
Abstract
Cervical cancer is one of the most common gynecologic malignancies worldwide, necessitating the identification of novel biomarkers and therapeutic targets. This study aimed to investigate the significance of MKRN1 in cervical cancer and explore its potential as a diagnostic marker and therapeutic target. The results indicated that MKRN1 expression was up-regulated in cervical cancer tissues and correlated with advanced tumor stage, higher grade, and poor patient survival. Functional studies demonstrated that targeting MKRN1 effectively inhibited cell proliferation, migration, and invasion, highlighting its critical role in tumor progression and metastasis. Moreover, the knockdown of MKRN1 resulted in altered expression patterns of six transcription factor-encoding genes, revealing its involvement in gene regulation. Co-expression network analysis unveiled complex regulatory mechanisms underlying the effects of MKRN1 knockdown on gene expression. Furthermore, the results suggested that MKRN1 might serve as a diagnostic marker for personalized treatment strategies and a therapeutic target to inhibit tumor growth, metastasis, and overcome drug resistance. The development of MKRN1-targeted interventions might hold promise for advancing personalized medicine approaches in cervical cancer treatment. Further research is warranted to validate these findings, elucidate underlying mechanisms, and translate these insights into improved management and outcomes for cervical cancer patients.
Similar content being viewed by others


Introduction
Cervical cancer (CESC) is one of the most common gynecologic malignancies worldwide, accounting for a significant number of cancer-related deaths in women1. It arises from transforming cervical epithelial cells due to persistent infection with high-risk human papillomavirus (HPV) types2. Despite advances in screening programs and the availability of prophylactic HPV vaccination, the incidence and mortality rates of cervical cancer remain alarming in certain regions, particularly in low-resource settings where access to healthcare services is limited3.
The molecular mechanisms underlying cervical cancer development and progression are complex and multifactorial. Numerous genetic and epigenetic alterations have been identified in cervical cancer, including aberrant expression of oncogenes and tumor suppressor genes4, dysregulation of signaling pathways5, and disruption of cellular processes such as proliferation6, apoptosis7, and DNA repair8. Elucidating the key molecular players involved in cervical carcinogenesis is crucial for developing effective strategies for early detection, risk assessment, and targeted therapies9.
The makorin ring finger protein 1 (MKRN1) gene belongs to the makorin ring zinc finger protein family, which is characterized by a conserved ring finger domain10. The gene is located on human chromosome 7, contains eight exons and seven introns, and its full cDNA length is 1449 nt. MKRN1 has been implicated in diverse biological processes, including ubiquitin-mediated protein degradation, mRNA splicing, and regulation of gene expression11. Emerging evidence suggests that MKRN1 may function as an oncogene or tumor suppressor, depending on the context and cancer type. For instance, in breast cancer, MKRN1 has been reported to promote tumor growth and metastasis through its involvement in epithelial-mesenchymal transition and cell cycle regulation12. The previous reports suggested that MKRN1 might be a useful adjunct biomarker in liquid-based cervical cytology screening13, and it might play an important role as an E3 ligase in the occurrence and development of cervical cancer14.
Based on the potential role of MKRN1 in cancer development and progression, it is imperative to explore its expression patterns and functional significance in cervical cancer. Although previous studies have investigated the involvement of MKRN1 in various cancer types, limited information is available on its contribution to cervical carcinogenesis. Therefore, in this study, we aimed to investigate the expression levels and biological implications of MKRN1 in cervical cancer using a combination of bioinformatics analysis and in vitro experiments. By elucidating the role of MKRN1 in cervical cancer, we hope to uncover novel insights into the molecular mechanisms underlying this disease and identify potential therapeutic targets for intervention.
Results
MKRN1 was highly expressed in cervical cancer based on bioinformatics analysis
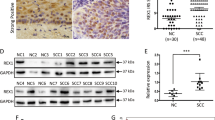
To investigate the correlation between the expression level of MKRN1 and the progression of cervical cancer, a comprehensive bioinformatics analysis was conducted using publicly available databases. The mRNA expression data of MKRN1 in both cervical tumor tissues and normal tissues were retrieved from the TNMplot web platform. The analysis revealed a significant upregulation of MKRN1 expression in non-paired cervical cancer samples (n = 189) compared to normal cervical samples (n = 56) (P = 0.00231; Fig. 1a). In the GSE3578 dataset, MKRN1 expression levels were found to be significantly down-regulated in CESC patients (n = 39) who received radiotherapy alone or radiotherapy plus concomitant chemotherapy (P = 0.0225; Fig. 1b). Moreover, a cohort of 306 CESC patients from The Cancer Genome Atlas (TCGA) datasets exhibited a significantly lower progression-free interval (PFI) survival probability in the high MKRN1 expression group compared to the low expression group (Fig. 1c).

Differential expression and survival analysis of MKRN1 gene in CESC. (a) The MKRN1 expression levels in normal cervical tissue samples (n = 56) and non-paired cervical cancer samples (n = 189). (b) The MKRN1 expression levels in cervical cancer samples prior to treatment and during treatment of the CESC patients (n = 39). (c) The CESC patients with high MKRN1 mRNA levels in CESC tumors, compared to those with low MKRN1 mRNA levels, had a significantly poorer progression-free interval (PFI) survival probability.
Down-regulation of MKRN1 could suppress HeLa cell proliferation
The impact of MKRN1 knockdown on cell proliferation in HeLa cells was assessed through the use of shRNA. The effectiveness of MKRN1 knockdown was confirmed by performing qRT-PCR analysis (Fig. 2a). The data demonstrated that sh-MKRN1-b (sh-b) exhibited the highest efficiency in suppressing MKRN1 gene expression, resulting in up to an 89% reduction compared to the control group (sh-control). Subsequent experiments were conducted using sh-b (named as sh-MKRN1). The results obtained from a CCK8 assay indicated a significant decrease in the proliferation ability of the sh-MKRN1 group compared to the control group at 24 h, 48 h, and 72 h time points (Fig. 2b). Furthermore, the colony formation assay revealed a notable decline of cell proliferation in the sh-MKRN1 group when compared with the control group (Fig. 2c). To gain further insights into the effects of MKRN1 on HeLa cell proliferation, flow cytometry was employed to examine changes in the cell cycle. The findings demonstrated that MKRN1 knockdown impeded the transition of HeLa cells from the G0/G1 phase to the S phase (Fig. 2d). These results collectively suggested that MKRN1 might play a crucial role in regulating the proliferation of HeLa cells.

Effects of MKRN1 knockdown on cell proliferation of HeLa cells. (a) MKRN1 knockdown by transfection with empty vector (sh-control) or with MKRN1 shRNA (sh-a, sh-b, and sh-c) was validated by qRT-PCR. (b) The effect of MKRN1 interference on HeLa cells was detected by a CCK8 assay. (c) The proliferation effect of MKRN1 interference in colony formation assay. (d) The cell cycle distribution of sh-control/sh-MKRN1 on HeLa cells.
Down-regulation of MKRN1 could suppress HeLa cell migration and invasion
In this study, the migration and invasion of HeLa cells were assessed through cell scratch and Transwell experiments. The results of the scratch assay indicated that migration in HeLa cells was significantly impaired when MKRN1 expression was down-regulated (sh-MKRN1), as compared to cells in the control group (sh-control), at 24 h and 48 h time points (Fig. 3a). Similarly, in the Transwell migration assay, a significant reduction in migrating HeLa cells was observed upon the knockdown of MKRN1 (Fig. 3b). Additionally, the Transwell invasion assay revealed a reduction in the number of HeLa cells passing through the Matrigel upon the down-regulation of MKRN1 (Fig. 3c). Taken together, these findings suggested that the migration and invasion capabilities of HeLa cells could be influenced by the expression of MKRN1.

Down-regulation of MKRN1 inhibited HeLa cell migration and invasion. (a) Scratch assay to detect HeLa cell migration ability. (b) Transwell assay to detect HeLa cell migration ability. (c) Transwell assay (containing matrix gel) to detect HeLa cell invasion ability.
Down-regulation of MKNR1 changed the gene expression profiles of Hela cell
To elucidate the role of MKNR1 in HeLa cells, an RNA-seq analysis was conducted to examine the expression patterns of the shRNA knockdown group (sh-MKRN1) and the control group (sh-control) of HeLa cells. The data files for RNA-seq have been deposited in the Sequence Read Archive (SRA) database under PRJNA987347. Despite having only three biological replicates in each group, a significant correlation between the sh-MKRN1 group and the sh-control group was observed (Fig. 4a). The result of principal component analysis (PCA) also showed samples were divided into two groups (Fig. 4b). The analysis of differential gene expression identified genes with |log2 (Fold Change)| > 1 and adjusted p < 0.05 as differentially expressed genes (DEGs). Among these, a total of 208 DEGs were induced by the shRNA knockdown of MKRN1, consisting of 145 up-regulated genes and 63 down-regulated genes (Fig. 4c). The complete list of DEGs can be found in Supplementary Table S1. Furthermore, hierarchical clustering analysis was performed on the differentially expressed genes, grouping genes with similar expression patterns together (Fig. 4d).

Down-regulation of MKNR1 could change the gene expression profiles of HeLa cells. (a) The correlation heatmap between MKNR1 knockdown (b1, b2, and b3) and control (con1, con2, and con3) samples. (b) Principal component analysis (PCA) plot. (c) Differentially expressed genes between the sh-MKRN1 group and the sh-control group. (d) Hierarchical cluster analysis of differentially expressed genes between the sh-MKRN1 group (b1, b2, and b3) and the sh-control group (con1, con2, and con3).
Overview of affected biological function by the shRNA knockdown of MKRN1
To investigate the potential biological functions of MKRN1-related genes, GO enrichment analyses and KEGG pathway annotation analyses were performed on the significantly differentially expressed genes. The top ten of enriched GO biological processes of the regulated genes were mainly related to the L-serine biosynthetic process, extracellular matrix organization, extracellular structure organization, and so on (Fig. 5a). The enriched GO cellular component terms were related to the I band and extracellular region, and so on (Fig. 5b). The enriched GO molecular function terms were primarily associated with integrin binding, guanyl-nucleotide exchange factor activity, extracellular matrix binding, and so on (Fig. 5c). The results of KEGG pathway enrichment showed that DGEs were involved in pathways associated with cellular processes, environmental information processing, genetic information processing, metabolism, and so on (Fig. 5d).

The potential biological functions of differentially expressed genes. (a) The enriched GO biological processes terms. (b) The enriched GO cellular component terms. (c) The enriched GO molecular function terms. (d) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment.
Analysis of potential MKRN1‑regulated transcription factor and enrichment biological function analysis
To gain a deeper understanding of the regulatory mechanisms involved in the MKRN1 knockdown, transcription factors (TFs) of DEGs were identified and annotated based on the Ami-nalTFDB database in this study. A total of 6 DEGs encoding TFs were identified with the shRNA knockdown of MKRN1, and these TFs belonged to 5 TF families (Table 1). Transcription factor activity was evaluated by using CoRegNet package. The co-operated TF interactions were listed in the Supplementary Table S2. To further investigate the regulatory networks between these identified TFs and their targeted genes, the h-Lincorn model in conjunction with CoRegNet to was used to construct a co-regulatory network. The co-expression network of transcription factors was shown in the following Fig. 6a. And the hierarchical clustering heatmap on TF activity was shown in the Fig. 6b.

Transcription Factor Activity Analysis. (a) Co-operation network of transcription factors. Nodes represent transcription factors (TFs). Edges represent co-operated relations. (b) Hierarchical clustering heatmap on TF activity between sh-MKRN1 group (b1, b2, and b3) and the sh-control group (con1, con2, and con3). Each column represents a sample, and each row represents a TF. The red ones represented the TFs with positive regulating effects on genes, and the blue ones represented that of negative ones.
Discussion
In this study, a comprehensive bioinformatics analysis was conducted to investigate the correlation between MKRN1 expression and the progression of cervical cancer. A significant upregulation of MKRN1 expression in non-paired cervical cancer samples compared to normal cervical tissue samples was observed, suggesting a crucial role of MKRN1 in the development and progression of cervical cancer. Improved treatment response was associated with down-regulation of MKRN1 expression in CESC patients who underwent radiotherapy alone or radiotherapy plus concomitant chemotherapy, indicating the potential of MKRN1 as a predictive marker for treatment outcomes. Furthermore, a significant association was found between high MKRN1 expression and a lower progression-free interval (PFI) survival probability in cervical cancer patients, suggesting that MKRN1 overexpression might indicate poor prognosis. These novel findings provide valuable informationinto the potential therapeutic targeting and prognostic use of MKRN1 in cervical cancer.
The results demonstrated that knockdown of MKRN1 could significantly decrease in the proliferation ability of HeLa cells, as evidenced by CCK8 and colony formation assays. Furthermore, flow cytometry analysis revealed that MKRN1 knockdown impeded the transition of HeLa cells from the G0/G1 phase to the S phase, indicating a role for MKRN1 in regulating the cell cycle. Our findings were consistent with previous studies that have identified MKRN1 as a key regulator of cell proliferation in various cancer types, such as pancreatic cancer15, renal clear cell carcinoma16, and colorectal cancer17. Further research is needed to elucidate the molecular mechanisms underlying the role of MKRN1 in regulating cancer cell migration and invasion.
The results of RNA-seq analysis showed that the knockdown of MKRN1 led to significant changes in gene expression profiles. The enriched gene ontology (GO) analyses revealed that DEGs induced by MKRN1 knockdown were associated with various biological processes, cellular components, and molecular functions. The top term of biological processes was related to regulating the L-serine biosynthetic process. Serine metabolism might play a key role in maintaining metabolic homeostasis and health18. And serving as a primary source of one-carbon units, serine is a significant contributor to cancer metabolism19 (non-melanoma skin cancer20, pancreatic cancer21, and lung cancer22). It has been reported that drugs, targeting serine-related one-carbon metabolic pathway, could inhibit the proliferation of cancer cells23, such as bladder cancer24, and endometrial cancer25. In this study, the regulation of the L-serine biosynthetic process by MKRN1 suggested that it might have a role in cell proliferation and survival. Understanding the mechanism by which MKRN1 regulates the L-serine biosynthetic process could provide valuable insights into the potential role of MKRN1 in diseases where cell proliferation and survival are deregulated.
The results of GO enrichment analysis showed that extracellular matrix organization, extracellular region, and extracellular matrix binding were listed as the top ten terms of biological processes, cellular components, and molecular functions, respectively. The extracellular matrix (ECM) is a complex network of proteins and other macromolecules that provide structural support and regulate cell behavior. Dysregulation of extracellular matrix organization can lead to various diseases, including fibrotic diseases26, pancreatic cancer27, gastric cancer28, breast cancer29, and other cancer types30. Furthermore, the enrichment of terms related to the extracellular region underscores the importance of these genes in regulating the cellular microenvironment. The regulation of the extracellular microenvironment is crucial for cellular proliferation, differentiation, migration, and survival. Enriching molecular functions related to extracellular matrix binding provides further evidence for the involvement of these genes in ECM regulation31. Extracellular matrix binding proteins interact with ECM components, mediating cell-ECM interactions and regulating ECM assembly and remodeling32. The findings supported previous studies that had implicated MKRN1 in the modulation of cell–matrix adhesion and signaling pathways involved in tumor progression, as well as its role in inducing the degradation of multiple oncogenic proteins and inhibiting tumor growth through the proteasomal pathway33. Therefore, the role of MKRN1 in extracellular matrix organization, extracellular region, and extracellular matrix binding suggested that it might play a pivotal role in maintaining the structural and functional integrity of the ECM, which is essential for tissue homeostasis and development. Additionally, KEGG pathway analysis revealed that the DEGs were involved in multiple pathways, including the MAPK signaling pathway, spliceosome, and biosynthesis of amino acids. These results were consistent with previous studies showing that MKRN1 might play a critical role in regulating cell proliferation and apoptosis through the p53 pathway34. Overall, the findings of this study provided new insights into the molecular mechanisms underlying the role of MKRN1 in cervical cancer progression.
In the present study, the effects of MKRN1 knockdown on gene expression and regulatory mechanisms were also investigated. The results revealed that the knockdown of MKRN1 led to altered expression of six genes encoding transcription factors, indicating that MKRN1 might play an important role in regulating transcriptional activity. It was consistent with previous studies suggesting the involvement of TFs in the regulation of cellular processes and cancer progression35. Furthermore, the co-expression network analysis showed that these TF-encoding genes were closely interconnected, suggesting a complex regulatory cascade that governed the effects of MKRN1 knockdown on gene expression. It provided a systems-level view of the regulatory landscape, emphasizing the complexity of transcriptional regulation in cervical cancer. Taken together, the findings might enhance our understanding of the molecular mechanisms involved in regulating gene expression by MKRN1, offering valuable implications for comprehending cancer pathogenesis and devising innovative therapeutic approaches.
There are certain limitations in our study. Firstly, this study primarily relied on bioinformatics analysis and in vitro experiments, which might not fully capture the complexity of the tumor microenvironment and the interplay between cancer cells and their surrounding tissues. Further studies using in vivo models and clinical samples would be needed to validate our findings and assess the therapeutic potential of targeting MKRN1 in a more clinically relevant context. Secondly, although a significant correlation between MKRN1 expression and clinical parameters was observed, further investigation would be required to establish the causative relationship between MKRN1 and cervical cancer progression. Thirdly, although an extensive analysis of TFs and co-regulatory networks was performed, it is important to acknowledge that other regulatory mechanisms, such as non-coding RNAs, epigenetic modifications, and post-translational modifications, might also contribute to the observed changes in gene expression. More experimental validation would be needed to confirm the functional roles of the identified TFs and their interactions. Future studies should consider conducting functional assays, such as knockdown or overexpression experiments, to validate the effects of these TFs on gene expression and cellular processes, and to provide a more complete understanding of MKRN1-mediated transcriptional regulation.
In conclusion, this study highlighted the significance of MKRN1 in cervical cancer. MKRN1 might be up-regulated in cervical cancer tissues and be associated with advanced tumor stage, higher grade, and poor patient survival. The functional studies provided evidence that targeting MKRN1 could effectively inhibit cell proliferation, migration, and invasion, highlighting its crucial role in promoting tumor progression and metastasis. Moreover, MKRN1 could potentially serve as a diagnostic marker for tailoring personalized treatment strategies and as a therapeutic target for inhibiting tumor growth, metastasis, and overcoming drug resistance in cervical cancer. However, further research is required to validate these findings, elucidate the underlying mechanisms, and translate this knowledge into improved management and outcomes for cervical cancer patients. Developing MKRN1-targeted interventions might help advance personalized medicine approaches in cervical cancer treatment.
Methods
Bioinformatics analysis
In this study, a comprehensive bioinformatics analysis was conducted to investigate the correlation between the expression level of MKRN1 and the progression of cervical cancer. The expression data of MKRN1 in cervical samples were obtained from three publicly accessible databases: the TNMplot web platform (https://www.tnmplot.com/), the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo), and The Cancer Genome Atlas (TCGA) datasets (https://portal.gdc.cancer.gov). The TNMplot was a web tool for the comparison of gene expression in normal, tumor and metastatic tissues36. It provided expression analysis for one selected gene in one kind of tissue type using RNA-Seq based data or gene chip-based data, which included the data from paired tumor and adjacent normal tissues, or the data from non-paired tumor and normal tissues. For CESC analysis, there were only mRNA expression data of MKRN1 in non-paired cervical tumor tissues (n = 189) and normal cervical tissues (n = 56) from chip-based studies in the TNMplot database. A Mann–Whitney U test was used to compare the difference of MKRN1 between non-paired cervical tumor and normal cervical tissues. The dataset of GSE357837, which included the cervical squamous cell carcinoma patients who received radiotherapy alone or radiotherapy plus concomitant chemotherapy, was retrieved from. The expression levels of MKRN1 were analyzed to evaluate the correlation with different treatment regimens in CESC patients (n = 39). In the TCGA database, a total of 306 RNA-seq data from CESC patients with clinical records were available. However, there were only three samples of adjacent non-cancerous tissues of CESC. Because of the low numbers of adjacent non-cancerous tissue samples, the comparison of paired normal and adjacent cervical tumors was not analyzed in this study. The expression levels of MKRN1 in the cohort of 306 CESC patients from were obtained, and the patients were divided into two groups based on high and low MKRN1 expression levels. Kaplan–Meier survival analysis and log-rank test were conducted to assess the differences in progression-free interval (PFI) survival probability between the high and low MKRN1 expression groups. The data used in this study were obtained from the publicly available datasets. Therefor, ethical approval was not required.
Cell culture and lentivirus transfection
The human cervical cancer cell line HeLa was purchased from the cell bank of the Chinese Academy of Sciences in Shanghai and was cultured in appropriate growth media supplemented with fetal bovine serum and antibiotics were cultured in DMEM (Dulbecco's Modified Eagle Medium) (Procell, Wuhan, China) with 10% Gibco™ fetal bovine serum (Invitrogen, AUS), 1% Penicillin–Streptomycin (10,000 U/mL) (Invitrogen, USA), at 37 °C in a 5% CO2 humidified atmosphere. Cells were maintained under standard cell culture conditions. For the knockdown of MKRN1 expression, lentiviral particles encoding MKRN1-specific shRNA sequences were generated and used to transduce HeLa cells. The target sequence of sh-a was 5′-GCTAACTACAAAGTCATCCCT-3′, the target sequence of sh-b was 5′-GCAATTTGAGAGCAAGATCAT-3′, and the target sequence of sh-c was 5-GCGAGATGTTGCTTATGCTTT-3′. The empty vehicle was used as a control. The transfection flow was performed according to the reagent manufacturer's instructions using the Lipofectamine™ 2000 Transfection Reagent (Invitrogen, USA). The cells were selected with 1.5 μg/mL puromycin for two weeks. The efficiency of MKRN1 knockdown was validated by quantitative real-time polymerase chain reaction (qRT-PCR). GAPDH was used as the internal reference gene. The following primers were used for expression analysis: MKRN1-fw: 5′-CATGGGGTTTGTAAGGAAGGAG-3′. MKRN1-rv: 5′-GCACACTACACTATACGGACTGT-3′; GAPDH-fw: 5′-ATGGGTGTGAACCATGAGAAGTA-3′; GAPDH-rv: 5′-GAGTGGGTGTCGCTGTTGAAGTC-3′. Each sample was run in triplicate by qRT-PCR (Applied Biosystems™ 7500, USA). Relative gene expression level was measured by 2−ΔΔCt relative quantitative method.
Cell counting kit 8 (CCK8) assay
A cell counting kit 8 (CCK8) (Beyotime Biotechnology, China) assay was performed to assess the effect of MKRN1 knockdown on cell proliferation. The cells from the sh-MKRN1 group and the control group were plated in a 100 μL culture medium containing 1 × 103 into 96-well plates for cell culture 24, 48, and 72 h. Then, 10 μL of CCK8 reagent was added to each well. After incubated at 37 °C for 2 h, the absorbance at 450 nm (OD) of each well was measured.
Colony formation assay
A colony formation assay was conducted to investigate the effect of MKRN1 knockdown on cell proliferation. The cells from the sh-MKRN1 group and control group were seeded into culture plates at a low density (1 × 103 cells/well into 6-well plates), and were allowed to grow for 10 days. After that, the colonies were fixed in 4% paraformaldehyde, stained with 0.5% crystal violet, and gently rinsed three times with PBS. A spot with more than 50 cells was counted as one clone.
Cell cycle assay
To analyze changes in the cell cycle, the cells from two groups were harvested by trypsinization, washed with phosphate-buffered saline (PBS), and fixed in ice-cold 70% ethanol. Fixed cells were washed with PBS and stained with propidium iodide (PI) solution. After incubation at 37 °C for 30 min in a light-proof environment, flow cytometry analysis was performed using a flow cytometer (Backman Coulter, USA).
Cell scratch assay
For the scratch assay, the cells were seeded onto 6-well culture plates and allowed to grow until they reached approximately 90% confluency. A scratch was created on the cell monolayer using a sterile 200 μL pipette tip, ensuring consistent gap width and length. The plates were then gently washed with PBS to remove detached cells or debris. Fresh growth media was added to the wells to facilitate cell migration. The scratch area was imaged immediately after creating the scratch (0 h) and at the time points of 24 h and 48 h. The images were captured at the same location in each well to ensure consistency.
Cell migration and invasion assays
The impact of MKRN1 down-regulation on cell migration and invasion was evaluated using Transwell migration and invasion assays. The cells were seeded onto Transwell chambers (Costar, USA) with or without Matrigel (BD Biosciences) coating and incubated for 24 h. The cells that migrated or invaded through the membrane were fixed in 4% paraformaldehyde, stained with 0.5% crystal violet, and counted under a microscope. The number of invasive cells was counted in five random fields of view.
RNA sequencing
The RNA samples of the cells from the sh-MKRN1 group and the control group were subjected to sequencing analysis with three replicates per group at Biomarker Technologies Co., LTD (Beijing, China). Before library construction, the purity, concentration, and integrity of the RNA samples were assessed using NanoDrop and Agilent 2100. RNA samples that met the quality standards were further processed for library preparation. The resulting high-quality libraries were pooled according to pre-designed target data volume and subsequently subjected to sequencing on the Illumina NovaSeq6000 platform. Raw data underwent stringent filtering to obtain clean data with optimal quality. Subsequently, the clean data were aligned to the human reference genome employing the HISAT2 software38. The data files for RNA-seq have been deposited in the Sequence Read Archive (SRA) database under PRJNA987347.
Screening of differentially expressed genes
After processing and analyzing the RNA sequencing data, the alignment results were transferred to the program StringTie for transcript assembly39. The gene expression levels of RNA-seq were expressed as FPKM (Fragments Per Kilobase of transcript sequence per Millions base pairs sequenced). The differentially expressed genes were identified using the package of DESeq algorithm in the R software. The resulting p-values were adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate. The thresholds to screen differentially expressed genes (DEGs) were |log2Fold change (FC)| > 1 and adjust p value < 0.05.
Gene function enrichment analysis
To predict the potential functions of differentially expressed genes between the sh-MKRN1 group and the control group, gene ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional pathway analysis were employed. The GO analysis was used to annotate genes based on biological processes, molecular functions, and cellular components. The KEGG pathway analysis was utilized to identify significant pathways that were enriched with differentially expressed genes. Enrichment was considered significant when the adjusted p value was less than 0.05.
Transcription factor analysis
To identify potential transcription factors (TFs) associated with the differentially expressed genes, the gene sets were selected as candidate genes and subjected to prediction using the AnimalTFDB database via HMMsearch40. The activity of the identified TFs was assessed using the CoRegNet R package41. Subsequently, a co-regulatory network between the identified TFs and their targeted genes was constructed based on the h-Lincorn model using CoRegNet.
Statistical analysis
All the above analyses were carried out by using R software version 4.0.3. All experiments were repeated three times, and data were expressed as mean ± standard deviation. Differences between the two groups were analyzed using independent samples t-test and paired t-test. Correlation analysis was based on Pearson's correlation test. Statically significance was defined as p < 0.05 (*), and p < 0.01 (**).
Conmpeting interests
The authors declare no competing interests.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials. The raw sequence data of RNA were deposited in the NCBI SRA database with the accession number of PRJNA987347 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA987347). Publicly available datasets were analyzed in this study, that can be found in the TNMplot web platform (https://www.tnmplot.com/), GEO database (https://www.ncbi.nlm.nih.gov/geo), and The Cancer Genome Atlas (https://portal.gdc.cancer.gov).
References
Xu, J., Huang, Z., Wang, Y., Xiang, Z. & Xiong, B. Identification of novel tumor microenvironment regulating factor that facilitates tumor immune infiltration in cervical cancer. Front Oncol 12, 846786. https://doi.org/10.3389/fonc.2022.846786 (2022).
Schiffman, M., Castle, P. E., Jeronimo, J., Rodriguez, A. C. & Wacholder, S. Human papillomavirus and cervical cancer. Lancet 370, 890–907. https://doi.org/10.1016/S0140-6736(07)61416-0 (2007).
Bruni, L. et al. Cervical cancer screening programmes and age-specific coverage estimates for 202 countries and territories worldwide: a review and synthetic analysis. Lancet Glob Health 10, e1115–e1127. https://doi.org/10.1016/S2214-109X(22)00241-8 (2022).
Han, G. H. et al. The combination of transient receptor potential vanilloid type 1 (TRPV1) and phosphatase and tension homolog (PTEN) is an effective prognostic biomarker in cervical cancer. Int J Gynecol Pathol: Off J Int Soc Gynecol Pathol 40, 214–223. https://doi.org/10.1097/pgp.0000000000000677 (2021).
Lee, M. S. et al. PI3K/AKT activation induces PTEN ubiquitination and destabilization accelerating tumourigenesis. Nat Commun 6, 7769. https://doi.org/10.1038/ncomms8769 (2015).
Luna, A. J. et al. MEK/ERK signaling is a critical regulator of high-risk human papillomavirus oncogene expression revealing therapeutic targets for HPV-induced tumors. PLoS Pathogens 17, e1009216. https://doi.org/10.1371/journal.ppat.1009216 (2021).
Cho, H. et al. Apoptosis inhibitor-5 overexpression is associated with tumor progression and poor prognosis in patients with cervical cancer. BMC Cancer 14, 545. https://doi.org/10.1186/1471-2407-14-545 (2014).
Duenas-Gonzalez, A. et al. Epigenetics of cervical cancer. An overview and therapeutic perspectives. Mol Cancer 4, 38. https://doi.org/10.1186/1476-4598-4-38 (2005).
Balasubramaniam, S. D., Balakrishnan, V., Oon, C. E. & Kaur, G. Key molecular events in cervical cancer development. Medicina (Kaunas). https://doi.org/10.3390/medicina55070384 (2019).
Gray, T. A. et al. The ancient source of a distinct gene family encoding proteins featuring RING and C(3)H zinc-finger motifs with abundant expression in developing brain and nervous system. Genomics 66, 76–86. https://doi.org/10.1006/geno.2000.6199 (2000).
Zhang, Y. et al. MKRN1 promotes colorectal cancer metastasis by activating the TGF-beta signalling pathway through SNIP1 protein degradation. J Exp Clin Cancer Res 42, 219. https://doi.org/10.1186/s13046-023-02788-w (2023).
Lee, E. W. et al. Ubiquitination and degradation of the FADD adaptor protein regulate death receptor-mediated apoptosis and necroptosis. Nat Commun 3, 978. https://doi.org/10.1038/ncomms1981 (2012).
Lee, M. et al. Makorin ring finger protein 1 as adjunctive marker in liquid-based cervical cytology. Medicine 95, e2425. https://doi.org/10.1097/md.0000000000002425 (2016).
Zhai, F., Wang, J., Yang, W., Ye, M. & Jin, X. The E3 ligases in cervical cancer and endometrial cancer. Cancers https://doi.org/10.3390/cancers14215354 (2022).
Liu, M. et al. Hypoxia-induced feedback of HIF-1α and lncRNA-CF129 contributes to pancreatic cancer progression through stabilization of p53 protein. Theranostics 9, 4795–4810. https://doi.org/10.7150/thno.30988 (2019).
Yang, Y. et al. MKRN1/2 serve as tumor suppressors in renal clear cell carcinoma by regulating the expression of p53. Cancer Biomark: Sect A Dis Mark 36, 267–278. https://doi.org/10.3233/cbm-210559 (2023).
Zhang, Y. et al. MKRN1 promotes colorectal cancer metastasis by activating the TGF-β signalling pathway through SNIP1 protein degradation. J Exp Clin Cancer Res: CR 42, 219. https://doi.org/10.1186/s13046-023-02788-w (2023).
He, L., Ding, Y., Zhou, X., Li, T. & Yin, Y. Serine signaling governs metabolic homeostasis and health. Trends Endocrinol Metabol: TEM 34, 361–372. https://doi.org/10.1016/j.tem.2023.03.001 (2023).
Pacold, M. E. et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol 12, 452–458. https://doi.org/10.1038/nchembio.2070 (2016).
Cappello, A. et al. Serine and one-carbon metabolism sustain non-melanoma skin cancer progression. Cell Death Discov 9, 102. https://doi.org/10.1038/s41420-023-01398-x (2023).
Lan, C. et al. The prognostic role of serine racemase in patients with pancreatic cancer: a new marker in cancer metabolism. Pancreas 52, e101–e109. https://doi.org/10.1097/mpa.0000000000002210 (2023).
Zhou, X., Tian, C., Cao, Y., Zhao, M. & Wang, K. The role of serine metabolism in lung cancer: from oncogenesis to tumor treatment. Front Genet 13, 1084609. https://doi.org/10.3389/fgene.2022.1084609 (2022).
Newman, A. C. & Maddocks, O. D. K. Serine and functional metabolites in cancer. Trends Cell Biol 27, 645–657. https://doi.org/10.1016/j.tcb.2017.05.001 (2017).
Shi, T. et al. Competition between p53 and YY1 determines PHGDH expression and malignancy in bladder cancer. Cell Oncol (Dordrecht) 46, 1457–1472. https://doi.org/10.1007/s13402-023-00823-8 (2023).
Zhai, L., Yang, X., Cheng, Y. & Wang, J. Glutamine and amino acid metabolism as a prognostic signature and therapeutic target in endometrial cancer. Cancer Med 12, 16337–16358. https://doi.org/10.1002/cam4.6256 (2023).
Sinha, S. et al. Transcriptional analysis reveals evidence of chronically impeded ECM turnover and epithelium-to-mesenchyme transition in scar tissue giving rise to Marjolin’s ulcer. J Burn Care Res: Off Publ Am Burn Assoc 38, e14–e22. https://doi.org/10.1097/bcr.0000000000000432 (2017).
Khatri, I. et al. Systems biology approach to identify novel genomic determinants for pancreatic cancer pathogenesis. Sci Rep 9, 123. https://doi.org/10.1038/s41598-018-36328-w (2019).
Sun, H. et al. The involvement of collagen family genes in tumor enlargement of gastric cancer. Sci Rep 13, 100. https://doi.org/10.1038/s41598-022-25061-0 (2023).
Curty, G. et al. Human endogenous retrovirus expression is upregulated in the breast cancer microenvironment of HIV Infected women: A pilot study. Front Oncol 10, 553983. https://doi.org/10.3389/fonc.2020.553983 (2020).
Harikrishnan, K., Prabhu, S. S. & Balasubramanian, N. A pan-cancer analysis of matrisome proteins reveals CTHRC1 and a related network as major ECM regulators across cancers. PLoS ONE 17, e0270063. https://doi.org/10.1371/journal.pone.0270063 (2022).
Ma, H. et al. MFG-E8 alleviates intervertebral disc degeneration by suppressing pyroptosis and extracellular matrix degradation in nucleus pulposus cells via Nrf2/TXNIP/NLRP3 axis. Cell Death Discov 8, 209. https://doi.org/10.1038/s41420-022-01002-8 (2022).
Halper, J. Basic components of connective tissues and extracellular matrix: fibronectin, fibrinogen, laminin, elastin, fibrillins, fibulins, matrilins, tenascins and thrombospondins. Adv Exp Med Biol 1348, 105–126. https://doi.org/10.1007/978-3-030-80614-9_4 (2021).
Fang, Y. C. et al. Identification of MKRN1 as a second E3 ligase for Eag1 potassium channels reveals regulation via differential degradation. J Biol Chem 296, 100484. https://doi.org/10.1016/j.jbc.2021.100484 (2021).
Huang, Y. et al. SF3B1 deficiency impairs human erythropoiesis via activation of p53 pathway: implications for understanding of ineffective erythropoiesis in MDS. J Hematol Oncol 11, 19. https://doi.org/10.1186/s13045-018-0558-8 (2018).
Guo, H. et al. Identification of stemness-related genes for cervical squamous cell carcinoma and endocervical adenocarcinoma by integrated bioinformatics analysis. Front Cell Dev Biol 9, 642724. https://doi.org/10.3389/fcell.2021.642724 (2021).
Bartha, Á. & Győrffy, B. TNMplot.com: A web tool for the comparison of gene expression in normal, tumor and metastatic tissues. Int J Mol Sci https://doi.org/10.3390/ijms22052622 (2021).
Iwakawa, M. et al. The radiation-induced cell-death signaling pathway is activated by concurrent use of cisplatin in sequential biopsy specimens from patients with cervical cancer. Cancer Biol Ther 6, 905–911. https://doi.org/10.4161/cbt.6.6.4098 (2007).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915. https://doi.org/10.1038/s41587-019-0201-4 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295. https://doi.org/10.1038/nbt.3122 (2015).
Shen, W. K. et al. AnimalTFDB 4.0: A comprehensive animal transcription factor database updated with variation and expression annotations. Nucleic Acids Res 51, D39–D45. https://doi.org/10.1093/nar/gkac907 (2023).
Nicolle, R., Radvanyi, F. & Elati, M. CoRegNet: Reconstruction and integrated analysis of co-regulatory networks. Bioinform (Oxford, England) 31, 3066–3068. https://doi.org/10.1093/bioinformatics/btv305 (2015).
Funding
This work was supported by the Research Foundation for Advanced Talents of Bengbu Medical College (Grant No. 15190016), and Graduate Innovation Project of Bengbu Medical College (No. Byycxz22003). The funders had no involvement in the design, conduct, or interpretation of this study.
Author information
Authors and Affiliations
Contributions
Conception and design: Y.G.; Data acquisition: X.D., Y.Z., S.L. and M.Y.; Statistical analysis and interpretation of data: X.D., Y.Z. and Y.G.; Drafting the manuscript: X.D. and Y.G.; Figures and tables: X.D., Y.Z. and Y.G.. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dong, X., Zhan, Y., Li, S. et al. MKRN1 regulates the expression profiles and transcription factor activity in HeLa cells inhibition suppresses cervical cancer cell progression. Sci Rep 14, 6129 (2024). https://doi.org/10.1038/s41598-024-56830-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-56830-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
