
Abstract
Naturally occurring DNA gaps have been observed in eukaryotic DNA, including DNA in nondividing cells. These DNA gaps are found less frequently in chronologically aging yeast, chemically induced senescence cells, naturally aged rats, d-galactose-induced aging model rats, and older people. These gaps function to protect DNA from damage, so we named them youth-associated genomic stabilization DNA gaps (youth-DNA-gaps). Type 2 diabetes mellitus (type 2 DM) is characterized by an early aging phenotype. Here, we explored the correlation between youth-DNA-gaps and the severity of type 2 DM. Here, we investigated youth-DNA-gaps in white blood cells from normal controls, pre-DM, and type 2 DM patients. We found significantly decreased youth-DNA-gap numbers in the type 2 DM patients compared to normal controls (P = 0.0377, P = 0.0018 adjusted age). In the type 2 DM group, youth-DNA-gaps correlate directly with HbA1c levels. (r = − 0.3027, P = 0.0023). Decreased youth-DNA-gap numbers were observed in patients with type 2 DM and associated with increased HbA1c levels. Therefore, the decrease in youth-DNA-gaps is associated with the molecular pathogenesis of high blood glucose levels. Furthermore, youth-DNA-gap number is another marker that could be used to determine the severity of type 2 DM.
Similar content being viewed by others

Introduction
Type 2 DM has been a public health issue for several decades. Currently, an estimated 537 million people are at risk of developing type 2 DM, and that number is expected to rise to 592 million by 2035 and 783 million by 20451,2. A previous study demonstrated that patients with type 2 DM often have relatively impaired genetic function, and genome instability is believed to be a factor that contributes to type 2 DM3,4,5,6,7,8,9,10. Type 2 DM patients are more likely to develop various conditions that affect elderly people, causing type 2 DM to be one of the world's most major health problems11,12,13,14,15,16,17,18,19,20. Tissue damage and multiorgan dysfunction, such as cardiovascular disease, stroke, foot ulcers, chronic kidney failure, and diabetic retinopathy, can occur in type 2 DM patients21,22,23,24,25,26,27,28,29. However, the mechanism underlying cellular senescence in DM needs more research.
We previously reported the discovery of naturally occurring DNA gaps called youth-associated genome-stabilizing DNA gaps (youth-DNA-gaps)30. Youth-DNA-gap complexes are composed of Box A of HMGB1, which produces the DNA gap, SIRT1 deacetylated histones, and AGO4 methylated interspersed repetitive sequences31,32,33,34,35. Naturally occurring DNA gaps in the genome maintain genomic integrity, and these gaps decrease in aging yeast, rats, and human and chemically induced senescent cells31,36,37,38. The formation of DNA gaps is one of the cellular mechanisms that maintains DNA integrity. As a result, the role of youth-DNA-gaps is similar to that of gaps that remain between successive rails in a railway track, that is, relieving torsion force to prevent damage33. We previously demonstrated that reducing the DNA gap resulted in DNA shearing37. Moreover, introducing new Box A-produced DNA gaps increased DNA resistance to radiation-induced DNA breaks, rejuvenated senescent cells, and rejuvenated both naturally and d-galactose-induced aging rats33. Two types of DNA breaks occur in eukaryotic cells. The most common type is pathologic breaks. This type of break is associated with H2AX foci and exists is small numbers under normal circumstances39,40,41,42,43,44. Youth-DNA-gaps are another type of DNA break that plays a physiologic role and is produced by cellular enzymes. Therefore, while a pathologic break represents DNA damage, youth-DNA-gaps represent epigenetic markers. Although both types are detectable by PCR, we proved by high throughput sequencing that the majority of PCR-detected DNA breaks under physiologic conditions are youth-DNA-gaps38,40. In aging cells, pathologic breaks are actively produced; however, the overall number of DNA gaps that could be detected by DNA gap PCR is reduced37. Therefore, DNA gap PCR results can indicate the youthfulness of eukaryotic DNA33,37. Type 2 DM accelerates the aging process both at the molecular level and in terms of physical phenotypes. Patients with type 2 DM have a higher risk of developing common geriatric syndromes, including frailty, arteriosclerosis, kidney failure, and dementia 45,46,47,48,49,50,51,52. Similar to elderly, type 2 DM patients have increased telomere shortening, mitochondrial DNA depletion, Alu hypomethylation, and DNA methylation age epigenetic clock53,54,55,56,57,58,59,60,61,62. Since the number of youth-DNA-gaps is inversely associated with the degree of aging and the degree of aging phenotype is directly associated with the severity of type 2 DM, the number of youth-DNA-gaps should be low in patients with severe type 2 DM.
Presently, blood tests are used to diagnose type 2 DM by measuring fasting blood sugar (FBS) and hemoglobin A1c (HbA1c) levels 63,64,65,66,67,68,69; guidelines are used to design good diabetes care primarily based on HbA1c levels 70,71. However, HbA1c and FBS levels represent the excess sugar levels in blood 71,72; another type of molecular marker that indicates the severity of organ degeneration will help determine the prognosis of type 2 DM patients. The assessment of youth-DNA-gaps by DNA gap PCR provides information on the biological aging of the individual.
We hypothesized that a decrease in youth-DNA-gap numbers promotes the pathogenesis of type 2 DM by inducing DNA damage, leading to the cellular senescence process, and directly correlating with the disease. Therefore, reducing the youth-DNA-gap number should inversely correlate with HbA1C and might serve as a promising biomarker for monitoring and treating tissue degeneration associated with type 2 DM in the future.
Results
Youth-DNA-gaps in patients with type 2 DM
The 240 samples were divided into three groups based on their HbA1c levels: 63 normal controls, 78 pre-DM, and 99 type 2 DM patients. Additionally, fasting blood sugar (FBS) levels were used to classify these patients, and as a result, several patients changed groups (74 normal controls, 93 pre-DM, and 73 type 2 DM patients) compared to the grouping based on HbA1c values (Table 1). The youth-DNA-gap numbers in each sample was then measured. We found that the number of youth-DNA-gaps was decreased in type 2 DM patients when the patients were classified by HbA1c (P = 0.0377) (Fig. 1a). We found no significant difference in youth-DNA-gap number in type 2 DM patients compared with normal subjects when the patients were grouped according to FBS levels (Fig. 1b).

The percentage of each DNA-gap number in normal and type 2 DM patients when grouped by HbA1c (a) and FBS levels (b). The values of independent age-matched pairs when grouped by HbA1c (c) and FBS levels (d). The boxes represent the interquartile ranges (25th to 75th percentile), while the median lines represent the 50th percentile. The whiskers represent the minimum and maximum values, and all individual data points are displayed. Significance levels are denoted as follows: *P < 0.05, **P < 0.01 (Mann–Whitney U test).
In our data, the average age of the normal and type 2 DM groups was different (Table 1). Age-matched pairs were created by matching normal to type 2 DM patients of the same age. Age-matched pairs revealed a significantly lower youth-DNA-gap numbers in the type 2 DM group than in the normal group when the patients were grouped according to HbA1c and FBS levels (P = 0.0018 and P = 0.0051, respectively) (Fig. 1c and d, respectively).
Correlations between youth-DNA-gap numbers and HbA1c levels in all samples, normal, pre-DM, and type 2 DM, were reported (Fig. 2a–d) (r2 = − 0.2199, P = 0.0006, r2 = − 0.3683, P = 0.0030, r2 = − 0.0380, P = 0.7410, and r2 = − 0.3027, P = 0.0023, respectively). The results showed a correlation between youth-DNA-gap number and FBS level in all samples, normal, pre-DM, and type 2 DM (Fig. 3a–d). There was a significant negative relationship between youth-DNA-gap number and FBS level in all sample (r2 = − 0.1452, P = 0.0245) (Fig. 3a).

Association between the percentage of each DNA-gap with HbA1c levels in all samples (a), normal (b), pre-DM (c), and type 2 DM patients (d). Spearman's rank correlation (r) with P values are indicated. (**P < 0.01). The age ranges are represented by the following colors: violet (31–40 years), blue (41–50 years), green (51–60 years), yellow (61–70 years), red (71–80 years), and black (81–90 years).

Correlation of DNA-gap number with FBS level in all samples (a), normal (b), pre-DM (c) and type 2 DM patients (d). Spearman's rank correlation (r) with P values are indicated. (*P < 0.05). The age ranges are represented by the following colors: violet (31–40 years), blue (41–50 years), green (51–60 years), yellow (61–70 years), red (71–80 years), and black (81–90 years).
Correlation between youth-DNA-gap number and age in all in samples, normal, pre-DM and type 2 DM
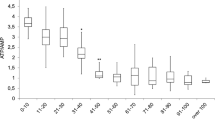
We investigated the relationship between youth-DNA-gap number and age in the the HbA1c and FBS groups. The results showed a significant negative correlation between youth-DNA-gap numbers and age in all groups (all samples, normal, pre-DM, and type 2 DM) when using HbA1c levels (Fig. 4a–d) (r2 = − 0.3618, P < 0.0001, r2 = − 0.4377, P = 0.0003, r2 = − 0.2946, P = 0.0088, and r2 = − 0.3338, P = 0.0007, respectively) and FBS levels (Fig. 5a–d) (r2 = − 0.2544, P < 0.0001, r2 = − 0.2211, P = 0.0583, r2 = − 0.3130, P = 0.0023, and r2 = − 0.1183, P = 0.3187, respectively).

Association between the percentage of each DNA-gap and age when patients were grouped according to HbA1c levels. Correlation between % DNA-GAPs and age in all samples (a), normal (b), pre-DM (c), and type 2 DM (d). Each plot represents youth-DNA-gap levels of the patients. Spearman's rank correlation (r) with P values are indicated. **P < 0.01.

Correlation between the percentage of each DNA-gap and age in all samples (a), normal (b), pre-DM (c), and type 2 DM patients (d) when grouped according to FBS level. Spearman's rank correlation (r) with P values are indicated. **P < 0.01.
Here, we separated males and females into normal, pre-DM, and type 2 DM groups. The gender distribution was determined by voluntary participation, resulting in a higher number of females, which accounts for the greater representation of females in this study. Despite the difference in the number of genders, the Youth-DNA-gap numbers in males and females in the HbA1c group (Fig. 6a) and FBS group (Fig. 6b) did not differ significantly. This demonstrates that gender has no impact on the youth-DNA-gaps.

Comparisons of the percentage of DNA-gap levels between males and females in the normal, pre-DM and DM groups when grouped using the HbA1c level (a) and FBS level (b). The boxes represent the interquartile ranges (25th to 75th percentile), while the median lines represent the 50th percentile. The whiskers represent the minimum and maximum values, and all individual data points are displayed. (Mann–Whitney U test).
Discussion
Previously, we established that youth-DNA gaps are epigenetic marks distributed genome-wide under the influence of AGO4, commonly binding to interspersed repetitive sequences34,37,38. Youth-DNA-gaps prevent DNA damage, DNA damage response, and cellular senescence33. By utilizing Box A of the HMGB1 plasmid to create DNA gaps, we not only reversed aging phenotypes but also successfully cured age-associated diseases in two rat aging models. Our research demonstrated that HMGB1-produced DNA gaps effectively cured conditions such as senile dementia, poor liver function, liver fibrosis, and reversed symptoms of insulin resistance, increased visceral fat, and the size of islets of Langerhans33. As a result, we hypothesize that the reduction of youth-DNA gaps may facilitate age-associated diseases, including type 2 DM.
In this study, we observed a reduction in the number of youth-DNA gaps in patients with type 2 DM. Naturally occurring youth-DNA gaps in the genome play a crucial role in maintaining genomic integrity and tend to decrease with aging in yeast, rats, humans, and in cells induced into senescence by chemicals33,36,37,38. Therefore, to determine whether the association between youth-DNA-gap and type 2 DM was influenced by age, we adjusted for age before comparing normal and type 2 DM patients, and the results remained significant after adjusting for age. The number of youth-DNA gaps displayed an inverse correlation with age when classified by HbA1c levels, which was more pronounced than when classified by FBS levels. Additionally, Youth-DNA-gap number inversely correlated with HbA1c levels more than FBS levels. FBS can be reduced in the short term through diet and exercise before testing, and patients only need to fast overnight for at least 8 to 16 h before the test73. In contrast, HbA1c is proportional to the average glucose, and erythrocytes have a lifespan of approximately 120 days74. As a result, HbA1c values cannot be changed in the short term, making HbA1c levels better than FBS levels for confirming type 2 DM75. More importantly, HbA1c levels represent polymer structure alteration due to the molecular pathogenesis of type 2 DM. The strong correlation between high HbA1c levels and low youth-DNA-gap numbers may imply a connection between reduced youth-DNA-gap numbers and molecular pathology of type 2 DM.
Interestingly, HbA1c levels are associated with other molecular aging markers. HbA1c levels are independently and inversely correlated with telomere length in peripheral blood, genome-wide methylation, and DNA methylation markers of youth58,61,76. Previously, our results showed the roles of the youth-DNA-gap in genomic stability and senescence prevention, and lower numbers of youth-DNA-gaps indicates progressive DNA aging33. Our result showed the association between a low youth-DNA-gap and high HbA1c confirmed that HbA1c represents the cellular aging biomarker of type 2 DM and supported a hypothesis that a low youth-DNA-gap may drive cellular senescence in type 2 DM. Youth-DNA-gap reduction in type 2 DM may be caused by hyperglycemia. High blood glucose (hyperglycemia) is the most important factor in the progression of type 2 DM77,78,79,80,81,82,83,84,85,86. Prolonged hyperglycemia significantly promotes HMGB1 release, causing an upregulation of proinflammatory cytokines and increasing the expression of oxidative production and suppression87,88. Glucose promotes HMGB1 release, resulting in a lack of intranuclear HMGB1. Youth-DNA-gap complexes are composed of Box A of HMGB1, which produces the DNA gap, SIRT1 deacetylated histones, and AGO4 methylated interspersed repetitive sequences31,32,33,34,35. HMGB1 produces the youth-DNA-gap, so HMGB1 release that cause decreases in intranuclear HMGB1 results in decreases in youth-DNA-gaps.
Moreover. Increased hyperglycemia is triggered in response to various cellular stresses, such as oxidative stress, inflammation, and DNA damage89,90,91. Accumulating evidence suggests that oxidative stress can directly damage DNA, resulting in the formation of DNA adducts and strand breaks92,93. The presence of DNA damage initiates p53 activation as a protective response94,95. Furthermore, p53 can directly induce tyrosine phosphorylation of insulin receptor substrate 1 (IRS1). This effect has been found to inhibit insulin signaling and impede glucose uptake. Consequently, increased DNA damage-inducing p53 activity can lead to a decrease in IRS1 expression, modifications in IRS1 phosphorylation, disruption of downstream insulin signaling pathways, and contributing to insulin resistance96,97,98,99. Therefore, hyperglycemia leads to an imbalance in oxidative production and suppression. This imbalance induces p53 activation, contributing to impaired insulin signaling, compromised glucose metabolism, and a decrease in intranuclear HMGB1 levels, ultimately resulting in fewer youth-DNA-gaps. Youth-DNA-gaps play a crucial role in genomic stability and senescence prevention33. Lower numbers of youth-DNA-gaps indicate an adverse effect on genomic stability and cell aging, which may lead to the pathogenesis of type 2 DM.
Decreased youth-DNA-gap numbers may drive DM pathogenesis. Youth-DNA-gap reduction causes DNA damage and consequences that lead to senescence, inflammation, and tissue degeneration33. Several studies have found that DNA damage accumulates and leads to genomic instability during the aging process and in type 2 DM patients100,101,102,103,104,105,106,107,108,109,110. A study showed that the loss of SIRT1 accelerates DNA damage in diabetes111. Loss of SIRT1 function also results in decreased youth-DNA-gap numbers36. One of the pathogenic mechanisms of type 2 DM has been proposed to be cellular dysfunction caused by senescence112,113,114,115. These pieces of evidence support a hypothesis that decreased youth-DNA-gap numbers promote type 2 DM pathogenesis via a DNA damage-induced senescence cascade. Therefore, we hypothesize that decreased youth-DNA-gap numbers may be associated with the molecular pathogenesis of high blood glucose, so youth-DNA-gap levels are another biomarker for monitoring the HbA1c levels in type 2 DM. Previously, we showed that Box A of the HMGB1 expression plasmid could revitalize aging cells and rats by producing Box A-produced DNA gaps. As a result, Box A increased DNA durability and stopped the DNA damage-induced senescence cascade33. Therefore, it is exciting and crucial to investigate whether Box A can reverse the pathogenesis of type 2 DM-associated degenerative diseases.
Conclusions
Youth-DNA-gaps in the genome help maintain genomic stability and prevent cell aging. Our findings revealed a significant decrease in youth-DNA-gap numbers in type 2 DM patients compared to normal controls and an inverse relationship between youth-DNA-gap numbers and HbA1c levels in type 2 DM patients. Decreased youth-DNA-gap numbers may promote the deterioration of cellular function in type 2 DM patients, so youth-DNA-gaps are a promising biomarker and drug target for monitoring and treating type 2 DM tissue degeneration.
Materials and methods
Participants
In this study, we included 240 samples with blood glucose levels monitored using HbA1c. HbA1c values below 5.7% were considered normal, while values between 5.7 and 6.4% indicated pre-DM, and values of 6.5% or higher were indicative of type 2 DM. We divided the samples into three groups: 63 were classified as normal, 78 as pre-DM, and 99 as type 2 DM patients. Additionally, we classified the same samples based on fasting blood sugar (FBS) levels: less than 100 mg/dL was considered normal, 100 to 125 mg/dL indicated pre-DM, and 126 mg/dL or higher indicated type 2 DM. Consequently, some patients changed groups, resulting in 74 normal controls, 93 pre-DM, and 73 type 2 DM patients. All participants in these groups were monitored at the Tambon Health Promoting Hospital in Thailand, with ages ranging from 31 to 85 years. Patients with kidney or cancer conditions were excluded from the experiment. The Ethics Clearance Committee on Human Rights Related to Research Involving Human Subjects, Walailak University, Nakorn Sri Thammarat, Thailand, reviewed and approved the study. Written informed consent was provided by each participant. The study was voluntary for all the participants, and all methods were performed in accordance with the relevant guidelines and regulations.
High-molecular-weight DNA (HMWDNA) preparation
HMWDNA was prepared to preserve the integrity of genomic DNA. Cells (approximately 1 × 106 cells) were collected as previously described33. In brief, cells were mixed and embedded in 70 µl of 1% low-melting-point agarose (MO BIO, CA, USA), lysed and digested in a 400 µl lysis buffer (50 mM Tris pH 8.0, 20 mM EDTA, 1% sodium lauryl sarcosine, and 1 mg/ml proteinase K), and then incubated overnight at 37 °C. The next day, the digested plugs were washed with Tris–EDTA buffer 6 times for 40 min each. The cohesive end-DNA was polished using T4 DNA polymerase and dNTPs (New England Biolabs, MA, USA)37.
HMWDNA preparation for DNA-GAP PCR
Plugs were incubated at 37 °C for 60 min before being washed six times with Tris–EDTA buffer for 20 min each. Ligation-mediated PCR (LM-PCR) linkers (5′-AGGTAACGAGTCAGACCACCGATCGCTCGGAAGCTTACCTCGTGGACGT-3′ and 5′-ACGTCCACGAG-3′) were prepared and ligated to polished the DNA in the plugs with T4 DNA ligase (New England Biolabs), and the plugs were incubated at room temperature for two nights. Using a QIAquick gel extraction kit, HMWDNA was extracted from the agarose plugs (Qiagen, Basel, Switzerland). HMWDNA from each plug was diluted to 20 ng/µl for DNA-GAP PCR.
DNA-GAP measurement
The IRS-EDSB-LM-PCR or DNA-GAP PCR was performed as previously described33. HMWDNA was used for DNA-GAP PCR to identify EDSBs in cells using a QuanStudioTM 6 Flex Real-Time PCR system (Thermo Fisher Scientific, MA, USA). The following components were included in the PCR mix: 1× TaqmanTM Universal PCR Master mix (Applied Biosystems, CA, USA), 0.5 U HotStarTaq DNA polymerase (Qiagen, Hilden, Germany), 0.3 µM probe homologous to the 3′-linker sequence (6-fam) ACGTCCACGAGGTAAGCTTCCGAGCGA (tamra) (phosphate), 0.5 µM IRS primer (LINE-1) (5'-CTCCCAGCGTGAGCGAC-3'). EcoRV and AluI (Thermo Fisher Scientific, MA, USA) were used to digest the control DNA, which was then ligated with linkers. The following were the PCR cycle conditions: 1 cycle of 50 °C for 2 min followed by 95 °C for 10 min and 60 cycles of 95 °C for 15 s and then 60 °C for 2 min. The amount of DNA-GAP PCR in each reaction was compared to the ligated control digested DNA and reported as a percentage of EcoRV and AluI-digested genome DNA-GAP PCR amplicons (%DNA-GAP PCR of control DNA).
Statistical analyses
The data were analyzed using SPSS statistical software. The mean ± SD and median represent the average and distributions of characteristic data in all the samples. T tests were used to determine differences in blood glucose levels between groups in matched cases using a P value threshold of 0.05. To investigate the relationship between two continuous variables, Spearman's correlation coefficient was used.
Ethics approval and consent to participate
The study was reviewed and approved by the Ethics Clearance Committee on Human Rights Related to Researched Involving Human Subjects, Walailak University, Nakorn Sri Thammarat, Thailand. Written informed consent was obtained from each participant.
Data availability
All data generated or analyzed during this study are included in this published article. Further inquiries can be directed to the corresponding author.
Abbreviations
- DM:
-
Diabetes mellitus
- FBS:
-
Fasting blood sugar
- Hyperglycemia:
-
High blood glucose levels
- HbA1c:
-
Hemoglobin A1c
- Youth-DNA-gaps:
-
Youth-associated genome-stabilizing DNA gaps
References
Sun, H. et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 183, 109119 (2022).
Guariguata, L. et al. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res. Clin. Pract. 103, 137–149 (2014).
Chehadeh, S. E. et al. Genetic variants and their associations to type 2 diabetes mellitus complications in the United Arab Emirates. Front. Endocrinol. https://doi.org/10.3389/fendo.2021.751885 (2021).
Prasad, R. B. & Groop, L. Genetics of type 2 diabetes—pitfalls and possibilities. Genes 6, 87–123 (2015).
Ali, O. Genetics of type 2 diabetes. World J. Diabetes 4, 114 (2013).
DeForest, N. & Majithia, A. R. Genetics of type 2 diabetes: Implications from large-scale studies. Current diabetes reports 22, 227–235 (2022).
Curtis, D. Analysis of rare coding variants in 200,000 exome-sequenced subjects reveals novel genetic risk factors for type 2 diabetes. Diabetes/Metab. Res. Rev. 38, e3482 (2022).
Lyssenko, V. & Laakso, M. Genetic screening for the risk of type 2 diabetes: Worthless or valuable?. Diabetes Care 36, S120–S126 (2013).
Hansen, T. In Annales Universitatis Mariae Curie-Sklodowska. Sectio D: Medicina. 544–549.
Laakso, M. & Fernandes Silva, L. Genetics of type 2 diabetes: Past, present, and future. Nutrients 14, 3201 (2022).
Elhefnawy, M. E., Ghadzi, S. M. S. & Noor Harun, S. Predictors associated with type 2 diabetes mellitus complications over time: A literature review. J. Vasc. Dis. 1, 13–23 (2022).
Yen, H.-Y. et al. Complications and comorbidities as influencing factors of health outcomes in older adults with type 2 diabetes mellitus. Collegian 30, 230–235 (2023).
Chen, L., Magliano, D. J. & Zimmet, P. Z. The worldwide epidemiology of type 2 diabetes mellitus—Present and future perspectives. Nat. Rev. Endocrinol. 8, 228–236 (2012).
Zheng, Y., Ley, S. H. & Hu, F. B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 14, 88–98 (2018).
Bayani, M. A., Shakiba, N., Bijani, A. & Moudi, S. Depression and quality of life in patients with type 2 diabetes mellitus. Caspian J. Intern. Med. 13, 335 (2022).
Nikpour, S. et al. Challenges of type 2 diabetes mellitus management from the perspective of patients: Conventional content analysis. Interact. J. Med. Res. 11, e41933 (2022).
Horikawa, Y. et al. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat. Genet. 26, 163–175 (2000).
Liang, B., Li, R., Zhang, P. & Gu, N. Empagliflozin for patients with heart failure and type 2 diabetes mellitus: Clinical evidence in comparison with other sodium-glucose co-transporter-2 inhibitors and potential mechanism. Journal of Cardiovascular Translational Research 16, 327–340 (2023).
Seyoum, T., Tesfaye, S., Shiferaw, Y., Hailu, R. & Tefera, D. Knowledge of behavioural risk factors for types 2 diabetes mellitus and its associated factors among reproductive-age women in Arba Minch town, Gamo zone, 2022. medRxiv (2022).
Zhang, J., Chen, Y., Zou, L. & Gong, R. Prognostic nutritional index as a risk factor for diabetic kidney disease and mortality in patients with type 2 diabetes mellitus. Acta Diabetologica 60, 235–245 (2023).
Kovesdy, C. et al. Predictors of cardio-kidney complications and treatment failure in patients with chronic kidney disease and type 2 diabetes treated with SGLT2 inhibitors. BMC Med. 20, 1–15 (2022).
Lee, J., Yun, J.-S. & Ko, S.-H. Advanced glycation end products and their effect on vascular complications in type 2 diabetes mellitus. Nutrients 14, 3086 (2022).
Zhou, J. et al. Metformin versus sulphonylureas for new onset atrial fibrillation and stroke in type 2 diabetes mellitus: A population-based study. Acta Diabetol. 59, 697–709 (2022).
Ma, Z.-Y., Wu, Y.-Y., Cui, H.-Y.-L., Yao, G.-Y. & Bian, H. Factors influencing post-stroke cognitive impairment in patients with type 2 diabetes mellitus. Clin. Interv. Aging 17, 653–664 (2022).
Chai, K.-C., Chen, W.-M., Chen, M., Shia, B.-C. & Wu, S.-Y. Association between Preexisting Sarcopenia and Stroke in Patients with Type 2 Diabetes Mellitus. The journal of nutrition, health & aging 26, 936-944 (2022).
Thomas, M. C., Cooper, M. E. & Zimmet, P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat. Rev. Nephrol. 12, 73–81 (2016).
Yao, X. et al. Relationship between renal and liver function with diabetic retinopathy in patients with type 2 diabetes mellitus: A study based on cross-sectional data. Sci. Rep. 12, 1–9 (2022).
Raja, S. A., Chong, V. H., Rahman, N. A., Shakir, L. M. & Knights, J. Prevalence and associated factors of diabetic retinopathy among type 2 diabetes mellitus patients in Brunei Darussalam: A cross-sectional study. Korean J. Ophthalmol. 36, 26 (2022).
Eid, M., Mounir, A., El Etriby, S., Al Taher, A. & Ezzat, M. A. Diabetic retinopathy as a predictor of angiographic coronary atherosclerosis severity in patients with type 2 diabetes mellitus. Diabetes Metab. Syndr. Obes. Targets Ther. 15, 1485 (2022).
Mutirangura, A. Is global hypomethylation a nidus for molecular pathogenesis of age-related noncommunicable diseases?. Epigenomics 11, 577–579 (2019).
Pornthanakasem, W. et al. LINE-1 methylation status of endogenous DNA double-strand breaks. Nucl. Acids Res. 36, 3667–3675 (2008).
Kongruttanachok, N. et al. Replication independent DNA double-strand break retention may prevent genomic instability. Mol. Cancer 9, 1–14 (2010).
Yasom, S. et al. The roles of HMGB1-produced DNA gaps in DNA protection and aging biomarker reversal. FASEB BioAdvances 4, 408–434 (2022).
Watcharanurak, P. & Mutirangura, A. Human RNA-directed DNA methylation methylates high-mobility group box 1 protein-produced DNA gaps. Epigenomics 14, 741–756 (2022).
Chalertpet, K. et al. Argonaute 4 as an effector protein in RNA-directed DNA methylation in human cells. Front. Genet. 10, 645 (2019).
Thongsroy, J. et al. Replication-independent endogenous DNA double-strand breaks in Saccharomyces cerevisiae model. PLoS ONE 8, e72706 (2013).
Thongsroy, J., Patchsung, M., Pongpanich, M., Settayanon, S. & Mutirangura, A. Reduction in replication-independent endogenous DNA double-strand breaks promotes genomic instability during chronological aging in yeast. FASEB J. 32, 6252–6260 (2018).
Pongpanich, M., Patchsung, M., Thongsroy, J. & Mutirangura, A. Characteristics of replication-independent endogenous double-strand breaks in Saccharomyces cerevisiae. BMC Genomics 15, 750 (2014).
Vilenchik, M. M. & Knudson, A. G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. 100, 12871–12876 (2003).
Pongpanich, M., Patchsung, M. & Mutirangura, A. Pathologic replication-independent endogenous DNA double-strand breaks repair defect in chronological aging yeast. Front. Genet. 9, 501 (2018).
Kuo, L. J. & Yang, L.-X. γ-H2AX-a novel biomarker for DNA double-strand breaks. In vivo 22, 305–309 (2008).
Celeste, A. et al. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 5, 675–679 (2003).
Katsuta, E. et al. H2AX mRNA expression reflects DNA repair, cell proliferation, metastasis, and worse survival in breast cancer. Am. J. Cancer Res. 12, 793 (2022).
Oberdoerffer, P. & Miller, K. M. In Seminars in Cell & Developmental Biology. 59–72 (Elsevier).
Yang, Y. C. et al. Geriatric syndromes and quality of life in older adults with diabetes. Geriatr. Gerontol. Int. 19, 518–524 (2019).
Yanase, T., Yanagita, I., Muta, K. & Nawata, H. Frailty in elderly diabetes patients. Endocr. J. 65, 1–11 (2018).
Chao, C.-T., Lee, S.-Y., Wang, J., Chien, K.-L. & Huang, J.-W. Frailty increases the risk for developing urinary tract infection among 79,887 patients with diabetic mellitus and chronic kidney disease. BMC Geriatr. 21, 349 (2021).
Kong, L.-N., Lyu, Q., Yao, H.-Y., Yang, L. & Chen, S.-Z. The prevalence of frailty among community-dwelling older adults with diabetes: A meta-analysis. Int. J. Nurs. Stud. 119, 103952 (2021).
Schmidt, M. I. et al. Identifying individuals at high risk for diabetes: The Atherosclerosis Risk in Communities study. Diabetes Care 28, 2013–2018 (2005).
Arvind, K., Pradeepa, R., Deepa, R. & Mohan, V. Diabetes & coronary artery disease. Indian J. Med. Res. 116, 163 (2002).
Kaneyama, A. et al. Impact of hypertension and diabetes on the onset of chronic kidney disease in a general Japanese population. Hypertens. Res. 46, 311–320 (2023).
Xu, W., Qiu, C., Winblad, B. & Fratiglioni, L. The effect of borderline diabetes on the risk of dementia and Alzheimer’s disease. Diabetes 56, 211–216 (2007).
Qi Nan, W., Ling, Z. & Bing, C. The influence of the telomere-telomerase system on diabetes mellitus and its vascular complications. Expert Opin. Ther. Targets 19, 849–864 (2015).
Zhou, Y., Ning, Z., Lee, Y., Hambly, B. D. & McLachlan, C. S. Shortened leukocyte telomere length in type 2 diabetes mellitus: Genetic polymorphisms in mitochondrial uncoupling proteins and telomeric pathways. Clin. Transl. Med. 5, 1–6 (2016).
Murillo-Ortiz, B. et al. Telomere length and type 2 diabetes in males, a premature aging syndrome. Aging Male 15, 54–58 (2012).
Zhang, Z. et al. The impact of oxidative stress-induced mitochondrial dysfunction on diabetic microvascular complications. Front. Endocrinol. https://doi.org/10.3389/fendo.2023.1112363 (2023).
Cojocaru, K.-A. et al. Mitochondrial dysfunction, oxidative stress, and therapeutic strategies in diabetes, obesity, and cardiovascular disease. Antioxidants 12, 658 (2023).
Thongsroy, J., Patchsung, M. & Mutirangura, A. The association between Alu hypomethylation and severity of type 2 diabetes mellitus. Clin. Epigenet. 9, 93 (2017).
Willmer, T., Johnson, R., Louw, J. & Pheiffer, C. Blood-based DNA methylation biomarkers for type 2 diabetes: Potential for clinical applications. Front. Endocrinol. 9, 744 (2018).
Ahmed, S. A. H., Ansari, S. A., Mensah-Brown, E. P. & Emerald, B. S. The role of DNA methylation in the pathogenesis of type 2 diabetes mellitus. Clin. Epigenet. 12, 1–23 (2020).
Vetter, V. M. et al. DNA methylation age acceleration is associated with risk of diabetes complications. Commun. Med. 3, 21 (2023).
Fraszczyk, E. et al. DNA methylation trajectories and accelerated epigenetic aging in incident type 2 diabetes. GeroScience 44, 2671–2684 (2022)
Chung, J.K.-O., Xue, H., Pang, E.W.-H. & Tam, D.C.-C. Accuracy of fasting plasma glucose and hemoglobin A1c testing for the early detection of diabetes: A pilot study. Front. Lab. Med. 1, 76–81 (2017).
Florkowski, C. HbA1c as a diagnostic test for diabetes mellitus—reviewing the evidence. Clin. Biochemist Rev. 34, 75 (2013).
Moghadam, A. R. S., Hozhabr-Araghi, F., Vashani, H. B., Moonaghi, H. K. & Bazzi, A. Evaluation of the effect of self-care education based on VARK learning style on HbA1c and FBS levels in patients with type II diabetes. Med.-Surg. Nurs. J. 5, 58–65 (2016).
Rasoulinejad, S. A., Meftah, N., Maniati, M. S. & Maniati, M. High levels of FBS and HbA1c and their association with diabetic retinopathy: A study in the north of Iran. J. Diabetes Metab. Disord. 21, 399–406 (2022).
Jeon, J. Y. et al. Prevalence of diabetes and prediabetes according to fasting plasma glucose and HbA1c. Diabetes Metab. J. 37, 349–357 (2013).
Association, A. D. Standards of medical care in diabetes—2019 abridged for primary care providers. Clin. Diabetes 37, 11 (2019).
Lipska, K. J. et al. Elevated HbA1c and fasting plasma glucose in predicting diabetes incidence among older adults: Are two better than one?. Diabetes Care 36, 3923–3929 (2013).
Inzucchi, S. E. et al. Management of hyperglycemia in type 2 diabetes: A patient-centered approach: Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 35, 1364–1379 (2012).
Sherwani, S. I., Khan, H. A., Ekhzaimy, A., Masood, A. & Sakharkar, M. K. Significance of HbA1c test in diagnosis and prognosis of diabetic patients. Biomark. Insights 11, BMI. S38440 (2016).
Organization, W. H. Use of Glycated Haemoglobin (HbA1c) in Diagnosis of Diabetes Mellitus: Abbreviated Report of a WHO Consultation (World Health Organization, 2011).
Mathew, T. K. & Tadi, P. StatPearls (StatPearls Publishing, 2021).
Sacks, D. B. A1C versus glucose testing: A comparison. Diabetes Care 34, 518–523 (2011).
Swetha, N. Comparison of fasting blood glucose & post prandial blood glucose with HbA1c in assessing the glycemic control. Int. J. Healthc. Biomed. Res. 2, 134–139 (2014).
Tamura, Y. et al. β-cell telomere attrition in diabetes: Inverse correlation between HbA1c and telomere length. J. Clin. Endocrinol. Metab. 99, 2771–2777 (2014).
Beck, R. W. et al. The relationships between time in range, hyperglycemia metrics, and HbA1c. J. Diabetes Sci. Technol. 13, 614–626 (2019).
Davies, M. J. et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 41, 2669–2701 (2018).
Giugliano, D., Ceriello, A. & Esposito, K. Glucose metabolism and hyperglycemia. Am. J. Clin. Nutr. 87, 217S-222S (2008).
Suh, S. & Park, M. K. Glucocorticoid-induced diabetes mellitus: An important but overlooked problem. Endocrinol. Metab. 32, 180–189 (2017).
Association, A. D. Diagnosis and classification of diabetes mellitus. Diabetes Care 32, S62–S67 (2009).
Chaudhury, A. et al. Clinical review of antidiabetic drugs: Implications for type 2 diabetes mellitus management. Front. Endocrinol. 8, 6 (2017).
Association, A. D. Diagnosis and classification of diabetes mellitus. Diabetes Care 36, S67 (2013).
Association, A. D. Diagnosis and classification of diabetes mellitus. Diabetes Care 33, S62–S69 (2010).
Godinjak, A. et al. Hyperglycemia in critically ill patients: Management and prognosis. Med. Arch. 69, 157 (2015).
Kovatchev, B. P., Otto, E., Cox, D., Gonder-Frederick, L. & Clarke, W. Evaluation of a new measure of blood glucose variability in diabetes. Diabetes Care 29, 2433–2438 (2006).
Newsholme, P., Cruzat, V. F., Keane, K. N., Carlessi, R. & de Bittencourt Jr, P. I. H. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 473, 4527–4550 (2016).
Chen, Y., Qiao, F., Zhao, Y., Wang, Y. & Liu, G. HMGB1 is activated in type 2 diabetes mellitus patients and in mesangial cells in response to high glucose. Int. J. Clin. Exp. Pathol. 8, 6683 (2015).
Giri, B. et al. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed. Pharmacother. 107, 306–328 (2018).
Gheitasi, I. et al. Molecular mechanisms of hawthorn extracts in multiple organs disorders in underlying of diabetes: A review. Int. J. Endocrinol. 2022, 1–14 (2022).
Yang, X. et al. Stress hyperglycemia is independently associated with persistent organ failure in acute pancreatitis. Digest. Dis. Sci. 67, 1879–1889 (2022).
Cadet, J. & Wagner, J. R. Oxidatively generated base damage to cellular DNA by hydroxyl radical and one-electron oxidants: Similarities and differences. Arch. Biochem. Biophys. 557, 47–54 (2014).
De Bont, R. & Van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 19, 169–185 (2004).
Williams, A. B. & Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 6, a026070 (2016).
Wang, Y. H. & Sheetz, M. P. Transcription-independent functions of p53 in DNA repair pathway selection. BioEssays 45, 2200122 (2023).
Xi, G., Shen, X., Wai, C., White, M. F. & Clemmons, D. R. Hyperglycemia induces vascular smooth muscle cell dedifferentiation by suppressing insulin receptor substrate-1-mediated p53/KLF4 complex stabilization. J. Biol. Chem. 294, 2407–2421 (2019).
Peng, P. et al. Inhibition of p53 relieves insulin resistance in fetal growth restriction mice with catch-up growth via activating IGFBP3/IGF-1/IRS-1/Akt signaling pathway. J. Nanosci. Nanotechnol. 18, 3925–3935 (2018).
Machado-Neto, J. A. et al. Insulin Substrate Receptor (IRS) proteins in normal and malignant hematopoiesis. Clinics 73, e566s (2018).
Cui, D. et al. The cross talk between p53 and mTOR pathways in response to physiological and genotoxic stresses. Front. Cell Dev. Biol. 9, 3320 (2021).
Lodovici, M. et al. Oxidative DNA damage and plasma antioxidant capacity in type 2 diabetic patients with good and poor glycaemic control. Mutat. Res./Fundam. Mol. Mech. Mutagen. 638, 98–102 (2008).
Blasiak, J. et al. DNA damage and repair in type 2 diabetes mellitus. Mutat. Res./Fundam. Mol. Mech. Mutagen. 554, 297–304 (2004).
Pitozzi, V., Giovannelli, L., Bardini, G., Rotella, C. M. & Dolara, P. Oxidative DNA damage in peripheral blood cells in type 2 diabetes mellitus: Higher vulnerability of polymorphonuclear leukocytes. Mutat. Res./Fundam. Mol. Mech. Mutagen. 529, 129–133 (2003).
Ibarra-Costilla, E. et al. DNA damage evaluated by comet assay in Mexican patients with type 2 diabetes mellitus. Acta Diabetol. 47, 111–116 (2010).
Al-Aubaidy, H. A. & Jelinek, H. F. Oxidative DNA damage: Antioxidant response in postprandial hyperglycaemia in type 2 diabetes mellitus. Br. J. Diabetes Vasc. Dis. 11, 87–91 (2011).
Binici, D., Karaman, A., Coşkun, M., Oğlu, A. U. & Uçar, F. Genomic damage in patients with type-2 diabetes mellitus. Genet. Couns 24, 149–156 (2013).
Çalışkan, Z. et al. SIRT6 expression and oxidative DNA damage in individuals with prediabetes and type 2 diabetes mellitus. Gene 642, 542–548 (2018).
Hinokio, Y. et al. Oxidative DNA damage in diabetes mellitus: Its association with diabetic complications. Diabetologia 42, 995–998 (1999).
Oguntibeju, O. O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 11, 45 (2019).
Milic, M. et al. DNA damage in non-communicable diseases: A clinical and epidemiological perspective. Mutat. Res./Fundam. Mol. Mech. Mutagen. 776, 118–127 (2015).
Lee, S. C. & Chan, J. C. Evidence for DNA damage as a biological link between diabetes and cancer. Chin. Med. J. 128, 1543–1548 (2015).
Bartoli-Leonard, F. et al. Loss of SIRT1 in diabetes accelerates DNA damage-induced vascular calcification. Cardiovasc. Res. 117, 836–849 (2021).
Khosla, S., Farr, J. N., Tchkonia, T. & Kirkland, J. L. The role of cellular senescence in ageing and endocrine disease. Nat. Rev. Endocrinol. 16, 263–275 (2020).
Palmer, A. K. et al. Cellular senescence in type 2 diabetes: A therapeutic opportunity. Diabetes 64, 2289–2298 (2015).
Chukwuma, C. Sr. Aging, cellular senescence and diabetes mellitus; Clinicopathological correlates, trends and targets. Arch. Health Sci. 10, 31829–32641 (2020).
Xiong, Y. & Zhou, L. The signaling of cellular senescence in diabetic nephropathy. Oxid. Med. Cell. Longev. 2019, 1–16 (2019).
Acknowledgements
We express our gratitude to the staff of the Ban Ton Riang Tambon Health Promoting Hospital in Nakorn Sri Tummarat, Thailand, for their invaluable assistance in sample collection.
Funding
This s was financially supported by Office of the Permanent Secretary, Ministry of Higher Education, Science, Research and Innovation Grant No. RGNS 63-213, and the National Science and Technology Development Agency, Thailand [Research Chair Grant, P-19-50189 to A.M.].
Author information
Authors and Affiliations
Contributions
J.T. isolated DNA from plasma samples and processed the data. J.T. performed the HMWDNA preparation for DNA-GAP PCR. J.T. and A.M. discuss the results part. J.T. and A.M. wrote the first draft of the manuscript and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thongsroy, J., Mutirangura, A. The inverse association between DNA gaps and HbA1c levels in type 2 diabetes mellitus. Sci Rep 13, 18987 (2023). https://doi.org/10.1038/s41598-023-46431-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-46431-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
