N content of 4.8 wt%, which matches that of the original catalyst, before the reaction, within experimental error. The absence of N oxidation is presumably due to the favourable kinetics of oxygen evolution over dinitrogen formation on the oxide-based catalyst at elevated temperatures. Subsequently, other pH-neutral electrolytes were also studied, all of which showed enhanced POWS performance at 270 °C compared to pure water (Supplementary Fig. 7 and Supplementary Table 2). However, the enhancement varied among the different electrolytes—this will be discussed in detail later.

a,c, POWS activity of the Pt/N–TiO2 tested in NaCl aqueous solutions (a) and artificial seawaters (c) at 270 °C. The POWS activity is defined as the H2 evolution rate (mmol h−1) per unit mass of photocatalyst, presented in units of mmol g−1 h−1. The central data points are the average of three independent measurements (n = 3). Error bars represent s.d. Yellow dots represent the data points. b,d, TRPL spectra of the 1 wt% Pt/N–TiO2 tested in NaCl aqueous solutions at 25 °C (the concentrations are 0, 1, 2, 3, 4, 5 and 6 mol l−1, respectively, increasing along the arrow in b) (b) and artificial seawaters (d). In TRPL measurements, the intervals between the consecutive measured laser pulses are tuned to ∼100 ns by a pulse picker to ensure that the interval is long enough to allow the excited charge carriers to fully relax to the ground states before the next pulse arrives.
TRPL spectroscopy was next used to investigate the separation of the photo-generated charge carriers in the photocatalyst (Fig. 1b). Two different decay components were observed in each case on fitting the TRPL spectra (Supplementary Table 3). The fast component can be attributed to the intrinsic recombination process of TiO2 in the bulk region, which is barely influenced by the salty solution. The slow decay component shows a positive correlation with the concentration of NaCl solution, so it is attributed to suppressed charge recombination due to the LEF near the surface, introduced by the ionic species. All the TRPL fittings are shown in Supplementary Fig. 8, and the fitting parameters are provided in Supplementary Table 3. In addition, the instrument response function (IRF) is shown in Supplementary Fig. 9, which shows an IRF width of 48 ps. Several artificial seawaters were subsequently prepared to simulate seawater in different areas of the world (Supplementary Table 4). The Dead Sea water, which had the highest salt content, showed the best photocatalytic activity of 34.04 mmol g−1 h−1, followed by Lop Nor water, which exhibited an activity of 29.97 mmol g−1 h−1. Red Sea water, which had the lowest salt concentration, corresponding to the global average (3.5 wt%, 0.6 mol l−1), gave a relatively lower activity of 9.98 mmol g−1 h−1. TRPL studies again showed that the charge-carrier lifetimes are prolonged in artificial seawaters, and the longest average electron/hole pair lifetime of 10.28 ns was observed in the artificial Dead Sea water (Fig. 1d and Supplementary Table 3). Obviously, the ionic species in the solutions and artificial seawaters substantially prolong the lifetime of the photo-generated charge carriers in the Pt/N–TiO2 photocatalyst, and lead to greatly enhanced photocatalytic performance. Natural seawater was also tested for POWS activity; excitingly, this showed an improved activity of 11.05 mmol g−1 h−1 (Fig. 1c).
Mechanism of charge separation
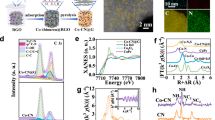
Figure 2a–c presents HAADF–STEM images of facet-controlled anatase N–TiO2 nanostructures. The percentages of exposed (101) and (001) facets were estimated to be 40% and 60%, respectively, according to the Wulff construction29. Pt or CoOx NPs were loaded onto the N–TiO2 through facet-selective photo-deposition of their soluble Pt(II) or Co(II) precursors, respectively30,31. It has been demonstrated that, upon light irradiation, photoexcited electrons/holes migrate to the surfaces of semiconductor particles and reduce/oxidize the soluble precursors in the solution, forming deposited NPs31,32. Therefore, these NPs could act as indicators of the photo-generated electrons and holes resident on these catalyst surfaces. The STEM images, together with energy X-ray dispersive analysis (STEM–EDS), clearly show that CoOx NPs are dominantly deposited on the exposed (001) facets (Fig. 2e and Supplementary Fig. 10a,b). On the other hand, statistical analysis shows that 85 ± 7% of the Pt NPs are preferentially found on the (101) facets (Supplementary Fig. 10 and Fig. 2d). These results clearly show that the photo-generated electrons and holes are preferentially trapped on the (101) and (001) facets of the N–TiO2, respectively. This was also supported by our DFT-calculated band-edge positions for the two facets of N–TiO2 (Supplementary Fig. 11): the energy difference of the VB is 1.16 eV and that of the conduction band is 0.79 eV, and this could effectively facilitate anisotropic migration of the charge carriers towards different facets. Therefore, it is anticipated that Na+ and Cl− ions will be preferentially adsorbed on the respective facets by electrostatic attractions, similar to the behaviour observed for the Pt(II) and Co(II) precursors.

a–c, Low-magnification (a) and high-magnification (b,c) HAADF–STEM images showing the morphology of N–TiO2. The high-magnification images also exhibit lattice fringes with d-spacings of 0.237 and 0.352 nm, in accordance with the [004] and [101] crystallographic planar directions of anatase TiO2, respectively, indicating the preferential exposure of the (001) and (101) facets. d,e, HAADF–STEM images of N–TiO2 loaded with Pt NPs and CoOx NPs via the facet-selective photo-deposition method. Schematic illustrations are given as insets. STEM–EDS mapping of the CoOx/N–TiO2 sample is also given in e (scale bars, 5 nm). More microscopic images and EDS mappings are provided in Supplementary Fig. 10.
AP-XPS studies were carried out at 270 °C to investigate the surface electronic features of the N–TiO2 using trimethylphosphine (TMP) as a surface probe (Supplementary Note 3)33. When TMP is adsorbed on N–TiO2, the Ti 2p signal shifts to a lower binding energy (BE; Fig. 3a), because TMP is a Lewis base, rendering the surface Ti4+ electron-rich by forming adducts, while the O 1s signal remains the same (Fig. 3b). Under LED white-light illumination, the Ti 2p signal shifts slightly to a lower BE and that of O 1s to a higher BE (Fig. 3a,b, yellow lines), in accordance with the literature34, because the electrons are excited from the O-dominated VB to the Ti-dominated conduction band. The shifts in these signals are recovered when the light is off, suggesting that the BE shifts are due to photoexcitation rather than any sample damage or irreversible surface reconstruction. It should be noted that the Ti 2p and O 1s spectra include both surface and bulk electronic features, given the XPS sampling depth of several nanometres into the bulk region35. In contrast, the P 2p spectra only reveal surface features, because the TMP probe molecules are adsorbed only on the topmost surface of the photocatalyst particle. Doublet signals are identified in the P 2p spectra, which are attributed to the P 2p3/2 and P 2p1/2 of the surface-adsorbed TMP molecules when illumination is absent (Fig. 3c). Upon illumination, two sets of doublet peaks appear through deconvolution of the spectrum: one set shifts to a higher BE, and the other to a lower BE (Supplementary Table 5). Again, this change of P 2p spectra is fully reversible when the illumination is removed (Fig. 3c, green line). The N 1s spectra remain almost the same throughout the measurements, indicating that the N doping is stable against TMP and illumination. Obviously, the above results clearly indicate that the (001) facets become positively charged under illumination due to hole trapping, shifting the P 2p signal to a higher BE; in contrast, those adsorbed on the electron-rich (101) facets shift to a lower BE. Quantitative analysis suggests that the area ratio of the two sets of doublet peaks is ∼40:60, which matches the ratio of (101) and (001) facets, confirming the attribution of the signals. Control experiments were performed on N–TiO2 powders with 95% (101) facet exposure: the AP-XPS results did not indicate any observable surface polarization effect (Supplementary Fig. 12). Similar AP-XPS experiments were performed at different temperatures, and all showed the same phenomenon of charge-carrier polarization on the facets (Supplementary Fig. 13).

a–d, Ti 2p (a), O 1s (b), P 2p (c) and N 1s (d) spectra under different conditions. All spectra were obtained at 270 °C. The measurements began by acquiring the spectra under vacuum in the dark (red line), then 0.2 mbar of TMP vapour was directed into the measurement chamber and the spectra were obtained without (blue line) and with (yellow line) LED white-light irradiation, respectively. Finally, the illumination was turned off again (green line) to ensure there was no permanent change to the material. Deconvoluted XPS components are presented in grey lines and shaded with orange and blue colours for a clearer view.
DFT calculations were next carried out to understand the effects of the ionic species in seawater. The electronic structures of the surface models are provided in Supplementary Figs. 14–17. Dipole correction was not considered in the calculations as it has a trivial effect on the total energy and electrostatic potential (Supplementary Fig. 18). Anions and cations were placed on the N-doped anatase TiO2(001)-(1 × 4) and (101) surfaces (Fig. 4a,b), respectively, to investigate their adsorption behaviours. We systematically investigated the possible sites for ion adsorption on the N–TiO2 surfaces. For example, we found that Na+ prefers to adsorb at the bridge sites of two adjacent O atoms on the N–TiO2(101) surface, and the adsorption energies of Na+ on these sites are almost identical. Cl− prefers to adsorb at the topmost four-coordinated Ti site of the N-doped TiO2(001)-(1 × 4) surface (Supplementary Fig. 19). The findings for Na+ and Cl− adsorption also apply to other cations and anions (Supplementary Figs. 20–25). The coordinates of the systems studied are provided as Supplementary Data 1. The adsorption energy (Eads) of the electrolyte can be calculated by taking the sum of the adsorption energies of the corresponding anion and cation. Holes and electrons are added to the surface models deliberately to simulate charge polarization upon excitation. Apparently, the intensity of the charge polarization effect induced by electrolytes can be evaluated by the change in the Eads of the electrolytes upon excitation, herein defined as the electrolyte-assisted charge polarization energy (Eecp). The calculated Eecp values show that all the considered electrolytes can exert a charge polarization effect to different extents, depending on the ionic radius and charge, because the nature of this effect is the electrostatic interaction between charge carriers (Supplementary Table 8). The electrostatic interaction energies between the ions and the N dopant that can trap the charge carriers, that is, electrons on (101) or holes on (001)-(1 × 4), were calculated. The electrostatic interaction energy differences of the corresponding charged and charge-neutral systems were all negative and dependent on the valence state of the ions (Supplementary Table 9), consistent with the calculated ion-induced charge polarization energies (Supplementary Table 8). We also calculated charge density difference plots for the ion adsorptions on the N-doped TiO2(101) and N-doped TiO2(001)-(1 × 4) surfaces. The plots show charge transfer between the ions and the surfaces, and the charge transfer in a charged system is generally more notable than that in a charge-neutral system (Supplementary Figs. 26–31), consistent with the calculated Bader charges and spin-density differences. All these results clearly indicate mutual stabilization between the charge carriers and the ions. In other words, the charge polarization effect leads to the accumulation of cations and anions on the (101) and (001) surfaces, respectively, and the adsorbed ions in turn further promote the charge polarization. Figure 4c,d shows that, as the polarization effect becoming stronger (that is, more negative Eecp), the charge carriers’ lifetime is greatly prolonged, resulting in enhanced POWS activity, and linear correlations are observed. In the case of the artificial seawaters, both the POWS activity and the charge carriers’ lifetime, again, exhibit linear correlations with the Eecp (Fig. 4e,f). Undoubtedly, the strong interaction between the ionic species and the polarized surfaces plays an important role in our POWS system at elevated temperatures, which facilitates surface trapping of the photo-generated charge carriers, resulting in a prolonged lifetime of the charge carriers (also supported by TRPL in Fig. 1). In addition, the activity exhibits a positive linear correlation with the charge carriers’ lifetime (Fig. 5a,b). The prolonged lifetime allows more charge carriers to react with the H+ and OH− to promote H2 and O2 evolutions, hence leading to promoted POWS activity.

a,b, Structures of the N-doped anatase TiO2(001)-(1 × 4) (a) and N-doped anatase TiO2(101) surfaces (b) used for DFT calculations. The lattice parameters of anatase TiO2 were optimized using a dense Γ-centred 6 × 6 × 3 k-point mesh. The calculated values of the lattice parameters are a = b = 3.878 Å and c = 9.530 Å, in good agreement with the experimentally obtained values (a = b = 3.784 Å; c = 9.512 Å)60. More details are shown in Supplementary Tables 6 and 7. c,d, Correlations between POWS activity (c) and lifetime of the slow decay component (d) with the calculated Eecp for different electrolytes. e,f, Correlations between POWS activity (e) and lifetime of the slow decay component (f) with the calculated total Eecp for different artificial seawaters. The central data points are the average of three independent measurements (n = 3). Error bars represent s.d. Details of the Eecp calculations are provided in Supplementary Table 8.

a,b, Relationship between POWS activity and the lifetime of the photo-generated charge carriers in NaCl solutions (a) and artificial seawaters (b). M represents molarity. c, Schematic illustration of the electrolyte-assisted charge polarization among different facets of the facet-controlled N–TiO2 in seawater upon photoexcitation. The reconstruction of the (001) surfaces is not indicated, for simplicity, but is demonstrated by DFT calculations. According to the HAADF–STEM, AP-XPS and DFT results, surface polarization takes place upon photoexcitation, which facilitates the adsorption of cations on the exposed (101) facets and anions on the (001) facets. Such facilitated surface adsorption, in turn, attracts electrons and holes by the Coulomb force, suppressing charge recombination and prolonging the charge carriers’ lifetime. d, QE evaluations of the 1 wt% Pt/N–TiO2 in pure water and the artificial Dead Sea water at different wavelengths at 270 °C. Error bars indicate s.d. A high average QE over the whole visible regime was achieved in artificial Dead Sea water, without the typical sharp decline at long wavelengths. e, IQE enhancement percentage [(IQEDead Sea − IQEpure water)/IQEpure water] and IQE difference (IQEDead Sea − IQEpure water) for different wavelengths at 270 °C, derived from d. f, Five-cycle stability test of 1 wt% Pt/N–TiO2 in artificial Dead Sea water. g, Light-concentrated furnace mimicking a solar furnace to provide heat and light without energy input from other electrical heating devices, showing an enhanced activity of 40.24 mmol g−1 h−1 over 20 h, which equates to a H2 evolution rate of ∼246.9 l h−1 m−2 at standard temperature and pressure, given that the illuminated area was 0.785 cm−2 and 20 mg of the photocatalyst was used in this experiment. In this figure, all error bars indicate s.d., and the central data points for the error bars are the average of three independent measurements (n = 3). Yellow dots represent data points.
Clearly, all the above results demonstrate that, in our seawater splitting system—where the photocatalyst particles are surrounded by plenty of ionic species—the facet-selective surface adsorption can take place readily due to facet polarization under constant illumination. In turn, the selectively adsorbed ionic species promote the separation of the photo-generated charge carriers via electrostatic attraction, resulting in an enhanced POWS performance (Fig. 5c). We also studied the effect of ion adsorption on the workfunction and band alignment of the N-doped TiO2 surfaces, and we found that water splitting can be further accelerated after the adsorption of electrolytes (Supplementary Figs. 32–35).
Photocatalytic performance evaluation
Figure 5d shows that the IQE of Pt/N–TiO2 is also remarkably enhanced by the polarization effect. A high IQE is obtained in both artificial Dead Sea water and pure water at 385 nm and 440 nm, but it drops sharply in pure water at longer wavelengths from 575 nm to 850 nm, in accordance with previous reports5,6,25. Such sharp IQE attenuation is attributed to wavelength-dependent electron–hole pair generation: the absorbed photons at a longer wavelength may not generate excited electron–hole pairs, but instead excite local transitions36,37. In addition, short-wavelength excitation leads to better charge separation and stronger electron–phonon coupling38,39,40, while the long-wavelength excitation leads to enhanced local polaron formation, decreasing the mobility of the charge carriers and resulting in fewer carriers reaching the catalyst surface41,42. When artificial seawater is used, it is noted that the IQE at short wavelengths is influenced less, because a substantial number of charge carriers with intrinsic excess energy can reach the surface. However, a higher IQE at longer wavelengths (575–850 nm) can be preserved due to the above-demonstrated polarization effect induced by electrolytes. Consequently, a high IQE of ∼50% was recorded even at 850 nm (Fig. 5e). It is also shown that there is not much difference in the UV–vis absorption of pure water and artificial Dead Sea water (Supplementary Fig. 36). Details of the QE calculations and external QE (EQE) are given in Supplementary Table 10 and Supplementary Note 4. The ηSTH was then evaluated, and a ηSTH of 20.2 ± 0.5% was achieved from this particulate POWS system in artificial Dead Sea water at 270 °C (Supplementary Note 4 and Supplementary Table 11); to the best of our knowledge, this is among the best reported for particulate POWS systems, and even exceeds those reported for many photoelectrochemical (PEC) and photovoltaic-electrolysis systems (Supplementary Table 12). This ηSTH is also well over the goal (10%) for practical applications of POWS systems imposed by the United States Department of Energy43. The Pt/N–TiO2 photocatalyst shows a stable evolution of H2 and O2, without any obvious activity drop over five cycles (Fig. 5f).
We then evaluated the overall energy conversion efficiency (ηOEC), in which the energy input for heating the reaction system is also considered. A very promising ηOEC of 15.9 ± 0.4% was demonstrated over 8 h (details are provided in Supplementary Note 5). Although this is a little lower than the ηSTH after considering the energy input for heating and maintaining the reactor temperature, this ηOEC of 15.9 ± 0.4% is still superior to all the recently reported values in the literature in related fields (Supplementary Table 12)44,45,46. Also, in a future design that we anticipate, energy for heating could be provided by concentrated solar light (Supplementary Note 2), so the energy consumption for heating the water and reactor could be minimized or even excluded in that scenario. In addition, superheated steam in a flow system could be fed into a steam turbine generator for energy recovery. By considering all these potential designs, a higher ηOEC could be expected.
Furthermore, we attempted to demonstrate the practical feasibility of the above-mentioned solar heating. A laboratory-scale light-concentrated furnace was used to mimic a solar furnace (Fig. 5g), showing that stable and efficient POWS activity at 270 °C could be maintained solely by the concentrated light without any other light sources or heating devices. A more exceptional activity of 40.24 mmol g−1 h−1 was observed over 20 h due to the high intensity of the concentrated light. This value is equivalent to a H2 evolution rate of ∼246.9 l h−1 m−2 at standard temperature and pressure, which is comparable in order of magnitude to the values reported for alkaline electrolysers (Supplementary Table 12), but this particulate POWS system is a direct single-step light-harnessing process with all thermal and photon energy inputs coming from the solar concentrator. Control experiments were carried out by covering the windows of the reactor with carbon paste to block light irradiation from illuminating the photocatalyst; these did not exhibit any H2 or O2 evolution, indicating that the water splitting reaction in the light-concentrated furnace is photocatalytic, and this reaction cannot be accomplished without light irradiation.
Discussion
In summary, we have demonstrated a facile and versatile technique of electrolyte-assisted charge polarization in water with high salt content, which facilitates the separation of photo-generated charge carriers on different facets of N–TiO2, leading to a dramatically enhanced stoichiometric evolution of H2 and O2 through thermal-assisted photocatalytic water splitting without a sacrificial reagent. This electrolyte-assisted charge polarization has been investigated using various techniques, and the quantitative analyses showed that both the photocatalytic performance and the charge-carrier lifetime linearly correlate with the electrolyte-assisted charge polarization energy. The obtained steady H2 evolution in the laboratory-scale solar furnace demonstrates an evolution rate of 40.24 mmol g−1 h−1. A recent life-cycle assessment has warned of environmental concerns about water electrolysis for large-scale H2 production9. Therefore, this direct single-step light-harnessing POWS system could provide an alternative option. It is believed that this study represents a pragmatic strategy for harvesting solar energy by splitting the more abundant saltwater resource.
Methods
Synthesis of N-doped TiO2 photocatalysts
The synthesis of the facet-controlled TiO2 nanocrystals was adopted from our previous work47: 5.0 ml of titanium butoxide, Ti(OC4H9)4, was mixed with 0.6 ml of hydrofluoric acid (48 wt%) in a 50-ml Teflon-lined autoclave and subsequently heated to 180 °C at a rate of 5 °C min−1 and maintained for 24 h. The obtained white precipitate was washed with ethanol and deionized water three times, respectively, and then dried in an oven at 80 °C overnight. For nitrogen doping, 200 mg of the facet-controlled TiO2 was typically placed in a quartz boat, which was then transferred to a tubular furnace. The sample was then heated to 600 °C at a rate of 5 °C min−1 and kept for 2 h under NH3 flow, after which it was allowed to cool naturally and a dark blue powder of N–TiO2 was collected. The N–TiO2 photocatalyst, unless stated otherwise, was loaded (via a photo-deposition method) with 1 wt% Pt NPs as the H2 evolution co-catalyst, before photocatalytic tests, as described in the next section.
Facet-selective photo-deposition on N–TiO2
CoOx and Pt NPs were loaded onto the facet-controlled N–TiO2 via a photo-deposition method adopted from the literature30,31. Typically, 0.5 g of the above-prepared N–TiO2 powder was suspended in 80 ml of NaIO3 solution (0.01 M), after which a certain amount of Co(NO3)2 solution (1 mg ml−1) was added into the suspension, followed by irradiation (300 W, Xe arc lamp) under vigorous stirring for 3 h. The suspension was filtered, washed with deionized water, and finally dried at 80 °C overnight. For Pt deposition, 0.05 g of the as-prepared N–TiO2 powder was suspended in a methanol (20 ml) and water (60 ml) mixed solution, after which H2PtCl6 solution (1 mg ml−1) was added into the suspension under continuous magnetic stirring, followed by irradiation (300 W, Xe arc lamp) for 0.5 h. The suspension was filtered, washed with distilled water, and finally dried at 80 °C overnight.
Material characterizations
Atomic-resolution STEM–HAADF images were obtained on a double spherical aberration-corrected S/TEM FEI Titan G2 60–300 system at 300 kV with a field-emission gun. The probe convergence angle on the Titan electron microscope was 24.5 mrad, and the angular range of the HAADF detector was from 79.5 mrad to 200 mrad. STEM–EDS mapping results were obtained by a Bruker Super-EDX 4 detector system.
The study of charge separation and distribution on the surface was performed by AP-XPS measurements using TMP as a surface probe, at the Diamond Light Source B07 beamline. The powdered samples were dispersed in ethanol and deposited on a silicon wafer, then heated by a heating plate at 140 °C to remove the solvent. Measurements were performed at different temperatures ranging from 150 to 270 °C. TMP vapour (0.2 mbar) was introduced into the experimental chamber as the surface probe when required. Spectra were acquired with and without LED white-light illumination (50 mW cm−2) to study the effect of photoexcitation. Ti 2p, O 1s, P 2p and C 1s spectra were recorded using a fixed photon energy of 950 eV. The BE was then calibrated with respect to the Fermi level.
Details of DFT calculations
All spin-polarized DFT calculations were carried out using the Vienna ab-initio Simulation Package (VASP)48. Electronic exchange and correlation were treated within the generalized gradient approximation (GGA) by using the Perdew–Burke–Ernzerhof (PBE) functional49. The projector-augmented wave (PAW) method50 with an energy cutoff of 400 eV was employed to describe the interactions between the core and valence electrons, with the Ti (3s, 3p, 3d, 4s), O (2s, 2p), N (2s, 2p), Na (3s), Mg (3s), K (3s, 3p, 4s), Ca (3s, 3p, 4s), S (3s, 3p) and Cl (3s, 3p) shells being treated as valence electrons. For all calculations, we applied the on-site Coulomb interaction correction with an effective U value of 4.2 eV on the Ti 3d orbitals to describe the localized electronic states accurately51,52.
The N-doped anatase TiO2(101) and N-doped reconstructed anatase TiO2(001)-(1 × 4) surfaces were constructed53. The N-doped anatase TiO2(101) surface was extended as a (2 × 2) cell, and the slab contained four TiO2 layers. A 2 × 3 × 1 k-point mesh was used for calculations. The N-doped reconstructed anatase TiO2(001)-(1 × 4) surface was extended as a (2 × 1) cell, and the slab contained eight TiO2 layers. A 3 × 2 × 1 k-point mesh was used for calculations. We set a vacuum layer of ∼15 Å to avoid interactions between neighbouring slabs. Geometry optimizations were converged when the Hellman–Feynman forces of the relaxed atoms were less than 0.02 eV Å−1. The relative energies of the different N-doped TiO2(101) and N-doped TiO2(001)-(1 × 4) surfaces with varied positions of N were calculated to locate the corresponding most favourable doping site of N (Supplementary Tables 6 and 7).
To simulate the ionic species and the photo-generated electrons/holes in the system, the NELECT (number of electrons) parameter was set to control the number of electrons in such a system. This is common practice in the literature54,55,56,57,58. For example, one excess electron was injected into the system to simulate Cl−, and one electron was pulled out of the system to simulate Na+. To simulate the N-doped anatase TiO2(101) and the N-doped reconstructed anatase TiO2(001)-(1 × 4) surfaces upon photoexcitation, that is, the N-doped anatase TiO2(101) with photo-electrons and the N-doped reconstructed anatase TiO2(001)-(1 × 4) with holes (that is, charged cases), we respectively increased and decreased the number of electrons in the systems by one. Atomic charges and the spin-density difference of each system were carefully checked to ensure that the ionic species and photo-electrons/holes were correctly simulated (Supplementary Figs. 14–35).
The adsorption energy (Eads) of the electrolytes was calculated as follows:
where \({E}_{{\rm{anion}}/001}\), \({E}_{{\rm{anion}}/{\rm{water}}}\), \({E}_{{\rm{cation}}/101}\) and \({E}_{{\rm{cation}}/{\rm{water}}}\) are, respectively, the calculated total energies of the N-doped reconstructed anatase TiO2(001)-(1 × 4) surface slab with the anion (for example, Cl−) being adsorbed on the surface, the N-doped reconstructed anatase TiO2(001)-(1 × 4) surface slab with the anion being placed in the middle of the water region of the model, the N-doped anatase TiO2(101) surface slab with the cation (for example, Na+) being adsorbed on the surface, and the N-doped anatase TiO2(101) surface slab with the anion being placed in the middle of the water region of the model. Note that the water environment was treated implicitly using the VASPsol code with a dielectric constant of 78.4 (ref. 59). The first part on the right-hand side (that is, \({E}_{{\rm{anion}}/001}-{E}_{{\rm{anion}}/{\rm{water}}}\)) is the adsorption energy of an anion on the N-doped reconstructed anatase TiO2(001)-(1 × 4) surface, and the second part on the right-hand side (that is, \({E}_{{\rm{cation}}/101}-{E}_{{\rm{cation}}/{\rm{water}}}\)) is the adsorption energy of a cation on the N-doped anatase TiO2(101) surface. If the two surfaces are charge-neutral/charged, the \({E}_{{\rm{ads}}}\) of electrolytes before and after excitation (that is, \({E}_{{\rm{ads}}}({\rm{before\; excitation}})\) and \({E}_{{\rm{ads}}}({\rm{after\; excitation}})\)) can be obtained.
The electrolyte-assisted charge polarization energies (Eecp) can be calculated as
A negative Eecp indicates that the charge polarization effect is enhanced by the electrolyte.
Photocatalytic tests
The POWS reaction was carried out in a 20-ml stainless-steel batch reactor equipped with two quartz windows (10-mm inner diameter (i.d.) and 18-mm thickness each)4,25. In a typical experiment, 5 mg of catalyst was added to 5 ml of aqueous solution (pure water, artificial seawater or natural seawater) in a quartz lining (20-mm i.d. × 24-mm outer diameter × 52-mm height) under magnetic stirring (750 r.p.m.), then the batch reactor was purged with a continuous Ar gas flow for 5 min, after being well-sealed, to remove the dissolved O2. The batch reactor was then pressurized with 6 bar of inert Ar gas. The reactor was then allowed to heat up to a certain elevated temperature with saturated water vapour pressure. A VeraSol solar simulator (AM 1.5 G, 100 mW cm−2) was applied to provide simulated solar irradiation through the quartz windows. The batch reactor was cooled naturally to room temperature after the reaction, and the amounts of oxygen and hydrogen were measured by a gas chromatograph (GC) equipped with two thermoconductivity detectors (TCDs) with He and N2 as carrier gas, respectively, for better sensitivity. GC analysis was also carried out before the reactions to ensure that the air and dissolved O2 were completed removed. The GC-TCD system was calibrated using an external standard method, so the peak area of the GC signal could be converted to the amount of gas using the calibration curve.
When demonstrating the feasibility of using solely solar energy to provide both heating and irradiation, a four-mirror floating-zone light furnace (operated at 66.7 V, 15.58 A and 1039 W) from Crystal Systems Inc. equipped with four halogen lamps was used to mimic a solar concentrator without using any electrical heating devices. Photocatalyst (20 mg) was added to 5 ml of Milli-Q H2O under vigorous magnetic stirring (600 r.p.m.), then the same reactor was placed in the light-concentrated furnace. The reaction temperature was monitored by a thermocouple and maintained at 270 °C. The gaseous products were measured by a GC equipped with TCDs. The natural seawater used in this work was collected near Bournemouth Pier (coordinates: 50.715474° N, 1.876075° W) and used after filtration. As the oxidation of Cl− ions will lead to a change of pH (equations (3) and (4)), for each reaction, the pH of the suspension was measured before and after the POWS reaction to make sure that no observable side reaction was taking place:
The IQE was measured in the same reactor following the same procedure. Photocatalyst that contained 20 mg of N–TiO2 was added to 5 ml of artificial Dead Sea water under magnetic stirring (600 r.p.m.), then the batch reactor was irradiated by a 300-W Xe lamp (Newport) equipped with band-pass filters of 385 ± 20, 440 ± 10, 500 ± 25, 575 ± 25, 650 ± 20, 750 ± 20 and 850 ± 20 nm, respectively. Generally, the incident photons were corrected by subtracting the scattered and transmitted light from the incident light—two quartz windows were provided, in parallel, on both sides of the batch reactor, facing each other. The incident light was first measured using a light metre in the centre of the batch reactor, then the scattered and transmitted light were also measured outside the opposite window when the reaction suspension was present. Subsequently, light coming out of the reactor was subtracted from the incident light, and the attenuation in the light intensity was worked out. The light inside the reactor might also have been scattered by the photocatalyst particles, but most of it would have been reflected by the stainless-steel surface and finally absorbed by the photocatalyst. Subsequently, the relevant number of incident photons was calculated from the irradiation powers at each wavelength. The IQE can be calculated according to
The EQE (also known as apparent quantum efficiency) was also calculated as
An example of the QE calculation is provided in Supplementary Note 4.
The ηSTH was measured with a similar procedure. Photocatalyst that contained 20 mg of N–TiO2 was added to 5 ml of artificial Dead Sea water under magnetic stirring (600 r.p.m.), then the suspension was irradiated by a VeraSol solar simulator (AM 1.5G, 100 mW cm−2, 1 sun). The ηSTH could then be calculated as
where P is the power of the solar irradiation (100 mW cm−2), S is the illuminating area, and t is the time of the reaction.
The QE and ηSTH measurements were repeated at least three times, and the average values and s.d. values were calculated. Examples of QE and ηSTH calculations are provided in Supplementary Note 4.
Evaluation of the overall energy conversion efficiency
The overall energy conversion efficiency was evaluated in a different photoreactor with a larger illuminating area (round sapphire window with a radius of 2 cm). We discussed the reactor specifications with the supplier (Thoughtventions Unlimited) to ensure both operational safety and experimental accuracy. This reactor allowed a relatively large illumination area and an appropriate penetration of light into the aqueous suspension. We also managed to cover the experimental set-up with band heater and thermally insulating materials, including silica wool, thermal foil and so on, to minimize heat loss. The POWS performance was evaluated by adding 100 mg of the 1 wt% Pt/N–TiO2 to 3 ml of artificial Dead Sea water under vigorous magnetic stirring (600 r.p.m.), then the suspension was heated to 270 °C under an Ar atmosphere and irradiated by a VeraSol solar simulator (AM 1.5G, 100 mW cm−2, 1 sun). The heating process was controlled by a Parr 4838 thermo-controller under the proportional integral derivative (PID) mode and monitored with SpecView 3 software. The reaction was performed for 8 h in total, and the gaseous product was sampled every 2 h. The amounts of H2 and O2 were measured by a GC equipped with TCDs. The actual energy input could be calculated by integrating the power versus time curve, then ηOEC could be calculated using equation (8):
where nHydrogen is the amount of H2 generated from the water splitting reaction, and the Helmholtz free energy is used for this constant-volume system (Supplementary Note 4), ESolar is the solar energy input from the solar simulator, and QActual is the actual electrical energy input for heating up the reactor and maintaining the reaction temperature. More details of this calculation are presented in Supplementary Note 5.
Detection of chloride oxidation
The gaseous product was analysed by a GC equipped with TCDs. This showed no signal of Cl2 or any other Cl-containing compound. The concentration of Cl− in the seawater and artificial seawater samples was analysed before and after the POWS reaction by back titration (Volhard Method). Typically, 2 ml of the reaction solution was diluted into 15 ml in a volumetric flask, then 10 ml of the resulting solution was transferred into a conical flask using a pipette. An excess amount of 0.1 mol l−1 silver nitrate solution was precisely added by pipette, together with 10 ml of concentrated nitric acid, then 1 ml of saturated ferric ammonium sulfate solution was added as an indicator. The unreacted silver ions were then titrated with 0.1 mol l−1 potassium thiocyanate solution. The end point was the first appearance of a dark-red colour due to the ferric thiocyanate complex. The concentration of chloride ions could then be calculated.
In addition, we attempted to detect dissolved Cl2 and HClO, which are potentially formed due to the oxidation of Cl−. Twenty millilitres of 0.5 mol l−1 potassium iodide solution was added into 1 ml of the post-reaction solution, and 1 ml of starch solution was added as an indicator. If there is Cl2 or HClO in the solution, I− will be oxidized to I2, which will then react with starch to show an intense blue-black colour. The detection limit of this method is known to be 20 ppm. There was no colour change observed in this work, indicating there was no oxidative Cl2 or HClO in the solution.
Data availability
The data supporting the findings of this study are available within the Article and its Supplementary Information or from the corresponding authors upon reasonable request. The optimized structures of DFT calculations are provided in Supplementary Data 1. Source data are provided with this paper.
References
Luo, J. et al. Water photolysis at 12.3% efficiency via perovskite photovoltaics and Earth-abundant catalysts. Science 345, 1593–1596 (2014).
Jia, J. et al. Solar water splitting by photovoltaic-electrolysis with a solar-to-hydrogen efficiency over 30%. Nat. Commun. 7, 13237 (2016).
Takata, T. et al. Photocatalytic water splitting with a quantum efficiency of almost unity. Nature 581, 411–414 (2020).
Foo, C. et al. Characterisation of oxygen defects and nitrogen impurities in TiO2 photocatalysts using variable-temperature X-ray powder diffraction. Nat. Commun. 12, 661 (2021).
Li, Y. et al. 2D photocatalysts with tuneable supports for enhanced photocatalytic water splitting. Mater. Today 41, 34–43 (2020).
Li, Y. et al. Local magnetic spin mismatch promoting photocatalytic overall water splitting with exceptional solar-to-hydrogen efficiency. Energy Environ. Sci. 15, 265–277 (2022).
Zhang, J., Hu, W., Cao, S. & Piao, L. Recent progress for hydrogen production by photocatalytic natural or simulated seawater splitting. Nano Res. 13, 2313–2322 (2020).
Yao, Y., Gao, X. & Meng, X. Recent advances on electrocatalytic and photocatalytic seawater splitting for hydrogen evolution. Int. J. Hydrog. Energy 46, 9087–9100 (2021).
Osman, A. I. et al. Hydrogen production, storage, utilisation and environmental impacts: a review. Environ. Chem. Lett. 20, 153–188 (2022).
Jin, H. et al. Stable and highly efficient hydrogen evolution from seawater enabled by an unsaturated nickel surface nitride. Adv. Mater. 33, 2007508 (2021).
Culp, T. E. et al. Nanoscale control of internal inhomogeneity enhances water transport in desalination membranes. Science 371, 72–75 (2021).
Di Vincenzo, M. et al. Biomimetic artificial water channel membranes for enhanced desalination. Nat. Nanotechnol. 16, 190–196 (2021).
Hausmann, J. N., Schlögl, R., Menezes, P. W. & Driess, M. Is direct seawater splitting economically meaningful? Energy Environ. Sci. 14, 3679–3685 (2021).
Kibria, M. et al. Seawater electrolysis for hydrogen production: a solution looking for a problem? Energy Environ. Sci. 14, 4831–4839 (2021).
Lu, X. et al. A sea-change: manganese doped nickel/nickel oxide electrocatalysts for hydrogen generation from seawater. Energy Environ. Sci. 11, 1898–1910 (2018).
Crawford, S., Thimsen, E. & Biswas, P. Impact of different electrolytes on photocatalytic water splitting. J. Electrochem. Soc. 156, H346 (2009).
Li, Y., He, F., Peng, S., Lu, G. & Li, S. Photocatalytic H2 evolution from NaCl saltwater over ZnS1−x−0.5yOx(OH)y-ZnO under visible light irradiation. Int. J. Hydrog. Energy 36, 10565–10573 (2011).
Lee, C. T. et al. Solar hydrogen production from seawater splitting using mixed-valence titanium phosphite photocatalyst. J. Environ. Chem. Eng. 9, 104826 (2021).
Liu, S. et al. Hollow heterostructure CoS/CdS photocatalysts with enhanced charge transfer for photocatalytic hydrogen production from seawater. Int. J. Hydrog. Energy 47, 9220–9229 (2022).
Dang Van, H., Wang, Y. H. & Wu, J. C. S. Exploration of photocatalytic seawater splitting on Pt/GaP-C3N4 under simulated sunlight. Appl. Surf. Sci. 572, 151346 (2022).
Ji, S. M. et al. Photocatalytic hydrogen production from natural seawater. J. Photochem. Photobiol. A Chem. 189, 141–144 (2007).
Maeda, K., Masuda, H. & Domen, K. Effect of electrolyte addition on activity of (Ga1−xZnx)(N1−xOx) photocatalyst for overall water splitting under visible light. Catal. Today 147, 173–178 (2009).
Tian, B. et al. Supported black phosphorus nanosheets as hydrogen-evolving photocatalyst achieving 5.4% energy conversion efficiency at 353 K. Nat. Commun. 9, 1397 (2018).
Zhang, J. et al. Photocatalytic hydrogen production from seawater under full solar spectrum without sacrificial reagents using TiO2 nanoparticles. Nano Res. 15, 2013–2022 (2022).
Li, Y. et al. Photocatalytic water splitting by N-TiO2 on MgO(111) with exceptional quantum efficiencies at elevated temperatures. Nat. Commun. 10, 4421 (2019).
Zhang, N. & Chai, Y. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647–4671 (2021).
Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457–465 (2017).
Guan, X. et al. Efficient unassisted overall photocatalytic seawater splitting on GaN-based nanowire arrays. J. Phys. Chem. C 122, 13797–13802 (2018).
Hu, Y. et al. Facet-dependent acidic and catalytic properties of sulfated titania solid superacids. Chem. Commun. 51, 14219–14222 (2015).
Meng, A., Zhang, J., Xu, D., Cheng, B. & Yu, J. Enhanced photocatalytic H2-production activity of anatase TiO2 nanosheet by selectively depositing dual-cocatalysts on (101) and (001) facets. Appl. Catal. B Environ. 198, 286–294 (2016).
Zhang, H. et al. Facet-dependent interfacial charge transfer in Fe(III)-grafted TiO2 nanostructures activated by visible light. ACS Catal. 8, 9399–9407 (2018).
Shi, M. et al. Intrinsic facet-dependent reactivity of well-defined BiOBr nanosheets on photocatalytic water splitting. Angew. Chem. Int. Ed. 59, 6590–6595 (2020).
Held, G. et al. Ambient-pressure endstation of the Versatile Soft X-ray (VerSoX) beamline at Diamond Light Source. J. Synchrotron Radiat. 27, 1153–1166 (2020).
Krishnan, P. et al. Characterization of photocatalytic TiO2 powder under varied environments using near ambient pressure X-ray photoelectron spectroscopy. Sci. Rep. 7, 43298 (2017).
Lau, W. M. Effects of a depth-dependent specimen potential on X-ray photoelectron spectroscopic data. J. Appl. Phys. 65, 2047–2052 (1989).
Piekner, Y., Ellis, D. S., Grave, D. A., Tsyganok, A. & Rothschild, A. Wasted photons: photogeneration yield and charge carrier collection efficiency of hematite photoanodes for photoelectrochemical water splitting. Energy Environ. Sci. 14, 4584–4598 (2021).
Grave, D. A. et al. Extraction of mobile charge carrier photogeneration yield spectrum of ultrathin-film metal oxide photoanodes for solar water splitting. Nat. Mater. 20, 833–840 (2021).
Ellingson, R. J. et al. Excitation energy dependent efficiency of charge carrier relaxation and photoluminescence in colloidal InP quantum dots. J. Phys. Chem. B 106, 7758–7765 (2002).
Ye, Z. et al. Phonon-assisted up-conversion photoluminescence of quantum dots. Nat. Commun. 12, 4283 (2021).
Tautz, R. et al. Charge photogeneration in donor-acceptor conjugated materials: influence of excess excitation energy and chain length. J. Am. Chem. Soc. 135, 4282–4290 (2013).
Carneiro, L. M. et al. Excitation-wavelength-dependent small polaron trapping of photoexcited carriers in α-Fe2O3. Nat. Mater. 16, 819–825 (2017).
Lane, P. A., Cunningham, P. D., Melinger, J. S., Esenturk, O. & Heilweil, E. J. Hot photocarrier dynamics in organic solar cells. Nat. Commun. 6, 7558 (2015).
Hisatomi, T. & Domen, K. Reaction systems for solar hydrogen production via water splitting with particulate semiconductor photocatalysts. Nat. Catal. 2, 387–399 (2019).
Teitsworth, T. S. et al. Water splitting with silicon p-i-n superlattices suspended in solution. Nature 614, 270–274 (2023).
Li, Z. et al. Blocking the reverse reactions of overall water splitting on a Rh/GaN–ZnO photocatalyst modified with Al2O3. Nat. Catal. 6, 80–88 (2023).
Zhou, P. et al. Solar-to-hydrogen efficiency of more than 9% in photocatalytic water splitting. Nature 613, 66–70 (2023).
Peng, Y.-K. et al. Mapping surface-modified titania nanoparticles with implications for activity and facet control. Nat. Commun. 8, 675 (2017).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Deskins, N. A., Rousseau, R. & Dupuis, M. Distribution of Ti3+ surface sites in reduced TiO2. J. Phys. Chem. C 115, 7562–7572 (2011).
Yoon, Y. et al. Anticorrelation between surface and subsurface point defects and the impact on the redox chemistry of TiO2(110). ChemPhysChem 16, 313–321 (2015).
Lazzeri, M. & Selloni, A. Stress-driven reconstruction of an oxide surface: the anatase TiO2(001) − (1 × 4) surface. Phys. Rev. Lett. 87, 266105 (2001).
Li, F., Wang, D. & Gong, X. Q. Subtle structure matters: boosting surface-directed photoelectron transfer via the introduction of specific monovalent oxygen vacancies in TiO2. Phys. Chem. Chem. Phys. 23, 19854–19861 (2021).
Yan, L. & Chen, H. Migration of Holstein polarons in anatase TiO2. J. Chem. Theory Comput. 10, 4995–5001 (2014).
Ma, X. et al. Hydrogen-bond network promotes water splitting on the TiO2 surface. J. Am. Chem. Soc. 144, 13565–13573 (2022).
Pham, T. D. & Deskins, N. A. Efficient method for modeling polarons using electronic structure methods. J. Chem. Theory Comput. 16, 5264–5278 (2020).
Di Valentin, C. & Selloni, A. Bulk and surface polarons in photoexcited anatase TiO2. J. Phys. Chem. Lett. 2, 2223–2228 (2011).
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, T. A. & Hennig, R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 140, 084106 (2014).
Burdett, J. K., Hughbanks, T., Miller, G. J., Smith, J. V. & Richardson, J. W. Structural-electronic relationships in inorganic solids: powder neutron diffraction studies of the rutile and anatase polymorphs of titanium dioxide at 15 and 295 K. J. Am. Chem. Soc. 109, 3639–3646 (1987).
Acknowledgements
Support for this project from the EPSRC in the UK (EP/K040375/1) and the National Natural Science Foundation of China for modelling (22003016) is gratefully acknowledged. S.C. acknowledges support from the General Research Fund (no. 15306021) from the Hong Kong Research Grant Council and the National Natural Science Foundation of China (grant no. 12104381). This work was carried out with the support of the Diamond Light Source (instrument B07/VerSoX, proposal SI29265).
Author information
Authors and Affiliations
Contributions
Y.L. prepared and tested the catalysts and analysed the results. Y.L. and D.P. performed the light furnace experiments. S.C. carried out STEM imaging and analysis. Y.L. and R.A.T. collected and analysed the PL and TRPL data. Y.L., W.N., A.L. and G.H. performed AP-XPS at the Diamond Light Source. H.Z. and X.-P.W. worked on the computational studies. Y.L., X.-P.W. and S.C.E.T. wrote the paper in discussion with all authors. S.C.E.T. supervised the overall project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Catalysis thanks Robert Godin, Al Rey Villagracia, Yoshihiro Yamazaki, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–38, Tables 1–12, Notes 1–5, Methods and References 1–31.
Supplementary Data 1
Coordinates of DFT computations.
Source data
Source Data Fig. 1
Statistical source data of Fig. 1.
Source Data Fig. 3
Statistical source data of Fig. 3.
Source Data Fig. 4
Statistical source data of Fig. 4.
Source Data Fig. 5
Statistical source data of Fig. 5.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Zhou, H., Cai, S. et al. Electrolyte-assisted polarization leading to enhanced charge separation and solar-to-hydrogen conversion efficiency of seawater splitting. Nat Catal 7, 77–88 (2024). https://doi.org/10.1038/s41929-023-01069-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41929-023-01069-1
