
Abstract
Pre-clinically, the mTORC1/2 inhibitor sapanisertib restored sensitivity to platinums and enhanced paclitaxel-induced cancer cell killing. NCT03430882 enrolled patients with mTOR pathway aberrant tumors to receive sapanisertib, carboplatin and paclitaxel. Primary objective was safety and secondary objectives were clinical response and survival. One patient had a dose-limiting toxicity at dose level 4. There were no unanticipated toxicities. Grade 3–4 treatment-related adverse events included anemia (21%), neutropenia (21%), thrombocytopenia (10.5%), and transaminitis (5%). Of 17 patients evaluable for response, 2 and 11 patients achieved partial response and stable disease, respectively. Responders included a patient with unclassified renal cell carcinoma harboring EWSR1-POU5F1 fusion and a patient with castrate resistant prostate cancer harboring PTEN loss. Median progression free survival was 3.84 months. Sapanisertib in combination with carboplatin plus paclitaxel demonstrated a manageable safety profile, with preliminary antitumor activity observed in advanced malignancies harboring mTOR pathway alterations.
Similar content being viewed by others

Introduction
The aberrant activation of the phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling pathway is associated with malignant transformation and resistance to apoptosis1. Hence, mTOR, which is the catalytic protein that nucleates both the mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), has become a target of therapy development. The US Food and Drug Administration has approved the two rapamycin analogues (“rapalogs”), temsiromlimus and everolimus for the treatment of multiple solid cancers including breast cancer and renal cell carcinoma (RCC)2,3,4,5. Rapalogs, however, serve as allosteric inhibitors of mTOR within the mTORC1 complex only and lack of mTORC2 inhibition, which has been proposed as a mechanism of resistance6. Therefore, a new generation of dual mTORC1/2 inhibitors has been developed with promising activity in preclinical models including those with acquired rapamycin resistance7,8,9. Sapanisertib (CB-228,TAK-228, MLN0128) is an orally bioavailable potent inhibitor of both mTORC1 and mTORC2 that demonstrated a manageable safety profile, with preliminary antitumor activity observed in renal cell carcinoma (RCC) and endometrial cancer10.
Anti-angiogenic effects of mTOR inhibition were shown to synergize with the antitumor and antiangiogenic effects of weekly paclitaxel11. However, this synergy was observed with sequential, rather than simultaneous, mTOR blockade after chemotherapy11. In vivo modeling confirmed that the combination of paclitaxel on Day 1 and sapanisertib given on Days 2–4 resulted in improved tumor growth inhibition compared to concomitant administration of the two agents12. Furthermore, platinum resistance has been shown to be related to activating phosphorylation of AKT and preclinical data demonstrated that mTORC1/2 inhibition restores sensitivity to platinum chemotherapy13. Taken together, preclinical data support the sequential addition of sapanisertib to carboplatin plus paclitaxel. Clinical tolerability for different schedules of the combination is expected to be different from single agent sapanisertib. Here, we evaluate a range of dosing schedules in this phase 1 study (NCT03430882) of sapanisertib in combination with carboplatin plus paclitaxel in patients with advanced solid malignancies.
Results
Characteristics of patients
From May 2018 to March 2020, 19 patients with advanced or metastatic solid malignancies that were refractory to standard-of-care therapy were enrolled. Demographics and disease characteristics are summarized in Table 1. The median age was 59 (range 33–74) and 58% were male. At baseline, about a quarter of patients (26%) had more than 3 metastatic sites of disease. Breast adenocarcinoma was the most represented primary disease site (32%), the majority of which were hormone receptor positive and HER2/neu negative (5 of 6 patients), followed by lung carcinoma (16%) and sarcoma (16%). All patients had ECOG performance score of 1 and the majority (63%) had three or more prior lines of therapy.
DLTs and MTD determination
Of the 19 patients treated, 4 patients were treated in dose level 1 (Carboplatin AUC 5 mg/mL•min every 3 weeks, Paclitaxel 40 mg/m2 weekly, TAK-228 2 mg Day 2–4, 9–11, 16–18). All 4 patients treated in dose Level 1 were DLT-evaluable with no DLT observed. Four patients were treated in dose level 2 (Carboplatin AUC 5 mg/mL min every 3weeks, Paclitaxel 40 mg/m2 weekly, TAK-228 3 mg Day 2–4, 9–11, 16–18). One patient was not DLT-evaluable, 3 patients were DLT-evaluable and no DLT was observed. Four patients were treated in dose level 3 (Carboplatin AUC 5 every 3 weeks, Paclitaxel 40 mg/m2, weekly, TAK-228 4 mg Day 2–4, 9–11, 16–18). All 4 patients were DLT evaluable and no DLT observed. Seven patients were treated in dose level 4 (Carboplatin AUC 5 mg/mL min every 3 weeks, Paclitaxel 60 mg/m2, weekly, TAK-228 4 mg Day 2–4, 9–11, 16–18). Among these patients treated in dose level 4, 1 patient was not evaluable for DLT, 1 experienced DLT (grade 4 thrombocytopenia lasting for more than a week) and 5 experienced no DLT.
Treatment exposure and safety
There were no unanticipated safety issues associated with the use of study drugs. Grade 3–4 treatment-related AEs (TRAE) included anemia (4/19; 21%), neutropenia (4/19; 21%), thrombocytopenia (2/19; 10.5%), and transaminitis (1/19; 5%). There were no grade 5 AEs. Treatment-related AEs across study are summarized in Table 2. Grade 3–4 TRAEs were seen at dose levels 2, 3 and 4 (Table 3). Treatment-related serious adverse events (SAEs) were seen in one patient under dose level 2. AEs leading to discontinuation of trial therapy occurred in 3 (16%) patients. AEs requiring a dose interruption and dose reduction occurred in 11 (58%) and 7 (37%) patients, respectively.
Antitumor activity
At data cut-off (Feb 5, 2021), all patients are no longer receiving study treatment. Median PFS was 3.84 months. Tumor responses across study participants in correlation with molecular profile, dose level and histology are summarized in Fig. 1. Of the 19 patients treated, 11 patients’ best overall response (BOR) was stable disease (SD); 2 patients’ BOR was partial response (PR) in RCC (37% decrease of target lesions) and in prostate cancer (34% decrease of target lesions), and 4 patients’ BOR was progressive disease. Disease control rate occurred in 13 (68%) patients. 2 patients were not evaluable for response due to withdrawal of consent prior to restaging.

Highlighted in red are genes implicated in the mTOR pathway. Key: PR partial response, SD stable disease, PD progressive disease, HR hormone receptor, HER2 human epidermal growth factor receptor 2, CRPC castration resistant prostate cancer, NSCLC non-small cell lung cancer, RCC renal cell cancer, SCC squamous cell carcinoma.
Clinical response in a patient with unclassified RCC harboring EWSR1-POU5F1 fusion t(6;22) (p21;q12)
Here, we describe a patient (#22) who presented with metastatic unclassified RCC to bulky abdominal lymph nodes and numerous nodules in lung bases (Fig. 2). A lung nodule biopsy revealed poorly differentiated carcinoma. Patient underwent diagnostic upfront nephrectomy which showed 12 cm poorly differentiated carcinoma invasive into renal sinus, perinephric adipose tissue, and renal vein. The tumor showed positive staining for CD56, PAX-8, and CAM5.2. Diagnosis was made as unclassified RCC. Molecular profiling revealed an EWSR1-POU5F1 fusion. The patient received frontline nivolumab plus ipilimumab with lack of response. Upon treatment with trial regimen, tumors demonstrated a dramatic, deep PR within 5 cycles of a trial regimen (Fig. 2). Patient remained on therapy for more than 20 months then discontinued therapy due to declining performance status. Mutations in FANCL, PBRM1, NTRK1, MLL2, IKZF1, HDAC1, and FOXL2 were among the unique mutations found in this patient and not found in non-responder patients (Fig. 1).

Panels (a), (b), (c) show partial response per RECIST version 1.1 in mediastinal adenopathy (a), lung metastasis (b) and pelvic adenopathy (c) in Left and right panels reflect baseline and nadir after therapy, respectively.
Clinical response in a patient with metastatic castrate-resistant prostate cancer harboring PTEN loss
Furthermore, we describe a patient (#17) who had developed castrate-resistant prostate cancer (mCRPC) metastatic to mediastinal and abdominal lymph nodes. He had received prior enzalutamide, abiraterone, and cabazitaxel. Molecular profiling revealed PTEN loss, which triggered the interest in mTOR targeting therapy. Upon treatment with trial regimen, tumors demonstrated PR per RECIST v1.1 and PSA50 criteria (Fig. 3). Time to treatment failure was 7 months. A mutation in PIK3CB was among the unique mutations found in this patient and not found in non-responder patients (Fig. 1).

Panels (a), (b) show response in mediastinal (a) and retroperitoneal (b) nodal metastases. c XY plot of the PSA (ng/mL) levels (blue, left Y axis) and % change from baseline (RECIST v1.1) in relation to time. Plot was created using GraphPad Prism version 9.2.0 for Mac OS, GraphPad Software, San Diego, California, USA, www.graphpad.com.
Discussion
In this open-label, Phase 1 study, the safety profile of triplet sapanisertib plus carboplatin and paclitaxel was characterized and shown to be manageable and consistent with the toxicity anticipated of either sapanisertib or carboplatin plus paclitaxel. Sapanisertib showed preliminary antitumour activity in a patient with unclassified RCC harboring an EWSR1-POU5F1 fusion and a patient with prostate adenocarcinoma harboring a PTEN deletion. The triplet MTD was determined as carboplatin AUC 5 mg/mL•min every 3 weeks, Paclitaxel 60 mg/m2, weekly, and TAK-228 4 mg Day 2–4, 9–11, 16–18. Given that majority of patients in our study were white, 4 mg dose seems consistent with a prior study showing RP2D of sapanisertib in East Asian patients (3 mg QD) was lower than in Western patients (4 mg QD)14. The safety profile of sapanisertib in this phase 1 study was generally manageable across all schedules, and tolerability was greater with increased intermittence of dosing. Common AEs related to sapanisertib across all schedules in both phases included hyperglycemia, nausea, and vomiting, which are all well-known side effect of PI3K/mTOR pathway inhibition15.
EWSR1 fusions are pathognomonic features of Ewing sarcoma (most commonly with Fli-1 proto-oncogene, ETS transcription factor [FLI1] and occasionally with ETS transcription factor ERG [ERG])16. On the other hand, POU5F1 (OCT4) belongs to the homeobox gene superfamily and encodes a transcription factor that act as a master regulator of development, especially during embryogenesis17. POU5F1 has preserved homeodomains (HD) that bind DNA. The fusion peptide brings the HD of POU5F1 in proximity to N-terminal transcriptional activation domain (TAD) of EWSR1. This fusion peptide hypothetically (Fig. 4) leads to enrichment of EWSR1-related signaling pathways including the mTOR signaling pathway, p53 signaling pathway, and WNT signaling pathway18. Furthermore, EWSR1 fusion tumors have been shown to have constitutive activation of the mTOR pathway and respond to mTOR inhibition19,20, perhaps explaining the response seen to sapanisertib in our patient. The molecular similarity between our unclassified RCC case harboring EWSR1-POU5F1 fusion and Ewing sarcoma with EWSR1-FLI1 fusion was the rationale for the platinum-based combination21 for our patient with mRCC.

a Schema highlighting the EWSR1-POU5F1 fusion t(6;22)(p21;q12) peptide. POU5F1 (OCT4) belongs to the homeobox gene superfamily and encodes a transcription factor that act as a master regulator of development. POU5F1 has preserved homaeodomains (HD) that bind DNA. The fusion peptide brings the HD of POU5F1 in proximity to N-terminal transcriptional activation domain (TAD) of EWSR1. This fusion peptide hypothetically leads to enrichment of EWSR1-related signaling pathways including the mTOR signaling pathway, p53 signaling pathway, and WNT signaling pathway. b Schema depicting the downstream signaling pathways following the activation of receptor tyrosine kinase including the RAF/MERK/ERK and the PI3K/AKT/mTOR pathways. Figure was created using biorender.com.
Downstream signaling pathways following the activation of receptor tyrosine kinase include the activation of the well-known RAF/MERK/ERK and the PI3K/AKT/mTOR pathways which is controlled in part by the tumor-suppressor gene: PTEN. PTEN-deficient tumors have been reported to have enhanced sensitivity to the inhibition of mTOR pathways22. The hypothesized (Fig. 4) downstream activation of mTOR signaling in our patient with mCRPC was the rationale for choosing this trial regimen for his therapy. In a murine model, mTOR complex 2 seems to be required for the development of prostate cancer induced by Pten loss23, which further supports the use of sapanisertib. In addition, the observed clinical benefit despite his heavily pretreated disease with cabazitaxel suggests the plausible role of sapanisertib in his response.
Our study was limited due to the lack of correlative data due to the nature of the study and biopsies not being mandated. Secondly, in the platinum-naïve PTEN-loss mCRPC patient, we were unable to confirm if the response seen to the combination therapy would have occurred with single agent sapanisertib therapy alone or the role of the concurrent PIK3CB alteration. A recent study showed significant co-occurrence between PTEN and PIK3CA/PIK3CB/PIK3R1 alterations in prostate cancer24. Further in vitro work is necessary to better isolate the impact of each of these mutations on the potential sensitization to mTORC1/2 inhibition. Our study was a proof-of-concept trial to show that a dual mTOR1/2 inhibitor could be safely combined with chemotherapy.
In conclusion, sapanisertib plus chemotherapy was generally well tolerated with no unexpected safety signals across the various schedules studied. The triplet MTD was determined as carboplatin AUC 5 mg/mL•min every 3 weeks, Paclitaxel 60 mg/m2, weekly, and TAK-228 4 mg Day 2–4, 9–11, 16–18. Preliminary antitumour activity was observed in two patients with unclassified mRCC and mCRPC, respectively. Although single-agent sapanisertib has been preliminarily reported to have modest activity in patients with previously treated, metastatic clear-cell RCC (NCT03097328), our findings will help better inform sapanisertib plus chemotherapy combination dosing strategies in future advanced solid tumor clinical trials.
Methods
Study design and treatment
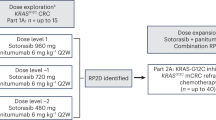
This Phase 1, open-label, dose-escalation study aimed to determine the safety, tolerability and preliminary efficacy of sapanisertib in combination with carboplatin plus paclitaxel in patients with advanced solid malignancies. Patients were treated with as many as six, 21-day cycles of the combination regimen (Fig. 5). The regimen was comprised of carboplatin (targeting area under the curve -AUC- 5 mg/mL•min) intravenously every 3 weeks on Day 1 of each cycle plus sapanisertib (TAK-228 or MLN0128) daily on Day 2–4 of each week at 2 mg, 3 mg and 4 mg orally and paclitaxel 40 mg/m2, 60 mg/m2, 80 mg/m2 QW IV on Day 1, 8, and 15 of each cycle. After the 6 cycles were completed, patients were treated to progression with sapanisertib 3 mg PO daily. The rationale behind limiting chemotherapy to six cycles was to avoid overlapping cumulative toxicity. Sapanisertib was continued as a maintenance therapy given the well-known resistance emerging from chemotherapy. This protocol utilized a standard 3 + 3 design. Three patients were treated per dose level. DLT was defined if the events occur between Day 1 and Day 21 of the first cycle. If none of the patients experienced DLT, the next cohort of three patients were treated at the next higher dose level. If one of three patients treated at a dose experiences DLT, then that cohort was expanded to a total of six patients. If the incidence of DLT among those six patients is one in six, then the next cohort was treated at the next higher dose level. If two or more of six patients treated at a dose level experience DLT, then the Maximum Tolerated Dose (MTD) was considered to have been exceeded. Two or three more patients (for a total of 6) were treated at the next lower dose as described above unless six patients have already been treated at that dose. In summary, the MTD is defined as the highest dose studied in which the incidence of DLT was less than 33%. The patient must have completed at least 66% of planned doses of all drugs to be evaluable for a DLT.

Patients were treated with as many as six, 21-day cycles of the combination regimen. The regimen is comprised of carboplatin AUC 5 intravenously every 3 weeks on Day 1 of each cycle plus sapanisertib (TAK-228 or MLN0128) daily on Day 2–4 of each week at 2 mg, 3 mg and 4 mg orally and paclitaxel 40 mg/m2, 60 mg/m2, 80 mg/m2 QW IV on Day 1, 8, and 15 of each cycle. Evaluation of the composite toxicity/efficacy endpoint was done at Day 21 of treatment. After the 6 cycles were completed, patients were treated to progression with sapanisertib 3 mg PO daily.
A DLT was defined, as predetermined in the study protocol, with the following: any grade ≥3 non-hematologic toxicity (except inadequately treated grade 3 nausea and/or vomiting and grade 3 diarrhea [all patients should have received optimal antiemetic and/or antidiarrheal prophylaxis and/or treatment], grade 3 hyperglycemia lasting ≤14 days [all patients should have received optimal anti-glycemic treatment, including insulin) and grade 3 rash lasting ≤3 days [all patients should have received topical steroid treatment, oral antihistamines and pulse oral steroids, if necessary]); grade 4 neutropenia lasting >7 days in the absence of growth factor support; grade 4 neutropenia of any duration accompanied with fever ≥38.5 °C and/or systemic infection; grade 3 thrombocytopenia with bleeding; any other grade ≥4 hematologic toxicity.
In the event of any adverse event defined as DLT attributable to the study drug(s), the drug(s) was(were) withheld. If the event resolved to Grade ≤ 2 within 28 days of interrupting therapy, the patient could resume study treatment at the next lower dose level. If sapanisertib (and other study agent(s), if applicable) dosing was delayed for >28 consecutive days for treatment-related toxicity, despite supportive treatment per standard clinical practice, or more than 2 dose reductions were required in a patient, sapanisertib (and other study agent(s), if applicable) therapy was stopped, the patient was discontinued from the study and the follow-up visit was completed within 30 days of the last administration of sapanisertib (and other agent if applicable), whichever is discontinued last.
Patients
Eligible patients were 18 years or older with solid tumor malignancy that is refractory to standard therapy, had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, and had adequate bone marrow reserve, hepatic, renal and metabolic (fasting serum glucose ≤ 130 mg/dL and fasting triglycerides ≤ 300 mg/dL) function. Patients were required to have evaluable or measurable disease by Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 criteria. Patients who have a history of brain metastases were eligible if their brain metastases had been treated (without evidence of progression or hemorrhage post-treatment), and if they had not taken dexamethasone 4 weeks prior to the first study drug administration and with no ongoing requirement for dexamethasone or antiepileptic drugs. Patients who had received carboplatin or paclitaxel within past 6 months, systemic corticosteroid therapy or proton pump inhibitors within past 7 days, dual PI3K/TORC1/2, mTORC1/2 inhibitors or mTORC1 inhibitors were not eligible. Additionally, patients with impaired cardiac function or significant active cardiovascular disease were also excluded.
Assessments
The safety population was defined as all enrolled patients who received at least 1 dose of any study drug. The DLT-evaluable population was defined as all patients who either experience DLT during the DLT window or those who receive at least 66% of the planned study drug administrations in Cycle 1 and 2 and have sufficient follow-up data to allow the investigators to determine whether DLT occurred. The response-evaluable population was defined as all patients who had measurable disease according to RECIST version 1.1 at baseline who have received at least 66% of the planned doses of any study drug, and who have at least 1 available post-baseline response assessment per RECIST version 1.1. Response was assessed according to the RECIST v1.125 after every two treatment cycles. Adverse events (AEs) were assessed using the National Cancer Institute Common Terminology Criteria for Adverse Events, v4.0. Patients were provided with a home blood glucometer to monitor their fasting blood glucose measurements to assess hyperglycemia as an on-target AE marker.
Objectives and statistical plan
Primary study objectives are to determine the safety and tolerability of the combination of sapanisertib plus paclitaxel and carboplatin, and to determine the optimal dose triplet, or MTD, in patients with advanced cancers refractory to standard therapy. Secondary study objectives are clinical tumor response of this combination, and progression free survival (PFS). PFS was calculated from the start of trial therapy till progression or last follow up where imaging did not demonstrate progression. Statistical analyses are primarily descriptive and graphical in nature, with no formal statistical hypothesis testing.
Molecular profiling
The molecular testing was part of clinical next generation sequencing (NGS) assays performed in the course of clinical care of the patient to identify targeted therapies for molecular matching. The biopsies were from sites that were considered safe by the interventional radiology. Samples were both from primary and metastatic sites. Biopsies were performed prior to trial therapy. NGS data were collected from five panels: FoundationOne (Foundation Medicine, Cambridge, MA) (N = 3), FoundationOne CDx (Foundation Medicine, Cambridge, MA) (N = 2), FoundationOne Liquid CDx (Foundation Medicine, Cambridge, MA) (N = 1), Guardant360 (Guardant Health, Palo Alto, CA) (N = 3), Liquid Biopsy Panel V1 (MD Anderson Cancer Center, Houston, TX) (N = 3), STGA-DNA 2018 (MD Anderson Cancer Center, Houston, TX) (N = 10), and Tempus xT (Tempus, Chicago, IL) (N = 2). A few patients had undergone multiple panel testing. The five panels included the following numbers of genes: FoundationOne (416), FoundationOne CDx (324), FoundationOne Liquid CDx (311), Guardant360 (74), Liquid Biopsy Panel V1 (70), STGA-DNA (147), and Tempus xT (648).
Data availability
The datasets supporting the results reported in this article, will be made available upon reasonable request from the corresponding author to researchers who provide a methodologically sound proposal. The data will be provided after its de-identification, in compliance with applicable privacy laws, data protection and requirements for consent and anonymization.
References
Vivanco, I. & Sawyers, C. L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2, 489–501 (2002).
Baselga, J. et al. Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer. N. Engl. J. Med. 366, 520–529 (2011).
Yao, J. C. et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet 387, 968–977 (2016).
Motzer, R. J. et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 372, 449–456 (2008).
Hudes, G. et al. Temsirolimus, Interferon Alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 356, 2271–2281 (2007).
O’Reilly, K. E. et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66, 1500–1508 (2006).
Zheng, B. et al. Pre-clinical evaluation of AZD-2014, a novel mTORC1/2 dual inhibitor, against renal cell carcinoma. Cancer Lett. 357, 468–475 (2015).
Korets, S. B., Musa, F., Curtin, J., Blank, S. V. & Schneider, R. J. Dual mTORC1/2 inhibition in a preclinical xenograft tumor model of endometrial cancer. Gynecol. Oncol. 132, 468–473 (2014).
Hassan, B. et al. Catalytic mTOR inhibitors can overcome intrinsic and acquired resistance to allosteric mTOR inhibitors. Oncotarget 5, 8544–8557 (2014).
Voss, M. H. et al. Phase 1 study of mTORC1/2 inhibitor sapanisertib (TAK-228) in advanced solid tumours, with an expansion phase in renal, endometrial or bladder cancer. Br. J. Cancer 123, 1590–1598 (2020).
Mondesire, W. H. et al. Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin. Cancer Res. 10, 7031–7042 (2004).
Kannan, K. et al. Abstract B189: MLN0128, an investigational mTORC1/2 inhibitor, demonstrates potent antitumor activity alone and in combination with paclitaxel in preclinical models of endometrial cancer. Mol. Cancer Therapeutics 12, B189 (2013).
David-West, G., Ernlund, A., Gadi, A. & Schneider, R. J. mTORC1/2 inhibition re-sensitizes platinum-resistant ovarian cancer by disrupting selective translation of DNA damage and survival mRNAs. Oncotarget 9, 33064–33076 (2018).
Shimizu, T. et al. A phase 1 study of Sapanisertib (TAK-228) in East Asian patients with advanced nonhematological malignancies. Target Oncol. 17, 15–24 (2022).
Fraenkel, M. et al. mTOR Inhibition by Rapamycin Prevents β-Cell Adaptation to Hyperglycemia and Exacerbates the Metabolic State in Type 2 Diabetes. Diabetes 57, 945 (2008).
Lessnick, S. L. & Ladanyi, M. Molecular pathogenesis of Ewing sarcoma: new therapeutic and transcriptional targets. Annu. Rev. Pathol. 7, 145–159 (2012).
Fogarty, N. M. E. et al. Genome editing reveals a role for OCT4 in human embryogenesis. Nature 550, 67–73 (2017).
He, D., Zhang, X. & Tu, J. Diagnostic significance and carcinogenic mechanism of pan-cancer gene POU5F1 in liver hepatocellular carcinoma. Cancer Med. 9, 8782–8800 (2020).
Subbiah, V. et al. Morphoproteomic profiling of the mammalian target of rapamycin (mTOR) signaling pathway in desmoplastic small round cell tumor (EWS/WT1), Ewing’s sarcoma (EWS/FLI1) and Wilms’ tumor(WT1). PLoS ONE 8, e68985 (2013).
Huang, H. J. et al. R1507, an anti-insulin-like growth factor-1 receptor (IGF-1R) antibody, and EWS/FLI-1 siRNA in Ewing’s sarcoma: convergence at the IGF/IGFR/Akt axis. PLoS ONE 6, e26060 (2011).
Pavarana, M. et al. Platinum and etoposide as second-line chemotherapy in advanced adult soft tissue sarcomas. J. Clin. Oncol. 22, 9063–9063 (2004).
Neshat, M. S. et al. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc. Natl Acad. Sci. USA 98, 10314–10319 (2001).
Guertin, D. A. et al. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 15, 148–159 (2009).
Mao, N. et al. Defining the therapeutic selective dependencies for distinct subtypes of PI3K pathway-altered prostate cancers. Nat. Commun. 12, 5053 (2021).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Acknowledgements
The authors thank all the patients who participated in these studies and their families, as well as all the investigators and site staff who helped conduct this study. We would like to acknowledge the late Dr. Kenneth R. Hess, PhD who made valuable contributions to statistical design of this study. V. Subbiah is an Andrew Sabin Family Foundation Fellow at The University of Texas MD Anderson Cancer Center. V. Subbiah acknowledges support of The Jacquelyn A. Brady Fund. V. Subbiah is supported by NIH grant R01CA242845. MD Anderson Cancer Center Department of Investigational Cancer Therapeutics is supported by the Cancer Prevention and Research Institute of Texas (RP1100584), the Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy (1U01 CA180964), the Center for Clinical and Translational Sciences (NCATS) Grant UL1 TR000371, and the MD Anderson Cancer Center Support Grant (P30 CA016672). Figure 4 was created with BioRender.com. This study was supported by Millennium Pharmaceuticals Inc., Cambridge, MA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. The funders had no role in the design of the study; the collection, analysis, and interpretation of the data; the writing of the manuscript; and the decision to submit the manuscript for publication. Final approval of manuscript: All authors. Agree to be accountable for all aspects of the work, which includes ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: All authors. Trial results were in part presented as an e-poster at the virtual AACR Annual Meeting 2021.
Author information
Authors and Affiliations
Contributions
Conception and design: R.G. and V.S. Collection and assembly of data: All authors. Data analysis and interpretation: O.A. and V.S. Drafting the manuscript: All authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. Institutional review boards (MD Anderson Cancer Center protocol 2016-0845) approved all aspects of the study. All participants provided written informed consent.
Competing interests
V.S. reports research funding and grant support for clinical trials: Roche/Genentech, Novartis, Bayer, GlaxoSmithKline, Nanocarrier, Vegenics, Celgene, Northwest Biotherapeutics, Berghealth, Incyte, Fujifilm, Pharmamar, D3, Pfizer, Multivir, Amgen, Abbvie, Alfa-sigma, Agensys, Boston Biomedical, Idera Pharma, Inhibrx, Exelixis, Blueprint medicines, Loxo oncology, Medimmune, Altum, Dragonfly therapeutics, Takeda and, National Comprehensive Cancer Network, NCI-CTEP and UT MD Anderson Cancer Center, Turning point therapeutics, Boston Pharmaceuticals Travel: Novartis, Pharmamar, ASCO, ESMO, Helsinn, Incyte, Consultancy/Advisory board: Helsinn, LOXO Oncology/Eli Lilly, R-Pharma US, INCYTE, QED pharma, Medimmune, Novartis. Other: Medscape. A.N. reports research funding from NCI, EMD Serono, MedImmune, Healios Onc. Nutrition, Atterocor, Amplimmune, ARMO BioSciences, Eli Lilly, Karyopharm Therapeutics, Incyte, Novartis, Regeneron, Merck, Bristol Myers Squibb, Pfizer, CytomX Therapeutics, Neon Therapeutics, Calithera Biosciences, TopAlliance Biosciences, Kymab, PsiOxus, Immune Deficiency Foundation (Spouse). Advisory board: CytomX Therapeutics, Novartis, Kymab, Genome. Travel and accommodation expenses: ARMO BioSciences. D.S.H. reports research funding from AbbVie, Adaptimmune, Aldi-Norte, Amgen, Astra-Zeneca, Bayer, BMS, Daiichi-Sankyo, Eisai, Fate Therapeutics, Genentech, Genmab, GSK, Ignyta, Infinity, Kite, Kyowa, Lilly, LOXO, Merck, MedImmune, Mirati, miRNA, Molecular Templates, Mologen, NCI-CTEP, Novartis, Pfizer, Seattle Genetics, Takeda, Turning Point Therapeutics. Travel, Accommodations, Expenses: Bayer, LOXO, miRNA, Genmab, AACR, ASCO, SITC. Consulting or Advisory Role: Alpha Insights, Amgen, Axiom, Adaptimmune, Baxter, Bayer, Genentech, GLG, Group H, Guidepoint, Infinity, Janssen, Merrimack, Medscape, Numab, Pfizer, Prime Oncology, Seattle Genetics, Takeda, Trieza Therapeutics, WebMD. Other ownership interests: Molecular Match (Advisor), OncoResponse (Founder), Presagia Inc (Advisor). S.P. reports research funds from Mirati Therapeutics, Inc., Eli Lilly, Red Hill Biopharma Ltd., Xencor, Five Prime Therapeutics, Novartis, Rgenix, Sanofi-Aventis, Arqule, Bristol-Myers Squibb, Onco Response, Sanofi US Services Inc., GlaxoSmith Kline. Financial Relationship/Speakers Bureau Consultant: Tyme, Inc., Zymeworks, Xencor and 4-D Pharma. F.M.B. reports consulting: Aduro BioTech Inc., DebioPharm, eFFECTOR Therapeutics, F. Hoffman-La Roche Ltd., Genentech Inc., IBM Watson, Jackson Laboratory, Kolon Life Science, OrigiMed, PACT Pharma, Parexel International, Pfizer Inc., Samsung Bioepis, Seattle Genetics Inc., Tyra Biosciences, Xencor, Zymeworks. Advisory Committee: Immunomedics, Inflection Biosciences, Mersana Therapeutics, Puma Biotechnology Inc., Seattle Genetics, Silverback Therapeutics, Spectrum Pharmaceuticals. Sponsored Research: Aileron Therapeutics, Inc. AstraZeneca, Bayer Healthcare Pharmaceutical, Calithera Biosciences Inc., Curis Inc., CytomX Therapeutics Inc., Daiichi Sankyo Co. Ltd., Debiopharm International, eFFECTOR Therapeutics, Genentech Inc., Guardant Health Inc., Millennium Pharmaceuticals Inc., Novartis, Puma Biotechnology Inc., Taiho Pharmaceutical Co. Honoraria: Chugai Biopharmaceuticals, Mayo Clinic, Rutgers Cancer Institute of New Jersey. T.Y. reports employment at University of Texas MD Anderson Cancer Center, where he is the Medical Director of the Institute for Applied Cancer Science, which has a commercial interest in DDR and other inhibitors (IACS30380/ART0380 was licensed to Artios. He also reports grant/research support (to Institution): Acrivon, Artios, AstraZeneca, Bayer, Beigene, BioNTech, Blueprint, BMS, Clovis, Constellation, Cyteir, Eli Lilly, EMD Serono, Forbius, F-Star, GlaxoSmithKline, Genentech, Haihe, ImmuneSensor, Ionis, Ipsen, Jounce, Karyopharm, KSQ, Kyowa, Merck, Mirati, Novartis, Pfizer, Ribon Therapeutics, Regeneron, Repare, Rubius, Sanofi, Scholar Rock, Seattle Genetics, Tesaro, Vivace and Zenith. He also reports consultancies from AbbVie, AstraZeneca, Acrivon, Adagene, Almac, Aduro, Amphista, Artios, Athena, Atrin, Avoro, Axiom, Baptist Health Systems, Bayer, Beigene, Boxer, Bristol Myers Squibb, C4 Therapeutics, Calithera, Cancer Research UK, Clovis, Cybrexa, Diffusion, EMD Serono, F-Star, Genmab, Glenmark, GLG, Globe Life Sciences, GSK, Guidepoint, Idience, Ignyta, I-Mab, ImmuneSensor, Institut Gustave Roussy, Intellisphere, Jansen, Kyn, MEI pharma, Mereo, Merck, Natera, Nexys, Novocure, OHSU, OncoSec, Ono Pharma, Pegascy, PER, Pfizer, Piper-Sandler, Prolynx, Repare, resTORbio, Roche, Schrodinger, Theragnostics, Varian, Versant, Vibliome, Xinthera, Zai Labs and ZielBio. He also reports stocks in Seagen. The rest of the authors report no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alhalabi, O., Groisberg, R., Zinner, R. et al. Phase I study of sapanisertib with carboplatin and paclitaxel in mTOR pathway altered solid malignancies. npj Precis. Onc. 7, 37 (2023). https://doi.org/10.1038/s41698-023-00369-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-023-00369-w
