
Abstract
A recent study has shown that highly crystalline graphene-based materials can be obtained from poorly organized carbon precursors using calcium as a non-conventional catalyst. XRD and TEM analyses of calcium-impregnated cellulose and lignin biochars showed the formation of well-ordered graphenic structures (Lc > 7 nm, d002 < 0.345 nm) above 1200 °C, far below the standard graphenization temperatures (T > 2000 °C). Herein, we propose new insights on the mechanism controlling the formation of highly graphenic biochars using Ca as a catalyst. We postulate that the calcium-catalyzed graphenization occurs through the formation of a metastable calcium carbide by reaction between CaO particles and amorphous carbon between 1000 and 1200 °C. CaC2 decomposes into calcium vapor and a graphenic shell covering the CaC2 particles as confirmed by TEM analysis. The thickness and planarity of the graphenic shell increase with the CaC2 initial particle size (between 20 and 200 nm), and its growth is controlled by the diffusion of the calcium vapor through the graphene layer. A much effective graphenization was obtained for the lignin biochars compared to cellulose, with Lc > 10 nm and d002 < 0.340 nm, attributed to the insertion of sulfur in the graphenic shells, which favors their ruptures and the decomposition of CaC2 into graphene. We believe that these findings would enable the reduction of costs and environmental impact of graphene-based materials synthesis using cheap and abundant renewable feedstocks and catalysts as well.
Similar content being viewed by others


Introduction
Graphene is a bidimensional carbon material composed of sp2 hybridized carbon atoms arranged in a hexagonal lattice. It is characterized by high electrical conductivity (~ 106 s m−1), mechanical resistance (tensile strength ≈ 130 GPa) and specific surface area (~ 2675 m2 g−1)1,2. Therefore, Graphene and graphene-based materials such as fullerene, carbon nanotube or graphite are considered for applications in various advanced fields such as supercapacitors3,4,5, electronics6, energy storage7 and medical devices8,9. Graphene is currently synthetized by either top-down or bottom-up processes that usually require complex mechanical, chemical or thermal treatments10,11,12. In addition, most of the standard carbon precursors for graphene synthesis involves non-sustainable fossil or petroleum-based resources, which deepens the environmental cost of graphene and graphene-based materials synthesis.
Over the last years, the use of lignocellulosic biomass as a precursor for graphene synthesis has attracted lot of attention due to its abundance, renewability, and cheap cost. However, lignocellulosic bioresources, which are a complex mix of cellulose, hemicelluloses and lignin with inorganic elements, are “non-graphitizing” carbons, which means that they produced short and irregularly stacked graphene sheets (turbostratic carbon) even after carbonization at high temperatures (T > 2000 °C)13,14,15,16. Therefore, graphenization corresponds to the formation and growth of graphene layers from disordered carbon to 2D carbon materials with a slight 3D character (turbostratic), while graphitization refers to a regular crystalline 3D structure17. Nonetheless, previous studies have reported that the doping of the lignocellulosic biomass precursor with carefully-selected species improves graphenization of non-graphitizing carbons to highly crystalline graphene-based materials at relatively low temperatures (T < 1000 °C). Hoekstra et al. and Sevilla et al. both obtained highly crystalline graphenic carbons from cellulose at 800 and 900 °C using nickel18,19, while Yan et al., Gong et al. and Thompson et al. used iron to produce multilayer graphenic materials below 1000 °C from kraft lignin, bamboo and softwood sawdust respectively20,21,22. Most of the articles about catalytic graphenization focused on transition metals, especially iron, cobalt and nickel for their graphenization efficiency and abundance19,20,21,22,23,24,25,26,27. Nonetheless, old research conducted in the 80’s, summarized by Oya et al.28, have revealed that multiple elements, in particular the alkaline earth metals such as calcium, can be efficient catalysts for the graphenization of various carbon ressources29,30. This latter route was no longer considered until a recent study from our team. We have confirmed the catalytic activity of calcium on the graphenization of cellulose biochar carbonized at 1800 °C31. In summary, impregnation of the bioresource with calcium led to the formation of a highly graphenic domain in the biochar, whose proportion rises with the calcium loading. As opposed to the standard transition metals catalysts (Fe, Ni, Co), calcium is more environmental-friendly, abundant and cheaper, and could therefore attract more attention in future works in the field of graphenic carbon materials.
The fundamental understanding of the mechanism of catalytic graphenization is a key issue to further optimize the process of green synthesis of graphene from bioresources and to obtain graphenic materials with tunable properties. Several mechanisms have been proposed for transition metals, and the most widely accepted ones are the carbide formation–decomposition and the dissolution–precipitation mechanisms. In the carbide formation–decomposition mechanism, the catalyst reacts with the amorphous carbon to form a metastable carbide intermediate that decomposes into graphene at high temperature28,32. In the dissolution–precipitation mechanism, the amorphous carbon dissolves in the molten metal that becomes supersaturated before precipitating as defect-free graphene during cooling18. Gomez-Martin et al. studied the iron-catalyzed graphenization of non-graphitizing carbon with in situ X-ray diffraction and ex situ total scattering experiments, and found the formation and decomposition into graphene of an iron carbide upon cooling at temperature as low as 800 °C24. More recently, Ghogia et al. investigated the catalyzed graphenization of iron-impregnated cellulose biochars and found that both the particles size and degree of reduction of the iron catalyst favored by thermal heating promote the biochar graphenization27.
To the best of our knowledge, the mechanism controlling the catalytic graphenization of cellulose and lignin using calcium has not been discussed in the literature. Hirano et al. reported in the 70s the formation of an unstable calcium carbide and its decomposition into crystalline graphenic structures above 1300 °C during the carbonization of polyvinylchloride coke with calcium oxide29. More recently, Yang et al. used melamine–formaldehyde resin as carbon precursor and CaCO3 nanoparticles as template to produce graphite-like nano-shells below 1300 °C, suggesting that graphenic structures develop around the catalyst particles33. Hence, the comprehensive mechanism driving the calcium-catalyzed graphenization lacks understanding.
In this context, this work aims at providing new insights on the comprehensive mechanism of calcium-catalyzed graphenization. To this end, we have carbonized calcium-impregnated commercial cellulose and lignin, two of the main polymers of a lignocellulosic bioresource, between 1000 and 1800 °C (Fig. 1). The carbon organization of the resulting biochars was characterized using X-ray diffraction (XRD) and transmission electron microscopy (TEM). From the results, we have highlighted the starting temperature of the catalytic activity of calcium, and we propose an approach describing the fundamental graphenization mechanism as a first contribution to the state of the art in the field.

Schematic representation of the synthesis of highly graphenic biochar using calcium.
Results
Temperature-dependance of the biochars graphenization
The XRD spectra of the calcium-impregnated cellulose biochars at different carbonization temperatures are presented in Fig. 2a. For the biochars prepared at 1000 and 1200 °C, multiple diffraction peaks of calcium species are observed, which are attributed to CaO (* symbols) and Ca(OH)2 (○ symbols) particles. As Ca(OH)2 decomposes into CaO and water around 500 °C34, the observed Ca(OH)2 likely results from the reaction of CaO particles with the room moisture after cooling. Application of the Scherrer equation indicates a slight decrease of the average particle diameter LCaO, going from LCaO = 21.70 nm at 1000 °C to 21.21 nm at 1400 °C. No distinctive carbon 002 peak is visible at these carbonization temperatures, which indicates that the carbon matter in these biochars is either amorphous or highly disorganized in this temperature range (1000–1200 °C).

XRD patterns (λ = 1.542 Å) of biochars carbonized at different temperatures: (a) calcium-impregnated cellulose biochars, (b) calcium-impregnated lignin biochars.
However, a sharp diffraction peak appears at the carbon 002 position (2θ ≈ 26°) and confirmed the formation of a graphenic domain from 1400 °C. From these observations, the graphenization starts between 1200 and 1400 °C, which is in agreement with previous observations from Hirano et al.29, who reported the formation of a graphenic domain in CaO-doped polyvinylchloride coke above 1300 °C. The biochar carbonized at 1400 °C still contains multiple calcium species, and the diffraction peaks of calcium cyanamide (CaCN2) are visible (red  symbols). Calcium cyanamide is also observed at 1200 °C, and should result from reactions of calcium species with the atmospheric nitrogen.
symbols). Calcium cyanamide is also observed at 1200 °C, and should result from reactions of calcium species with the atmospheric nitrogen.
At 1600 and 1800 °C, the diffraction peaks of the calcium species disappear. This could be attributed to the volatilization of the calcium species or their entrainment in the flue-gas stream35. The carbon 002 peak is sharp but asymmetric, which indicates the coexistence of two carbon phases, one highly turbostratic as observed for non-graphitizing carbons, and another highly crystalline. The 002 peak becomes thinner with the temperature, because of the improvement of the crystalline organization in the turbostratic domain, or because of stackings and junctions of the highly graphenic structures.
A similar trend is observed for the lignin biochars, with the apparition of a graphenic domain above 1400 °C (Fig. 2b). Calcium cyanamide is also detected at 1200 and 1400 °C, and most of the CaO and Ca(OH)2 particles have disappeared at 1400 °C unlike the cellulose biochar. The main difference with the cellulose biochars is the apparition of the diffraction peaks of calcium sulfide (CaS). Kraft lignin used in this study is indeed rich in sulfur (14.2 g kg−1) that comes from the industrial extraction process. It has previously been reported that calcium oxide can interact with sulfuric acids vapors formed during pyrolysis to produce calcium sulfate36, which is then reduced to calcium sulfide around 900 °C37,38.
Even though CaS is formed as soon as 900 °C, below the apparent temperature of formation of the graphenic structures, sulfur does not seem to have inhibit or reduce the catalytic activity of calcium. The sizes of the CaS and CaO particles in the lignin biochars significantly decrease with temperature, from LCaS = 34.53 nm to LCaS = 18.97 nm between 1000 and 1600 °C, and from LCaO = 31.50 nm to LCaO = 27.34 nm between 1000 and 1200 °C, but remained higher than for the CaO particles in the cellulose biochars in the same temperature ranges (LCaO ≈ 22 nm). This trend is opposed to the standard graphenization catalysts like iron, where sintering of the metal particles with temperature was reported39.
The crystallite sizes Lc and d002 of the highly crystalline graphenic domains in the biochars measured from the XRD spectra are summarized in Table 1. Higher crystallite size Lc and lower interlayer spacing d002 are obtained at 1400 °C. The high-intensities peaks of the calcium species in the XRD spectra may have hidden the contribution of the amorphous carbon to the apparent 002 peak, resulting in a thinner 002 peak. Lc increases and d002 decreases between 1600 and 1800 °C, which is consistent with the literature that indicates an improvement of the graphenization with the temperature39. The graphenic domain in the lignin biochars is more developed than in the cellulose biochar, with a greater Lc and lower d002. This enhancement could be explained by the slightly greater calcium loading in the initial lignin (4.72 wt.% vs 4.39 wt.%), from a positive effect of sulfur on the graphenization, or from the larger size of the calcium particles, as the metal particles size is known to improve the crystallite sizes of carbon39. To further investigate the effect of calcium on the carbon organization, the structure of the biochars was studied using TEM.
Observation of the carbon structure
TEM images of the calcium-impregnated cellulose biochars at different temperatures are presented in Fig. 3. The corresponding EDX analyses and additional images are presented in the Supplementary materials. Figure 3a, b, acquired at 1000 °C, show a poorly organized carbon matrix, typical of a non-graphitizing carbon, which confirms the XRD results that showed no catalytic effect of calcium at this temperature. Moreover, the TEM images reveal the presence of large amounts of CaO particles embedded in the carbon matrix.

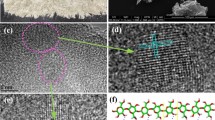
TEM images of the calcium-impregnated cellulose biochars at different temperatures: (a) 1000 °C dark field, (b) 1000 °C HRTEM, (c) 1400 °C dark field, (d) 1400 °C HRTEM, (e) 1800 °C dark field, (f) 1800 °C HRTEM.
However, the formation of a graphenic shell around a large calcium oxide particle is observed at 1400 °C (Fig. 3c, d). This graphenic shell is formed by the regular stacking of long-range graphene sheets and explains the high crystallite size Lc calculated from XRD. Even though the graphenic shell follows the edges of the particle, it is opened as it could be seen in the upper part of the image, which suggests that the CaO particle can partially slide outside of its shell, creating hollow spaces between the particle and the graphene sheets (blue arrow in Fig. 3c). Formation of a graphenic shell around a catalyst particle has already been observed with iron23,39. For this catalyst, the dimensions and orientations of the graphenic structures are directly linked to the geometry of the metal particle. Therefore, a large particle with planar facets, as observed in Fig. 3c, will form well-ordered and developed graphenic structures, while a small particle with an irregular shape will result in short and poorly ordered graphenic structures39 (see Fig. S3). The CaO particles are formed in situ during the carbonization (liquid impregnation), and therefore their sizes are not controlled, which results in heterogenous graphenic structures.
At 1800 °C, calcium particles are no longer observed due to their volatilization or entrainment in the flue-gas stream, and the carbon matrix has a crumpled-paper morphology composed of several cavity-shape graphenic structures (Fig. 3e, f). These structures have a diameter between 20 and 200 nm, in accordance with the dimensions of the graphenic shells observed at 1400 °C. Therefore, we can assume that these structures correspond to the graphenic shell leftovers after removal of the CaO core. Without a solid particle as a core, the graphenic shells can easily deform, which causes a broadening of the diffraction peaks and explain the higher crystallite size Lc at 1400 °C.
Figure 4 presents the TEM results for the calcium-impregnated lignin biochars. At 1000 °C, the carbon matrix is highly disordered with many calcium particles (CaO and CaS) embedded in (Fig. 4a, b). Some additional mineral elements in smaller proportions were detected, namely potassium, silica and iron. In opposition to the cellulose biochar, some crystalline graphenic structures are observed in addition to the disordered carbon. This can be attributed to an early start of the calcium-catalyzed graphenization for the lignin biochar triggers by sulfur, or to a catalytic effect of the inorganic species inherent to the lignin bioresource (Table S1). Indeed, the commercial lignin used in this study contains some iron (0.4 g kg−1), which is known to catalyze graphenization at temperature as low as 800 °C19. However, the biochar is mostly disorganized overall, as no crystalline graphenic structures 002 peak is observed in the XRD spectrum.

TEM images of the calcium-impregnated lignin biochars at different temperatures: (a) 1000 °C dark field, (b) 1000 °C HRTEM, (c) 1400 °C dark field, (d) 1400 °C HRTEM, (e) 1800 °C dark field, (f) 1800 °C HRTEM.
The TEM images of the lignin biochars carbonized at 1400 and 1800 °C both show peculiar carbon structures riddle with CaO and CaS nanoparticles (Fig. 4c, e, f, see Figs. S4 and S5 for the EDX analyses). They all present irregular and unique shapes and are few of hundred nanometers large. A zoom on one of these structures (Fig. 4f) show that they are composed of tens of stacked graphene sheets with embedded CaO and CaS nanoparticles. The graphene sheets present various orientations (highlighted by red lines) but remain mostly flat for tens of nanometers in width, despite some slight curvatures and U-shaped folds at the edges of the graphenic structure (blue arrows). The better stacking of the graphene sheets in these structures, unique to the lignin biochars, is in agreement with the greater crystallite size Lc and lower interlayer spacing d002 shown with the XRD analysis. The presence of CaS particles on the graphenic structures confirm that sulfur did not inhibit the catalytic activity of calcium and may have intervene in the graphenization mechanism. However, some graphenic structures without CaS particles (Fig. 4d), as well as lone CaO, particles were observed in the lignin biochars. This suggests that some calcium particles have catalyzed the lignin biochars graphenization without interaction with sulfur, in the same way than with the cellulose biochars. It is relevant to note that no sodium species were detected in the lignin biochars despite its high content (51.5 g kg−1) in the commercial lignin employed. Na is known to volatilize below 1000 °C during biomass pyrolysis40. It is likely that Na was already vaporized from the lignin biochars before the start of the graphenization, and therefore did not play any role in the Ca-catalyzed graphenization process.
Discussion
Highly graphenic structures can be formed by impregnation of cellulose and lignin with calcium at much lower temperatures than with the standard processes. As these main polymers of lignocellulosic biomass and calcium are both cheap and abundant resources, this novel approach would drastically reduce the cost and environmental impact of graphene-based materials production. That is why a precise understanding of the graphenization mechanism is required to control and improve the desired properties of the formed carbon materials. However, apart for scarce studies in the literature29,30,33, very limited information is known about the calcium-catalyzed graphenization mechanism. Based on our findings, we can propose some new insights on how calcium help to rearrange the carbon atoms into graphenic structures. As the cellulose and lignin biochars presented noticeably different graphenic structures, these two bioresources will be discuss separately, starting from cellulose.
From the XRD and TEM analyses, temperature appears to be the main factor driving the catalytic graphenization. Below 1200 °C, no catalytic effect of calcium is observed, and the calcium is mainly under the form of CaO particles embedded in an amorphous carbon matrix. However, formation of graphenic structures is observed around calcium oxide particles at 1400 °C on the TEM images. This observation is similar to the literature on iron-catalyzed graphenization. It is explained that the amorphous carbon dissolves in the metal catalyst to form an intermediary metastable carbide, and then precipitates into a graphenic shell around the catalyst upon cooling24,27. This mechanism fits well with calcium, as calcium oxide is known to react with carbon to form a calcium carbide around 1300 °C41:
CaC2 is known to be highly unstable42, and therefore it decomposes quickly at higher temperatures. Previous reports on the desulfurization of metal by CaC2 and on its thermal behavior suggests that CaC2 decomposes above 1200 °C to produce calcium vapor and a graphene layer around the CaC2 particle (reaction 2)42,43,44. The formation of calcium vapor may explain the disappearance of calcium in the biochars above 1200 °C. According to literature, this reaction is mainly driven by the diffusion of the Ca vapor through the graphenic shell43, and the reaction stops when the CaC2 particle is fully enveloped with a thick graphene layer.
However, neither CaC2 or metallic Ca were observed during the XRD and TEM analyses, which can be explained by their reactions with moisture and air when the samples have leaved the furnace to produce calcium oxide or calcium hydroxide33.
Nonetheless, the observation of calcium cyanamide can served as an undirect proof of CaC2 formation, as CaC2 was reported to react with atmospheric nitrogen above 1000 °C to form calcium cyanamide and a graphene phase according to the reaction30,33,45:
This suggests the formation of CaC2 in the biochars between 1000 and 1200 °C, slightly below the standard formation temperature reported in literature (T ≥ 1300 °C)41. As such, calcium catalyzes the biochars graphenization by formation of a metastable CaC2, followed by its decomposition either into calcium vapor and graphene or calcium cyanamide and graphene in case of a nitrogen atmosphere. As no graphenic carbon was observed at 1200 °C on the XRD spectra of the cellulose and lignin biochars despite apparent formation of calcium cyanamide, it is postulated that the graphenization mainly occurs by the first mechanism. Working with a different inert atmosphere, such as argon, can help to conclude on the preferential mechanism.
Based on previous reports using more conventional catalysts, the metal carbide can either decompose during the cooling phase (“dissolution–precipitation” mechanism) as for Fe3C24, or during the heating phase (formation–decomposition mechanism) as for SiC46. As the experiments were carried out ex situ, it is difficult to conclude between the two mechanisms, but the formation of graphenic structures between 1200 and 1800 °C despite the volatilization of the calcium particles in this temperature range and the high instability of CaC2 tends to indicate that the catalytic graphenization happens during the heating phase.
Above 1400 °C, the unreacted CaC2 and CaCN2 particles may slide out from their graphenic shells and are entrained out of the biochars in the flue-gas stream. The graphenic shells agglomerate, which confers to the carbon matrix a crumpled-paper morphology. It is possible that some stackings and junctions of the graphenic structures happen, but their high curvatures and random orientations limit this possibility. Based on our results and on the literature, a schematic description of the calcium-catalyzed mechanism is proposed on Fig. 5.

Schematic representation of the calcium-catalyzed graphenization.
In the case of the lignin biochars, the larger size of the CaO particles indicated by XRD (LCaO = 31.5 nm vs 22 nm for cellulose at 1000 °C) may have led to the formation of bulkier CaC2 particles and larger graphenic structures as a result. The fine CaS nanoparticles observed in the TEM images may come from the reaction of the calcium vapor formed by reaction 2 with the sulfur linked to the carbon matrix (reaction 4)43,44:
As explained before, reaction 2 is driven by the diffusion of calcium through the graphene layer around the CaC2 particle43, and its total encapsulation drastically limits the growth of the graphenic shell. It is possible that the graphenic shell may much easily break in the lignin biochars, possibly because of insertion of some sulfur in the graphene sheets, so that the CaC2 particle is always partially uncovered and ready to decompose. This could explain the shrinkage of the CaO and CaS particles with the temperature observed during the XRD analyses. Below 1200 °C, CaS is mainly formed from the reaction of initial large CaO particles with sulfuric acids vapors, but is produced at higher temperatures by reaction 4 from the calcium vapor produced by the CaC2 decomposition (reaction 2), which explains their fine dispersion near the graphenic structures. The CaC2 particles shrink as the graphenic structures develop and turn into small CaO particles after reaction with moisture when exiting the furnace. Additional research have to be carried out to better understand the role of sulfur in the mechanism of calcium-catalyzed graphenization.
Conclusion
The comprehensive mechanism of the catalytic graphenization of non-graphitizing carbons using calcium was investigated by carbonizing calcium-impregnated cellulose and lignin between 1000 and 1800 °C. The formation of highly crystalline graphenic structures (Lc > 7 nm, d002 < 0.345 nm) was observed for both bioresources when carbonized at 1400 °C. TEM analysis showed a graphenic shell around the catalyst particles. Above 1400 °C, the calcium particles volatilize and the graphenic shells agglomerate, which confers a crumpled-paper morphology to the carbon matrix. More ordered graphenic structures were found in the lignin biochars (Lc > 10 nm, d002 < 0.340 nm), suggesting a greater effect of calcium on this bioresource.
To explain the catalytic formation of the graphenic structures, new insights have been brought in this study. We have postulated that CaO reacts with amorphous carbon to produce a metastable calcium carbide around 1200 °C, which decomposes at higher temperatures into calcium vapor and a graphenic shell around the CaC2 particle (20 to 200 nm in width). The growth of the graphenic shell stops when the CaC2 particle is fully enveloped with a thick graphene layer and when the calcium vapor can no longer diffuse through the graphene layer. The sulfur in the lignin biochars has favored the rupture of the graphenic shells, which promotes the decomposition of CaC2 into graphene.
We believe that these new insights to the body of knowledge on the mechanism of calcium-catalyzed graphenization may contribute to widen research opportunities in the field of green synthesis of innovative graphenic material using cheap, abundant and renewable resources.
Methods
Impregnation protocol and determination of the inorganic composition
Microcrystalline cellulose (CAS: 9004-34-6) and kraft lignin (CAS: 8068-05-1) provided by Sigma Aldrich were used as carbon precursors and were loaded with calcium by liquid impregnation. 40 g of cellulose and lignin were immersed in 200 ml of a Ca(NO3)2 4H2O aqueous solution (respectively at 0.847 M and 0.381 M) and stirred for 6 h before filtration and drying at 105 °C for 24 h.
The inorganic compositions of the impregnated and non-impregnated bioresources were determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES, Horiba Ultima 2). The calcium concentration in the impregnated bioresources was respectively 4.39 wt.% and 4.72 wt.% for cellulose and lignin. The detailed inorganic compositions of the bioresources are available in supplementary Table S1.
Biochars preparation
The impregnated bioresources were first carbonized for 1 h at 800 °C under N2 with a heating ramp of 2 °C min−1 in a vertical tubular oven (carbolite vertical furnace) before uncontrolled cooling at room temperature. The resulting biochars were then carbonized for 1 h at a final temperature ranging from 1000 to 1800 °C (step of 200 °C) under N2 at 2 °C min−1 in a horizontal tubular furnace (Nabertherm RHTH 80/300/18).
X-ray diffraction
The crystalline organization of the biochars was studied by X-ray diffraction using a PANalytical X’pert Pro MPD with a Cu-Kα radiation source (λ = 1.542 Å) and operating at 45 kV and 40 mA. Diffraction peaks were recorded at 0.5 ° s−1 in the range 10°–70° in 2θ.
The carbon 002 diffraction peak (2θ ≈ 24°) was used for the evaluation of the out-of-plane organization in the graphenic domain. The 002 peak was fitted with one or two pseudo-Voigt functions depending on its asymmetry to account for poorly organized carbon. The most intense diffraction peaks of the inorganic species were also fitted with a pseudo-Voigt and used for the determination of the inorganic particles sizes. The average stacking height Lc of the graphenic domain and inorganic particles sizes LCaX (where CaX refers to a calcium specie) were all estimated using the Scherrer equation and the interlayer spacing d002 (average distance between two graphene layers in the graphenic domain) by Bragg’s law:
where λ is the radiation wavelength (0.1542 nm), θ is the peak position, K is a constant taken as 0.89 for both Lc and LCaX, β is the Full Width at Half Maximum (FWHM, in radians) of the studied diffraction peaks and s the FWHM of a standard specimen (silica) to adjust for instrumental broadening.
Transmission electron microscopy
The carbon structures were observed using High-Resolution Transmission Electron Microscopy (HRTEM, JEOL cold-FEG JEM-ARM200F) operated at 200 kV equipped with a probe Cs corrector reaching a spatial resolution of 0.078 nm. EDX spectra were recorded on a JEOL CENTURIO SDD detector. Biochars particles with a size below 50 μm were first dispersed in ethanol and then deposited on a copper grid with a carbon Lacey film.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Cooper, D. R. et al. Experimental review of graphene. ISRN Condens. Matter Phys. 2012, 1–56 (2012).
Zhu, Y. et al. Graphene and graphene oxide: Synthesis, properties, and applications. Adv. Mater. 22, 3906–3924 (2010).
Yang, S. et al. Nanographene-constructed hollow carbon spheres and their favorable electroactivity with respect to lithium storage. Adv. Mater. 22, 838–842 (2010).
de las Casas, C. & Li, W. A review of application of carbon nanotubes for lithium ion battery anode material. J. Power Sources 208, 74–85 (2012).
Wang, G., Shen, X., Yao, J. & Park, J. Graphene nanosheets for enhanced lithium storage in lithium ion batteries. Aust. Inst. Innov. Mater. Pap. https://doi.org/10.1016/j.carbon.2009.03.053 (2009).
Randviir, E. P., Brownson, D. A. C. & Banks, C. E. A decade of graphene research: production, applications and outlook. Mater. Today 17, 426–432 (2014).
Olabi, A. G., Abdelkareem, M. A., Wilberforce, T. & Sayed, E. T. Application of graphene in energy storage device—A review. Renew. Sustain. Energy Rev. 135, 110026 (2021).
Shen, H., Zhang, L., Liu, M. & Zhang, Z. Biomedical applications of graphene. Theranostics 2, 283–294 (2012).
Yang, Y., Asiri, A. M., Tang, Z., Du, D. & Lin, Y. Graphene based materials for biomedical applications. Mater. Today 16, 365–373 (2013).
Yan, Y. et al. Synthesis of graphene: Potential carbon precursors and approaches. Nanotechnol. Rev. 9, 1284–1314 (2020).
Choi, W., Lahiri, I., Seelaboyina, R. & Kang, Y. S. Synthesis of graphene and its applications: A review. Crit. Rev. Solid State Mater. Sci. 35, 52–71 (2010).
Lin, L., Peng, H. & Liu, Z. Synthesis challenges for graphene industry. Nat. Mater. 18, 520–524 (2019).
Mubari, P. K. et al. The X-ray, Raman and TEM signatures of cellulose-derived carbons explained. C J. Carbon Res. 8, 4 (2022).
Oberlin, A. Carbonization and graphitization. Carbon 22, 521–541 (1984).
Eom, Y. et al. Structure evolution mechanism of highly ordered graphite during carbonization of cellulose nanocrystals. Carbon 150, 142–152 (2019).
Handbook on Characterization of Biomass, Biowaste and Related By-products. (Springer, 2020). https://doi.org/10.1007/978-3-030-35020-8.
Bianco, A. et al. All in the graphene family—A recommended nomenclature for two-dimensional carbon materials. Carbon 65, 1–6 (2013).
Sevilla, M. & Fuertes, A. B. Graphitic carbon nanostructures from cellulose. Chem. Phys. Lett. 490, 63–68 (2010).
Hoekstra, J. et al. Base metal catalyzed graphitization of cellulose: A combined Raman spectroscopy, temperature-dependent X-ray diffraction and high-resolution transmission electron microscopy study. J. Phys. Chem. C 119, 10653–10661 (2015).
Yan, Q. et al. Catalytic conversion of Kraft lignin to bio-multilayer graphene materials under different atmospheres. J. Mater Sci. 53, 8020–8029 (2018).
Gong, Y., Li, D., Luo, C., Fu, Q. & Pan, C. Highly porous graphitic biomass carbon as advanced electrode materials for supercapacitors. Green Chem. 19, 4132–4140 (2017).
Thompson, E., Danks, A. E., Bourgeois, L. & Schnepp, Z. Iron-catalyzed graphitization of biomass. Green Chem. 17, 551–556 (2014).
Anton, R. In situ TEM investigations of reactions of Ni, Fe and Fe–Ni alloy particles and their oxides with amorphous carbon. Carbon 47, 856–865 (2009).
Gomez-Martin, A. et al. Iron-catalyzed graphitic carbon materials from biomass resources as anodes for lithium-ion batteries. Chemsuschem 11, 2776–2787 (2018).
Romero Millán, L. M., Ghogia, A. C., White, C. E. & Nzihou, A. Iron nanoparticles to catalyze graphitization of cellulose for energy storage applications. ACS Appl. Nano Mater. 6, 3549–3559 (2023).
Xia, S. et al. Synthesis and formation mechanism of biomass-based mesoporous graphitic carbon. Fuel Process. Technol. 209, 106543 (2020).
Ghogia, A. C., Romero Millán, L. M., White, C. E. & Nzihou, A. Synthesis and growth of green graphene from biochar revealed by magnetic properties of iron catalyst. Chemsuschem 16, e202201864 (2023).
Ōya, A. & Marsh, H. Phenomena of catalytic graphitization. J. Mater Sci. 17, 309–322 (1982).
Hirano, S., Saito, H. & Inagaki, M. Effect of coexisting minerals on the graphitization of carbon under pressure. IV. Accelerating effect of calcium oxide. BCSJ 43, 2599–2603 (1970).
Boehm, H. P. Formation of well-crystallized graphite in the calcium cyanamide process. Carbon 16, 385–387 (1978).
Béguerie, T., Weiss-Hortala, E. & Nzihou, A. Calcium as an innovative and effective catalyst for the synthesis of graphene-like materials from cellulose. Sci. Rep. 12, 21492 (2022).
Qiu, H., Han, L. & Liu, L. Properties and microstructure of graphitised ZrC/C or SiC/C composites. Carbon 43, 1021–1025 (2005).
Yang, G., Han, H., Li, T. & Du, C. Synthesis of nitrogen-doped porous graphitic carbons using nano-CaCO3 as template, graphitization catalyst, and activating agent. Carbon 50, 3753–3765 (2012).
Amin Alavi, M. & Morsali, A. Ultrasonic-assisted synthesis of Ca(OH)2 and CaO nanostructures. J. Exp. Nanosci. 5, 93–105 (2010).
Lachman, J. et al. An overview of slagging and fouling indicators and their applicability to biomass fuels. Fuel Process. Technol. 217, 106804 (2021).
Kuznetsov, G. V., Yankovskii, S. A., Tolokol’nikov, A. A. & Cherednik, I. V. Mechanism of the suppression of sulfur oxides in the oxidative thermolysis products of coals upon their combustion in a mixture with dispersed wood. Solid Fuel Chem. 54, 311–317 (2020).
Pánek, P., Kostura, B., Čepeláková, I., Koutník, I. & Tomšej, T. Pyrolysis of oil sludge with calcium-containing additive. J. Anal. Appl. Pyrol. 108, 274–283 (2014).
Jia, X., Wang, Q., Cen, K. & Chen, L. An experimental study of CaSO4 decomposition during coal pyrolysis. Fuel 163, 157–165 (2016).
Ghogia, A., RomeroMillán, L. M., White, C. E. & Nzihou, A. Synthesis and growth of green graphene from biochar revealed by magnetic properties of iron catalyst. Chemsuschem 16, e202201864 (2022).
Zhao, Y., Feng, D., Zhang, Y., Huang, Y. & Sun, S. Effect of pyrolysis temperature on char structure and chemical speciation of alkali and alkaline earth metallic species in biochar. Fuel Process. Technol. 141, 54–60 (2016).
Wang, J., Morishita, K. & Takarada, T. High-temperature interactions between coal char and mixtures of calcium oxide, quartz, and kaolinite. Energy Fuels 15, 1145–1152 (2001).
Li, G., Liu, Q. & Liu, Z. CaC2 production from pulverized coke and CaO at low temperatures—Reaction mechanisms. Ind. Eng. Chem. Res. 51, 10742–10747 (2012).
Condo, A. F. T., Lindström, D., Kojola, N. & Sichen, D. Study on the effect of aging on the ability of calcium carbide for hot metal desulfurization. Steel Res. Int. 87, 1137–1143 (2016).
Lindström, D. & Sichen, D. Kinetic study on desulfurization of hot metal using CaO and CaC2. Metall. Mater. Trans. B 46, 83–92 (2015).
Hirano, S., Inagaki, M. & Saito, H. Cooperative accelerating effect of calcium carbonate and gaseous nitrogen on graphitization of carbon. Carbon 17, 395–398 (1979).
Lin, Q. et al. Atomic scale investigations of catalyst and catalytic graphitization in a silicon and titanium doped graphite. Carbon 88, 252–261 (2015).
Acknowledgements
The authors thank Teresa Hungria for the acquisition of the HRTEM images. The graphenization equipment used for this work is supported by the French “Investissements d’Avenir” program under the Laboratory of Excellence, LABEX SOLSTICE, ANR-10-LABX-22-01 grant. This support is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
T.B. wrote the manuscript. N.L. acquired and helped analyzing the XRD data. E.W.H. and A.N., the supervisors of this work, helped discussing and put the results in a broader perspective. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Béguerie, T., Weiss-Hortala, E., Lyczko, N. et al. The mechanisms of calcium-catalyzed graphenization of cellulose and lignin biochars uncovered. Sci Rep 13, 11390 (2023). https://doi.org/10.1038/s41598-023-38433-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-38433-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
