
Abstract
Using Col2.3GFP transgenic mice expressing GFP in maturing osteoblasts, we isolated Col2.3GFP+ enriched osteoblasts from 3 sources. We performed RNA-sequencing, identified 593 overlapping genes and confirmed these genes are highly enriched in osteoblast differentiation and bone mineralization annotation categories. The top 3 annotations are all associated with endoplasmic reticulum and Golgi vesicle transport. We selected 22 trafficking genes that have not been well characterized in bone for functional validation in MC3T3-E1 pre-osteoblasts. Transient siRNA knockdown of trafficking genes including Sec24d, Gosr2, Rab2a, Stx5a, Bet1, Preb, Arf4, Ramp1, Cog6 and Pacs1 significantly increased mineralized nodule formation and expression of osteoblast markers. Increased mineralized nodule formation was suppressed by concurrent knockdown of P4ha1 and/or P4ha2, encoding collagen prolyl 4-hydroxylase isoenzymes. MC3T3-E1 pre-osteoblasts with knockdown of Cog6, Gosr2, Pacs1 or Arf4 formed more and larger ectopic mineralized bone nodules in vivo, which was attenuated by concurrent knockdown P4ha2. Permanent knockdown of Cog6 and Pacs1 by CRISPR/Cas9 gene editing in MC3T3-E1 pre-osteoblasts recapitulated increased mineralized nodule formation and osteoblast differentiation. In summary, we have identified several vesicle trafficking genes with roles in osteoblast function. Our findings provide potential targets for regulating bone formation.
Similar content being viewed by others


Introduction
Osteoblasts are major bone forming cells. Osteoblast dysregulation may cause inadequate or excessive mineralization of bones or ectopic calcification. Identification of novel genes and signaling pathways associated with osteoblast differentiation and function may lead to better understanding of bone development, homeostasis, and regeneration. Osteoblasts synthesize and secrete large amounts of type I collagen into the extracellular matrix that is subsequently mineralized to provide mechanical support and scaffolding for muscle attachment1. Proteins destined for secretion are synthesized in the endoplasmic reticulum (ER), then transported via vesicles to the Golgi apparatus, then in turn to the plasma membrane. During this process vesicle budding, cargo selection, transportation, and docking to destination compartments (such as the plasma membrane) are regulated by vesicle trafficking proteins such as Coat Protein Complex II (COPII) components, vesicular/target membrane proteins (v-SNARE/t-SNARE) and the Rab family of small GTPases2. In osteoblasts, mutations in COPII coat subunits (e.g. Sec23A and Sec24d) that regulate budding of collagen-containing vesicles from the ER are associated with skeletal defects in patients with cranio-lenticulo-sutural dysplasia3 and osteogenesis imperfecta (OI)4,5,6, respectively. A mutation variant of CREB3L1, which encodes ER-localized old astrocyte specifically induced substance (OASIS) has also been shown to be involved in altered cellular secretion in OI patients7.
In this study, we performed RNA sequencing of Col2.3GFP+ enriched osteoblasts, in which GFP expression marks mature osteoblasts8, and identified several vesicle trafficking genes that are involved in osteoblast differentiation and mineralization. Disruption of their expression in MC3T3-E1 pre-osteoblasts increased osteoblast differentiation and mineralized nodule formation, with enhanced collagen modification by prolyl 4-hydroxylase. Our study provides novel insight into the role of vesicle traffic proteins in osteoblast differentiation and mineralized nodule formation.
Results
Genes involved in vesicle trafficking are enriched in transcriptomes of mature osteoblasts
In Col2.3GFP transgenic mice that express green fluorescent protein (GFP) under the control of the 2.3 kb Col1a1 promoter, Col2.3GFP is expressed in maturing osteoblast lineage cells8. We reasoned that genome-wide gene expression profiling of the Col2.3GFP+ population might identify novel genes and signaling pathways critical for osteoblast function. We isolated Col2.3GFP+ enriched osteoblasts from three sources: (1) freshly isolated mouse bone GFP+ (lo) and GFP+ (hi) populations from long bones of Col2.3GFP transgenic mice, which represent early and late stage (mature) osteoblasts, respectively9,10; (2) GFP+ (lo) and GFP+ (hi) populations isolated from osteogenic differentiation of bone chips from Col2.3GFP transgenic mice11; and (3) GFP+ osteoblasts derived from osteogenic differentiation of mouse Col2.3GFP embryonic stem cell (ESC) lines9. We performed RNA-sequencing and confirmed that significantly upregulated genes in Col2.3GFP+ populations are enriched in the osteogenesis gene set (obtained from Qiagen, which includes 82 genes related to osteogenic differentiation) and in osteoblast differentiation, bone development, bone mineralization and ossification annotation categories (Fig. 1A and S1). We reasoned that overlapping genes expressed by Col2.3GFP+ populations from three sources of osteoblasts would further select for osteogenic candidate genes. We identified 593 overlapping genes that were highly enriched in Col2.3GFP+ osteoblasts from all three sources (Fig. 1B). Gene Ontology analysis reveals that the top 10 annotation terms include endochondral ossification, collagen fibril organization, positive regulation of osteoblast differentiation, and regulation of bone mineralization. Interestingly, the top 3 annotation terms are vesicle fusion with Golgi apparatus/Golgi vesicle transport; negative regulation of ER stress-induced intrinsic apoptotic signaling; and COPII-coated vesicle budding/ER to Golgi vesicle-mediated transport, all of which are associated with ER and Golgi vesicle transport (Fig. 1C). The endoplasmic reticulum unfolded protein response is also among the top 10 annotation terms (Fig. 1C). These gene annotations prompted us to focus on the function of vesicle trafficking genes in osteoblast cells.

Genes involved in vesicle trafficking are enriched in transcriptomes of mature osteoblasts. (A) Gene set enrichment analysis (GSEA) shows significantly upregulated genes in Col2.3GFP+ population are enriched in osteogenesis gene set. (B) 593 overlapping genes that highly enriched in Col2.3GFP+ osteoblasts from all three resources are identified. (C) Gene Ontology Analysis shows that the top 3 annotation terms are all associated with ER and Golgi vesicle transport.
Transient knockdown of vesicle trafficking genes increases MC3T3-E1 pre-osteoblast osteogenic differentiation and mineralized nodule formation
A total of 57 vesicle trafficking genes are included in the top 3 annotation terms (Supplementary File: Vesicle trafficking gene lists). We selected 22 trafficking genes that have not been well characterized in bone with relatively high expression reads to perform siRNA knockdown in MC3T3-E1 pre-osteoblast cells. Interestingly we found that knockdown of 15 trafficking genes (Sec24d, Aplp1, Gosr2, Rab2a, Stx5a, Bet1, Preb, Golph3, Golga2, Arf4, Ramp1, Golga4, Cog6, Myo18a and Pacs1) significantly increased mineralized nodule formation by Alizarin Red S staining (Fig. 2A), confirmed by Von Kossa staining (Fig. 2B). Alkaline phosphatase activity was mildly increased by knockdown of several trafficking genes (Supplementary Fig. S2). Expression of osteoblast gene markers (e.g., Ibsp and/or Bglap) was also significantly increased with knockdown of these 15 trafficking genes, except for Golga2 (Fig. 3). Knockdown efficiency was confirmed by qPCR (Supplementary Fig. S3). mRNA levels of these trafficking genes increased during osteogenic differentiation (Supplementary Fig. S4), consistent with a potential role in osteoblast differentiation and mineralized nodule formation.

Transient knockdown of vesicle trafficking genes increases mineralized nodule formation in MC3T3-E1 osteogenic differentiation. (A) Ten days after osteogenic differentiation, representative Alizarin red S (A) and Von Kossa staining (B) images in control and trafficking gene knockdown MC3T3-E1 osteoblast differentiation. All data represent with scatter plot with mean ± SD (N = 6–9; multiple t test; *P < 0.05; **P < 0.01; #P < 0.001; relative to siControl).

Transient knockdown of vesicle trafficking genes increases osteoblast marker expression in MC3T3-E1 osteogenic differentiation. Ten days after osteogenic differentiation, relative Ibsp and Bglap mRNA expression in control and trafficking gene knockdown MC3T3-E1 osteoblast differentiation were analyzed by qRT-PCR. Y axes show relative Ibsp and Bglap mRNA expression in trafficking gene knockdown normalized to siControl. All data represent with box-whiskers plot (N = 6–9; multiple t test; *P < 0.05; **P < 0.01; #P < 0.001; relative to siControl).
Osteoblasts secrete large amounts of collagen, especially type I collagen, into the extracellular matrix where they assemble into fibers forming a scaffold for the deposition of hydroxyapatite. We examined collagen gene expression in cells with knockdown of trafficking genes. With the exception of Preb, Golga2, Golga 4, and Myo18a knockdown in which Col1a1, Col1a2, Col4a1 or Col4a2 mRNA levels significantly increased, knockdown of most trafficking genes did not significantly increase expression of Col1a1, Col1a2, Col4a1 and Col4a2 (Supplementary Fig. S5).
Expression of prolyl 4-hydroxylase subunits increases with vesicle trafficking gene knockdown
Since these trafficking genes encode ER- and/or Golgi-associated proteins that participate in vesicular transport, we examined whether knockdown of these trafficking genes affected ER homeostasis. We found that expression of at least one of the ER stress sensors (Xbps1, GRP78/Bip and Chop) was significantly increased with knockdown of Sec24d, Aplp1, Gosr2, Rab2a, Stx5a, Bet1, Preb, Arf4, Ramp1, Golga4, Cog6 or Pacs1 (Fig. 4). Disruption of ER homeostasis may lead to accumulation of misfolded proteins and the unfolded protein response12,13.

Transient knockdown of vesicle trafficking genes increases ER stress sensor gene expression in MC3T3-E1 osteogenic differentiation. Ten days after osteogenic differentiation, relative Xbp1s, GRP78/Bip and Chop mRNA expression in control and trafficking gene knockdown MC3T3-E1 cell differentiation were analyzed by qRT-PCR. Y axes show relative Xbp1s, GRP78/Bip and Chop mRNA expression in trafficking gene knockdown normalized to siControl. All data represent with box-whiskers plot (N = 6–9; multiple t test; *P < 0.05; **P < 0.01; #P < 0.001; relative to siControl).
Hypermineralization is a hallmark of some forms of osteogenesis imperfecta (OI), the result of collagen over-modification during prolonged helical folding resulting in increased space for mineral crystal deposition between collagen molecules14. It has also been shown that metabolically regulated collagen modification (e.g., proline and lysine hydroxylation on collagen) renders the cartilaginous matrix more resistant to protease mediated degradation and thereby increases bone mass15. We examined expression of genes encoding several collagen-modifying enzymes including collagen prolyl 3-hydroxylase (P3H), prolyl 4-hydroxylase subunit alpha (P4HA, which forms tetramers with β subunit protein disulfide isomerase (PDI)), and lysine hydroxylases (PLOD) in MC3T3-E1 pre-osteoblast cells with knockdown of trafficking genes. Interestingly, we found that expression of at least one of collagen modifying isoenzyme was significantly increased with knockdown of 12 trafficking genes (Sec24d, Aplp1, Gosr2, Rab2a, Stx5a, Bet1, Preb, Arf4, Ramp1, Cog6, Myo18a and Pacs1) (Fig. 5A, Supplementary Fig. S6 and S7). Expression of one or both collagen prolyl 3-hydroxylase (P3h) isoenzymes was significantly increased with knockdown of Aplp1, Arf4, Myo 18a and Pacs1. Expression of at least one of the lysine hydroxylases (Plod) isoenzymes was significantly increased with knockdown of Sec24d, Gosr2, Bet1, Arf4 and Pacs1. Notably, expression of P4ha1 and/or P4ha2, which encode prolyl 4-hydroxylase subunit alpha (P4HA) was significantly increased with knockdown of 10 trafficking genes (Sec24d, Gosr2, Rab2a, Stx5a, Bet1, Preb, Arf4, Ramp1, Cog6 and Pacs1). Prolyl 4-hydroxylase plays a central role in collagen synthesis as 4-hydroxyproline residues are essential for the formation of triple helical molecules in vivo16. Therefore, we focused on P4ha1 and P4ha2 for further study.

Expression of prolyl 4-hydroxylase subunit alpha P4ha gene and hydroxyproline protein level increase with vesicle trafficking gene knockdown in MC3T3-E1 osteogenic differentiation. (A) Ten days after osteogenic differentiation, relative P4ha1 and P4ha2 mRNA expression in control and trafficking gene knockdown MC3T3-E1 cell differentiation were analyzed by qRT-PCR. Y axes show relative P4ha1 and P4ha2 mRNA expression in trafficking gene knockdown normalized to siControl. All data represent with box-whiskers plot (N = 6–9; multiple t test; *P < 0.05; **P < 0.01; #P < 0.001; relative to siControl). (B) Ten days after osteogenic differentiation, Western-blot analysis shows Hydroxyproline and GAPDH protein expression in control and trafficking gene knockdown MC3T3-E1 cell differentiation. Full gel blots are included in Supplementary Fig. S11. All data represent with scatter plots with mean ± SD (N = 3–6; multiple t test; *P < 0.05; **P < 0.01; #P < 0.001; relative to siControl).
Concurrent knockdown of P4ha1/P4ha2 rescues increased mineralized nodule formation associated with knockdown of vesicle trafficking genes
We examined total intracellular hydroxyproline protein levels and found they were significantly increased with knockdown of Sec24d, Gosr2, Rab2a, Stx5a, Bet1, Preb, Arf4, Ramp1, Cog6 and Pacs1 trafficking genes during osteogenic differentiation (Fig. 5B). To determine whether increased P4ha expression contributed to increased mineralized nodule formation with knockdown of trafficking genes, we performed concurrent siRNA knockdown of P4ha1 or P4ha2 with Sec24d, Gosr2, Rab2a, Stx5a, Bet1, Preb, Arf4, Ramp1, Cog6 or Pacs1 trafficking genes. Knockdown of P4ha1 and P4ha2 was confirmed by significantly decreased P4HA1 and P4HA2 protein levels (Supplementary Fig. S8). Concurrent knockdown of P4ha1 rescued the increased nodule formation phenotype associated with knockdown of Gosr2, Rab2a, Bet1, Arf4, Cog6 and Pacs1 trafficking genes (Fig. 6A). Concurrent knockdown of P4ha2 rescued the increased nodule formation phenotype associated with knockdown of Gosr2, Rab2a, Stx5a, Arf4, Ramp1, Cog6 and Pacs1 trafficking genes (Fig. 6B). These data suggest a role for prolyl 4-hydroxylation in the increased mineralized nodule formation results with knockdown of Gosr2, Rab2a, Stx5a, Bet1, Arf4, Ramp1, Cog6 and Pacs1 trafficking genes. Of note, neither osteoblast marker nor ER stress genes were significantly affected by concurrent knockdown P4ha1 or P4ha2 with Gosr2, Rab2a, Stx5a, Bet1, Arf4, Ramp1, Cog6 and Pacs1 (Supplementary Fig. S9), suggesting that prolyl 4-hydroxylation is directly involved in mineralized nodule formation rather than osteoblast differentiation and/or ER stress.

Concurrent knockdown of P4ha1/P4ha2 rescues increased mineralized nodule formation associated with knockdown of vesicle trafficking genes. Ten days after osteogenic differentiation, representative Alizarin red S staining images and quantification analysis in control and trafficking genes with/without concurrent knockdown of P4ha1 (A) and P4ha2 (B) MC3T3-E1 cell differentiation. All data represent with box-whiskers plot (N = 6–9; multiple t test; *P < 0.05; **P < 0.01; #P < 0.001).
Vesicle trafficking gene knockdown increases mineralized nodule formation in vivo
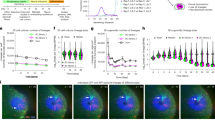
To examine the effects of vesicle trafficking gene knockdown on mineralized nodule formation in vivo, we knocked down Gosr2, Arf4, Cog6 and Pacs1 (selected for the most consistency of in vitro phenotypes) with and without concurrent knockdown of P4ha2 in MC3T3-E1 pre-osteoblasts, which were mixed with Matrigel and implanted subcutaneously into immunodeficient mice. Mice were euthanized at 8 weeks and implants were analyzed by X-ray and micro-CT. The number of mineralized nodules formed was significantly higher with knockdown of Gosr2, Arf4, Cog6 or Pacs1 compared to control (Fig. 7A, B). While concurrent knockdown of P4ha2 did not significantly reduce the numbers of mineralized nodules compared to knockdown of Gosr2, Arf4, Cog6 or Pacs1 alone, mineralized lump volume and micro-CT total intensity were significantly decreased (Fig. 7C). We confirmed mineralization by histological analysis of nodules stained with Alizarin Red S and Von Kossa (Fig. 7D). Collectively, these data suggest that knockdown of vesicle trafficking gene Gosr2, Arf4, Cog6 and Pacs1 increases mineralized nodule formation in vivo in a P4ha2-dependent manner.

Vesicle trafficking gene depletion increases mineralized nodule formation in vivo. MC3T3-E1 cells transfected with Control siRNA, trafficking gene siRNA with/without P4ha2 siRNA and Matrigel mixtures were subcutaneously injected into immunodeficient mice. Eight weeks later, mice were euthanized and dissected. (A) Representative micro-CT X-ray images of explanted nodules show significant mineralized structure in each cell group of implants. (B) Incidences of mineralized nodule formation in each group. *P < 0.05; Z proportion score. (C) Quantification of mineralized nodule volume and micro-CT total intensity in each group. All data represent with interleaved scatter plots with mean ± SD (multiple t test; *P < 0.05; **P < 0.01; #P < 0.001). (D) Serial cryosections were made in each explanted cell nodules. Mineralized structures were visualized using Alizarin red S and Von Kossa staining. Newly formed mineralized structures were visualized by Aniline Blue staining.
Cog6 −/− and Pacs1 −/− MC3T3-E1 cells exhibit increased osteogenic differentiation and mineralized nodule formation
siRNA-mediated knockdown of trafficking genes is transient. To test the effects of permanent deletion of vesicle trafficking genes and further study the mechanisms underlying increased mineralized nodule formation, we generated loss-of-function mutations in the Cog6 and Pacs1 genes used CRISPR-mediated editing. mRNA analysis confirmed the decreased mRNA in Cog6−/− and Pacs1−/− MC3T3-E1 CRISPR lines compared to control CRISPR line (Supplementary Fig. S10A). Deletion of Cog6 or Pacs1 had no effect on cell proliferation (Supplementary Fig. S10B). Congruent with transient siRNA knockdown results, Cog6−/− and Pacs1−/− MC3T3-E1 CRISPR lines displayed increased mineralized nodule formation compared to control CRISPR line subjected to osteogenesis (Fig. 8A and 8B). Expression of osteogenic markers Ibsp and Bglap were significantly higher compared to control CRISPR line, suggesting accelerated osteogenic differentiation (Fig. 8C). This data validates that lack of vesicle trafficking genes Cog6 or Pacs1 accelerated osteogenic differentiation and mineralized nodule formation in MC3T3 pre-osteoblasts.

Cog6−/− and Pacs1−/− MC3T3-E1 cells exhibit increased osteogenic differentiation and mineralized nodule formation. Cog6−/−, Pacs1−/− MC3T3-E1 CRISPR lines and control CRISPR line were subjected to osteogenesis for ten days. (A) Representative Alizarin red S staining images and quantification analysis in control and Cog6−/−, Pacs1−/− CRISPR line differentiation. All data represent with scatter plots with mean ± SD (N = 33; multiple t test; *P < 0.05; **P < 0.01; #P < 0.001). (B) Von Kossa staining in control and Cog6−/−, Pacs1−/− CRISPR line differentiation. (C) qRT-PCR analysis shows relative Ibsp and Bglap mRNA expression in control and Cog6−/−, Pacs1−/− CRISPR line differentiation. All data represent with box-whiskers plot (N = 11–13; multiple t test; *P < 0.05; **P < 0.01; #P < 0.001; relative to Control−/−).
Discussion
By performing RNA-sequencing on three sources of Col2.3GFP+ maturing osteoblasts, we identified 593 overlapping genes that are enriched among osteoblasts including genes encoding osteogenic markers (e.g., Ibsp, Bglap), osteogenic transcription factors (e.g., Runx2, Sp7, Dlx5) as well as genes in which mutations have been associated with skeletal defects (e.g., Sec24d, Creb3l1). Of note, the top three gene annotation terms were all related to vesicle mediated ER/Golgi transport.
We found that transient siRNA knockdown of 15 trafficking genes (Sec24d, Aplp1, Gosr2, Rab2a, Stx5a, Bet1, Preb, Golph3, Golga2, Arf4, Ramp1, Golga4, Cog6, Myo18a and Pacs1) increased mineralized nodule formation and osteoblast marker gene expression. Vesicle trafficking proteins are responsible for cargo selection, transportation, and secretion2. Disruption of vesicle trafficking gene expression and function can lead to the unfolded protein response, delayed collagen folding, prolonged collagen transportation, and increased collagen modification. In OI type I less than 50% of collagen is produced in an environment with a normal level of lysyl hydroxylating enzymes which results in an increased enzyme to collagen ratio and subsequent higher levels of hydroxylation and glycosylation as well14. Also, proline hydroxylation enhances the stability of the collagen triple helices and lysine modifications increase crosslinking16. In OI increased hydroxylation and glycosylation of collagen leads to greater distance between collagen molecules within the fibril. As a result of this steric hinderance more crystals can be deposited within collagen fibrils, leading to hypermineralization14. Increased expression of ER stress markers associated with knockdown of Gosr2, Rab2a, Arf4, Cog6, Pacs1, Bet1, Stx5a or Ramp1 suggest that ER homeostasis may have been disrupted. It has been shown that increased matrix deposition during endochondral ossification is associated with activation of endoplasmic reticulum stress sensors, which in turn changed the availability of the metabolic substrates and further increased gene expression levels of collagen-modifying enzymes including collagen prolyl hydroxylases, lysine hydroxylases and lysyl oxidase15. Therefore, one possible mechanism for the increased mineralized nodule formation seen with knockdown of trafficking genes Gosr2, Rab2a, Arf4, Cog6, Pacs1, Bet1, Stx5a or Ramp1 might be due to enhanced prolyl 4-hydroxylation caused by increased P4ha1 and/or P4ha2 expression, which in turn increases the space available for mineral deposition among the extracellular matrix. Consistent with this mode, increased mineralized nodule formation resulting from knockdown of Gosr2, Rab2a, Arf4, Cog6, Pacs1, Bet1, Stx5a and Ramp1 was associated with increased P4ha1 and/or P4ha2 expression, and double knockdown of these trafficking genes and P4ha1 and/or P4ha2 reduced mineralized nodule formation. In an ectopic bone formation model, knockdown of Gosr2, Arf4, Cog6 and Pacs1 increased mineralized nodule formation in vivo, while concurrent knockdown of P4ha2 rescued this phenotype.
Additional collagen-modifying enzymes may also play a role. In particular, expression of prolyl 3-hydroxylase genes P3h1 and/or P3h2 was increased by knockdown of Aplp1, Arf4, Myo18a or Pacs1 while expression of the lysine hydroxylase gene Plod was increased by knockdown of Sec24d, Gosr2, Bet1, Arf4 or Pacs1. Of the 15 vesicle trafficking genes we identified that exhibited increased mineralized nodule formation upon transient knockdown, knockdown of Golph3, Golga2 and Golga4 was not associated with increased P4ha, P3h, and Plod expression. Transient knockdown of trafficking genes was also associated with increased expression of osteoblast marker genes, suggesting enhanced osteoblast differentiation. Strikingly, double knockdown P4ha1 or P4ha2 with Gosr2, Rab2a, Stx5a, Bet1, Arf4, Ramp1, Cog6 and Pacs1 did not rescue increased osteoblast marker gene expression, suggesting that regulation of osteoblast differentiation by trafficking genes may be an independent mechanism contributing to mineralized nodule formation. Further investigation and elucidation of signal pathways and mechanisms involved in trafficking gene regulating osteoblast differentiation are important.
The specific function of each trafficking gene in osteoblast differentiation and function remains to be determined. Since siRNA-mediated knockdown is transient, we performed CRISPR/Cas-9-mediated gene editing to permanently knock down Cog6−/− and Pacs1−/− in MC3T3-E1 pre-osteoblasts and found that this similarly increases mineralized nodule formation and osteoblast marker gene expression in vitro.
We have further demonstrated that MC3T3 pre-osteoblasts with transient knockdown of Gosr2, Arf4, Cog6 and Pacs1 form more and larger ectopic bone nodules in vivo. However, conditional knockout models will be required to examine the role of trafficking genes in endogenous bone formation, and on bone mass, strength and quality. Cog6 (Component of Oligomeric Golgi Complex 6) belongs to the COG family which is an evolutionally conserved Golgi-associated tethering complex17. Cog6 is directly involved in both intra-Golgi and endosome to Golgi retrograde transport18. Interestingly, a recent mouse genome-wide association study (GWAS) identified a bone mineral density-associated locus containing Cog6 and showed that Cog6 was highly expressed both in bone and osteoblasts19. Pacs1 (Phosphofurin acidic cluster sorting protein 1) is a member of the trans-Golgi network (TGN) membrane protein family and directs the TGN localization of furin20. Pacs1 is critical for protein trafficking in vertebrates by binding to diverse cargo and trafficking machinery, including the adaptor protein-1 complex, to ensure the proper cytoplasmic localization of numerous proteins21,22,23. A function for Pacs1 in osteoblasts or bone has not been reported. Gosr2 (Golgi SNAP Receptor Complex Member 2) belongs to a group of SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins that are essential for budding, docking and fusion of COPII-coated vesicles during ER-Golgi membrane trafficking24,25. Gosr2 gene was identified as a causative gene for progressive myoclonus epilepsy with distinct clinical features include scoliosis26. It has also been shown that Gosr2 forms complex with Stx5 and Bet1 and the complex are responsible for fusion of endoplasmic reticulum derived vesicles with the ER-Golgi intermediate compartment and the cis-Golgi24,25,27. Similar to knockdown of Gosr2, we also found increased osteogenic differentiation and mineralized nodule formation with knockdown of Stx5, Bet1 and Rab2a in MC3T3-E1 pre-osteoblasts. Both Bet1 and Rab2a are members of the GTPase family and are involved in post-endocytic trafficking of membrane-bound MT1-MMP (MMP14, membrane type 1-matrix metalloproteinase), an essential metalloprotease for matrix remodeling, invasion, pericellular collagenolysis and skeletal/extraskeletal connective tissue modeling28,29,30. Arf4 (ADP Ribosylation Factor 4) is another vesicle trafficking protein belonging to GTPase family and functions an ADP-ribosyltransferase31. ARF4 regulates ciliary protein trafficking, dysfunction of which is a known cause of human genetic diseases and syndromic disorders known as ciliopathies31,32,33. Both Rab2a and Arf4 are highly expressed in osteoblasts and bone (data from BioGPS, http://biogps.org/)34.
In summary, we identified a set of vesicle trafficking genes that are highly expressed in Col2.3GFP+ osteoblast lineage cells and play a role in osteoblast differentiation and mineralized nodule formation. Osteoblasts synthesize, transport, and secrete large amounts of bone matrix components including collagen, which is tightly regulated. Any dysregulation would cause inadequate or excessive mineralization of bones. Elucidation of the specific function of vesicle traffic genes involving in osteoblasts function raises the possibility of developing novel therapeutics for bone diseases designed to target this process.
Methods
Tissue culture and osteogenic differentiation
Generation, maintenance and differentiation of mouse Col2.3GFP ESCs were performed as previously described9. Bone chips were harvested from Col2.3GFP transgenic mice and subjected to osteogenic differentiation as previously described35.
The MC3T3-E1 pre-osteoblast cell line (passage 35–45) was kindly provided by the David Feldman laboratory (Stanford University). MC3T3-E1 cells were maintained in basal medium (αMEM, no ascorbic acid, cat A1049001; Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (FBS; Gemini Bio Products, CA) and 1% penicillin–streptomycin. For osteogenic differentiation, the basal medium was replaced by osteogenic medium (basal medium supplemented by 50 μg/mL ascorbic acid (Sigma, St. Louis, MO)). For mineralization assays, 10 mM β-glycerol phosphate (Sigma, St. Louis, MO) and 100 nM dexamethasone (Sigma, St. Louis, MO) were added to the osteogenic medium. Osteogenic medium was changed every three days.
RNA interference
For transient knockdown target gene, siGENOME SMARTpool siRNA for specific target genes and non-Targeting siRNA for siControl were synthesized and purchased from Dharmacon, Inc. (Lafayette, CO). Cells were plated at a concentration of 1 X 104 cells/well in 24-well plates and transfected with the siRNA using Lipofectamine RNAiMax transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions on the following day after plating. 2.5 µl of 10 µM siRNA duplexes were mixed with 47.5 µl Opti-MEM (Thermo Fisher Scientific) and combined with 1 µl/well of transfection reagent and 49 µl Opti-MEM to a total volume of 100 µl and incubated for 20 min. After incubation, 100 µl siRNA-lipid complex were transfected to the adherent cells. Osteogenic differentiation was initiated with osteogenic medium on the second day after transfection. Transfected cells were analyzed at different time points.
Von Kossa, Alizarin Red S and alkaline phosphatase staining
For Von Kossa staining, cells were rinsed twice with PBS and fixed in 10% formaldehyde for 10 min, then washed with double distilled water (DDW). Cells were incubated in 5% silver nitrate solution under UV light for 30 min, washed with DDW, rinsed with 5% sodium thiosulfate for 5 min to remove unreacted silver, washed again with DDW and stored in PBS. For Alizarin Red S staining cells were fixed and washed, stained with 2% Alizarin Red S solution for 20 min, then washed with DDW to remove unincorporated excess dye. For quantification of Alizarin Red S staining, following the wash in DDW cells were incubated in 10% Acetic acid with shaking for 30 min at room temperature. Cells were collected by cell scraper and transferred to microcentrifuge tubes, heated for 10 min at 85 °C, and then put on ice for 5 min. After centrifugation at 20,000 g for 15 min the supernatant was transferred to a new microcentrifuge tube. Upon addition of 10% Ammonium hydroxide, 50 µl aliquot was transferred to a microplate reader well and absorbance was measured at 405 nm. Alkaline phosphatase staining was performed by using Stemgent® Alkaline Phosphatase Staining Kit II (Stemgent, San Diego, CA) according to the manufacturer’s instructions.
Quantitative reverse transcription PCR (qPCR) and RNA-sequencing
RNA was extracted by using PureLink® RNA Mini Kit (Invitrogen, Carlsbad, CA). RNA concentration and purity were measured by NanoDrop (Thermo Scientific, Wilmington, DE) and then used for reverse transcription using iScript Kit (Bio-Rad, Hercules, CA). Quantitative RT-PCR (qPCR) was performed following standard methods. Primer sequences are provided in Supplementary Table. mRNA levels were normalized to of the levels of β-Actin. Relative mRNA levels of trafficking gene knockdown cells were then normalized to siControl. PCR was performed in triplicate for each sample, and 3 independent experiments were carried out11.
For RNA-sequencing (RNA-seq), the integrity of extracted RNA was assayed by on-chip electrophoresis (Agilent Bioanalyzer) and only samples with a high RNA integrity (RIN) value were used for RNA-seq11. Poly-A mRNA was purified using Dynabeads® mRNA Purification Kit (Invitrogen) according to the manufacturer’s instructions. RNA sequencing libraries were prepared using the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA) according to the manufacturer’s instructions. Library quality was verified using the Agilent High Sensitivity DNA Kit on Agilent’s 2100 Bioanalyzer. For each library, an average of 420 bp fragments were sequenced using paired end reads (2 × 100 bp) on the Illumina HiSeq 2500 platform (Stanford Personalized Medicine Sequencing Core), with an average of 30 million reads per sample. Paired end sequencing reads (100 bp) were generated and aligned to mouse reference sequence NCBI Build 37/mm9 with the STAR (v2.4.2a) algorithm36. Normalization of RefSeq annotated genes, expression level and differential expression analysis were performed using the Bioconductor package DESeq2 in R (Version 3.2.2)37. Genes with False Discovery Rate (FDR) < 0.05 were defined as differentially expressed genes. Gene ontology analysis was performed using DAVID (david.abcc.ncifcrf.gov) and The Gene Ontology (GO) Consortium (http://geneontology.org/). For Gene Set Enrichment Analysis (GSEA, Broad Institute), gene set associated with osteogenesis were obtained from www.qiagen.com (Qiagen, Germany)38. All transcriptome raw data are publicly available in GEO (accession number GSE223192, Genome wide gene expression profiling of Col2.3GFP+ enriched osteoblasts).
Western blot analysis
Cells were lysed in Radioimmunoprecipitation (RIPA) assay buffer (Sigma, St. Louis, MO), total protein was extracted, and the protein concentration was measured by Bradford Protein Assay (Bio-Rad). Protein samples were loaded on 10% SDS-PAGE gel and the separated proteins were transferred by electroblotting to polyvinylidene fluoride (PVDF) membranes (Bio-Rad). The membranes were blocked with Odyssey® Blocking Buffer (LI-COR Biosciences, Lincoln, NE) and incubated with the primary antibody and secondary antibody. Immunolabeling was detected by using Odyssey® CLx system (LI-COR Biosciences). The following primary antibodies were used: Anti-Hydroxyproline antibody (ab37067, Abcam; 1:1000 dilution); Anti-P4HA1 antibody (ab59497, Abcam; 1:500 dilution); Anti-P4HA2 antibody (A4262, ABclonal Inc.; 1:500 dilution); Anti-GAPDH antibody (sc-32233, Santa Cruz Biotechnology, Inc.; 1:1000 dilution).
Subcutaneous implantation
Seven-week-old male CD-1 Nude Mice Crl:CD1-Foxn1nu purchased from Charles River (Wilmington, MA, USA) were housed in Innovive recyclable individually ventilated cages in a designated pathogen-free area facility and fed irradiated mouse chow and autoclaved water. The Veterinary Service Center (VSC) at Stanford University provides laboratory animal care and is administered by the Department of Comparative Medicine. The laboratory animal care program at Stanford University is fully accredited by the Association for Accreditation and Assessment of Laboratory Animal Care (AAALAC). All animal surgeries were approved by the Stanford University Administrative Panel on Laboratory Animal Care (APLAC). All methods were performed in accordance with relevant guidelines and regulations. This study is reported in accordance with ARRIVE guidelines (https://arriveguidelines.org). MC3T3-E1 cells were transfected siRNA and subjected to osteogenic differentiation for seven days. After anesthesia using inhaled isoflurane, cells (100 µl) were mixed with equal volume (100 µl) of Matrigel and then subcutaneously implanted into the back of 7-week-old male CD-1 Nude Mice Crl:CD1-Foxn1nu. Each implantation was conducted by injection of 1 × 106 cells and Matrigel mixture using 25G needles. Four implantations were injected on each side of the spine. Nodules were explanted after 12 weeks and analyzed by X-ray, microCT and histology. We implanted four single trafficking gene-knockdown samples in one spine side of mouse and implanted four trafficking gene and P4ha2 double knockdown samples on the other spine side of the mouse to minimize mouse variation.
Micro-computed tomography and histological analysis
Mineralization of subcutaneous implants was monitored by micro-computed tomography (micro-CT) using the vivaCT 40 Preclinical MicroCT Scanner (SCANCO Medical, AG)11. For small nodules formed after subcutaneous implantation, scanning was conducted with X-ray setting at Energy/intensity: 70 kVp,114 μA, 8w; with CT-Scan setting at high resolution, 21.5 mm FOV (field of view), 10.5 μm voxel size and 618 slices. The region of interest was selected to completely cover the nodules. X-ray images were analyzed by Inveon™ Research Workplace 4.2 software (IRW 4.2, Siemens Healthcare GmbH, Germany)11. For histological analysis, tissue samples were harvested and fixed for 24 h in 10% neutral-buffered formalin followed by incubation in 30% sucrose for 24 hours11. Tissue samples were embedded in SCEM embedding medium (Section-lab, Japan). 8-μm serial sections were made on a cryofilm (Section-lab, Japan) using tape-stabilized sectioning method for histology analysis. Aniline Blue staining was performed according to Masson’s trichrome method (Newcomer Supply, Inc., Middleton, WI)11. Von Kossa and Alizarin Red S staining were performed on sectioned slides11. Images were acquired by an EVOS FL Cell Imaging System (Live Technologies, South San Francisco, CA)11.
CRISPR/Cas9-mediated gene editing in MC3T3-E1 pre-osteoblast cells
Cog6−/− and Pacs1−/− MC3T3-E1 cells were generated by CRISPR-Cas9 gene editing technology as previously described39. Briefly, guide RNA sequences targeting Cog6 (5′-CGGACTCGAAGAAATTTACG-3′), Pacs1 (5′-CAGTGACGACCCATTGCAT-3′) and non-targeting control (5′-GAACTCGTTAGGCCGTGAAG-3′) were cloned into lentiCRISPR v2 plasmid (Addgene; cat. no. 52961). Lentivirus was generated and harvested in 293 T cells by transfecting cells with lentiviral vectors along with the packaging vectors psPAX2 and pMAD2.G (Addgene; cat. no. 16620 and 16,619, respectively) as previously described11. MC3T3-E1 cells were transduced with the lentivirus and selected with puromycin containing MEM (2 µg/ml) for ten days. Cog6−/− and Pacs1−/− pooled knockout cell lines were analyzed by qRT-PCR to obtain clones with permanent depletion of the gene product. CRISPR line proliferation rate was assessed by Cell Proliferation Assay (MTS) according to the manufacturer’s instructions (cat NO: G3582; Promega, Madison, WI).
Statistical analysis
Statistical analysis was performed using Microsoft Excel and GraphPad Prism 6 (GraphPad Software, San Diego, CA). Quantitative data are presented as scatter or interleaved scatter plots with mean ± SD, or box-whiskers plots with whiskers representing minimum and maximum values and box representing 1st to 3rd quartiles.
Shapiro–Wilk normality tests were performed to test whether data were normally distributed. If Shapero–Wilk tests were not satisfied, nonparametric tests were used. Statistical significance was assessed by multiple t test followed by correction for multiple comparisons using the Holm–Sidak method.
Data availability
The raw data required to reproduce these findings are available in the Open Data sectionGEO (accession number GSE223192, Genome wide gene expression profiling of Col2.3GFP+ enriched osteoblasts).
References
Long, F. Building strong bones: Molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol. 13, 27–38. https://doi.org/10.1038/nrm3254nrm3254[pii] (2012).
Stenbeck, G. & Coxon, F. P. Role of vesicular trafficking in skeletal dynamics. Curr. Opin. Pharmacol. 16, 7–14. https://doi.org/10.1016/j.coph.2014.01.003 (2014).
Boyadjiev, S. A. et al. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat. Genet. 38, 1192–1197. https://doi.org/10.1038/ng1876 (2006).
Zhang, H. et al. Novel mutations in the SEC24D gene in Chinese families with autosomal recessive osteogenesis imperfecta. Osteoporos. Int. 28, 1473–1480. https://doi.org/10.1007/s00198-016-3866-2 (2017).
Moosa, S. et al. Mutations in SEC24D cause autosomal recessive osteogenesis imperfecta. Clin. Genet. 89, 517–519. https://doi.org/10.1111/cge.12678 (2016).
Garbes, L. et al. Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am. J. Hum. Genet. 96, 432–439. https://doi.org/10.1016/j.ajhg.2015.01.002 (2015).
Keller, R. B. et al. Monoallelic and biallelic CREB3L1 variant causes mild and severe osteogenesis imperfecta, respectively. Genet. Med. 20, 411–419. https://doi.org/10.1038/gim.2017.115 (2018).
Kalajzic, I. et al. Use of type I collagen green fluorescent protein transgenes to identify subpopulations of cells at different stages of the osteoblast lineage. J. Bone Miner Res. 17, 15–25. https://doi.org/10.1359/jbmr.2002.17.1.15 (2002).
Zhu, H., Kimura, T., Swami, S. & Wu, J. Y. Pluripotent stem cells as a source of osteoblasts for bone tissue regeneration. Biomaterials 196, 31–45. https://doi.org/10.1016/j.biomaterials.2018.02.009 (2019).
Wu, J. Y. et al. Gsalpha enhances commitment of mesenchymal progenitors to the osteoblast lineage but restrains osteoblast differentiation in mice. J. Clin. Investig. 121, 3492–3504. https://doi.org/10.1172/JCI46406 (2011).
Zhu, H., Swami, S., Yang, P., Shapiro, F. & Wu, J. Y. Direct reprogramming of mouse fibroblasts into functional osteoblasts. J. Bone Miner Res. https://doi.org/10.1002/jbmr.3929 (2019).
Lemmer, I. L., Willemsen, N., Hilal, N. & Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 47, 101169. https://doi.org/10.1016/j.molmet.2021.101169 (2021).
Ghemrawi, R., Battaglia-Hsu, S. F. & Arnold, C. Endoplasmic reticulum stress in metabolic disorders. Cells 7, 63. https://doi.org/10.3390/cells7060063 (2018).
Nijhuis, W. H. et al. Current concepts in osteogenesis imperfecta: Bone structure, biomechanics and medical management. J. Child Orthop. 13, 1–11. https://doi.org/10.1302/1863-2548.13.180190 (2019).
Stegen, S. et al. HIF-1alpha metabolically controls collagen synthesis and modification in chondrocytes. Nature 565, 511–515. https://doi.org/10.1038/s41586-019-0874-3 (2019).
Myllyharju, J. & Kivirikko, K. I. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 20, 33–43. https://doi.org/10.1016/j.tig.2003.11.004 (2004).
Loh, E. & Hong, W. The binary interacting network of the conserved oligomeric Golgi tethering complex. J. Biol. Chem. 279, 24640–24648. https://doi.org/10.1074/jbc.M400662200 (2004).
Laufman, O., Hong, W. & Lev, S. The COG complex interacts directly with Syntaxin 6 and positively regulates endosome-to-TGN retrograde transport. J. Cell Biol. 194, 459–472. https://doi.org/10.1083/jcb.201102045 (2011).
Mesner, L. D. et al. Mouse genome-wide association and systems genetics identifies Lhfp as a regulator of bone mass. PLoS Genet. 15, e1008123. https://doi.org/10.1371/journal.pgen.1008123 (2019).
Wan, L. et al. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell 94, 205–216. https://doi.org/10.1016/s0092-8674(00)81420-8 (1998).
Dirk, B. S. et al. PACS-1 and adaptor protein-1 mediate ACTH trafficking to the regulated secretory pathway. Biochem. Biophys. Res. Commun. 507, 519–525. https://doi.org/10.1016/j.bbrc.2018.11.085 (2018).
Hinners, I. et al. AP-1 recruitment to VAMP4 is modulated by phosphorylation-dependent binding of PACS-1. EMBO Rep. 4, 1182–1189. https://doi.org/10.1038/sj.embor.7400018 (2003).
Crump, C. M. et al. PACS-1 binding to adaptors is required for acidic cluster motif-mediated protein traffic. EMBO J. 20, 2191–2201. https://doi.org/10.1093/emboj/20.9.2191 (2001).
Donkervoort, S. et al. BET1 variants establish impaired vesicular transport as a cause for muscular dystrophy with epilepsy. EMBO Mol. Med. 13, e13787. https://doi.org/10.15252/emmm.202013787 (2021).
Jepson, J. E. C., Praschberger, R. & Krishnakumar, S. S. Mechanisms of neurological dysfunction in GOSR2 progressive myoclonus epilepsy, a golgi SNAREopathy. Neuroscience 420, 41–49. https://doi.org/10.1016/j.neuroscience.2019.03.057 (2019).
Dibbens, L. M. & Rubboli, G. GOSR2: A progressive myoclonus epilepsy gene. Epileptic Disord. 18, 111–114. https://doi.org/10.1684/epd.2016.0848 (2016).
Volker, J. M. et al. Functional assays for the assessment of the pathogenicity of variants of GOSR2, an ER-to-Golgi SNARE involved in progressive myoclonus epilepsies. Dis. Model Mech. 10, 1391–1398. https://doi.org/10.1242/dmm.029132 (2017).
Miyagawa, T. et al. MT1-MMP recruits the ER-Golgi SNARE Bet1 for efficient MT1-MMP transport to the plasma membrane. J. Cell Biol. 218, 3355–3371. https://doi.org/10.1083/jcb.201808149 (2019).
Kajiho, H. et al. RAB2A controls MT1-MMP endocytic and E-cadherin polarized Golgi trafficking to promote invasive breast cancer programs. EMBO Rep. 17, 1061–1080. https://doi.org/10.15252/embr.201642032 (2016).
Holmbeck, K. et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 99, 81–92. https://doi.org/10.1016/s0092-8674(00)80064-1 (1999).
Deretic, D., Lorentzen, E. & Fresquez, T. The ins and outs of the Arf4-based ciliary membrane-targeting complex. Small GTPases 12, 1–12. https://doi.org/10.1080/21541248.2019.1616355 (2021).
Ezratty, E. J., Pasolli, H. A. & Fuchs, E. A presenilin-2-ARF4 trafficking axis modulates Notch signaling during epidermal differentiation. J. Cell Biol. 214, 89–101. https://doi.org/10.1083/jcb.201508082 (2016).
Follit, J. A. et al. Arf4 is required for Mammalian development but dispensable for ciliary assembly. PLoS Genet. 10, e1004170. https://doi.org/10.1371/journal.pgen.1004170 (2014).
Su, A. I. et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 101, 6062–6067. https://doi.org/10.1073/pnas.0400782101 (2004).
Panaroni, C. et al. PTH signaling in osteoprogenitors is essential for B-lymphocyte differentiation and mobilization. J. Bone Miner Res. 30, 2273–2286. https://doi.org/10.1002/jbmr.2581 (2015).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. https://doi.org/10.1093/bioinformatics/bts635bts635[pii] (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Decker, M. et al. Leptin-receptor-expressing bone marrow stromal cells are myofibroblasts in primary myelofibrosis. Nat. Cell Biol. 19, 677–688. https://doi.org/10.1038/ncb3530 (2017).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308. https://doi.org/10.1038/nprot.2013.143 (2013).
Acknowledgements
This work was supported by the National Institutes of Health Grant R21 AR081073 to J.Y.W. We thank Drs. Rajat Rohatgi, Ganesh Pusapati, Mandy Ma (Department of Biochemistry, Stanford University) for advice on CRISPR methods. We would like to thank Dr. Frezghi Habte and Dr. Edwin Chang (Stanford Small Animal Imaging Facility) for assistance with microCT.
Author information
Authors and Affiliations
Contributions
Experiments were conceived and designed by H.Z and J.Y.W. The experiments were performed by H.Z., Y.S., and J.Y.W., H.Z. analyzed and interpreted the data. The paper was written by H.Z. and J.Y.W., with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, H., Su, Y., Wang, J. et al. The role of vesicle trafficking genes in osteoblast differentiation and function. Sci Rep 13, 16079 (2023). https://doi.org/10.1038/s41598-023-43116-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-43116-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
