
Abstract
The aim of this study was to evaluate the expression of the TGFB1 gene encoding the TGF-β1 cytokine in 64 patients, and then to compare it with clinico-pathological features. The study also investigated whether the regulation of the gene expression is caused by methylation of the promoter region between − 235 and + 22 nucleotide from the start of transcription. The dependence of the relative level of the TGFB1 gene expression on the clinical advancement according to the TNM classifications was shown. Additionally, the individual grades of the T and M features of the TNM classification differed in the relative transcript levels of the TGFB1 gene. Moreover, the higher relative expression level of the studied gene was associated with a lack of vascular invasion by cancer cells and presence of lymphocytes in the neoplastic tissue. The obtained results may indicate a possible impact of the gene on the process of carcinogenesis in colorectal cancer and reduction of its expression level may be one of the factors contributing to progression of the disease.
Similar content being viewed by others

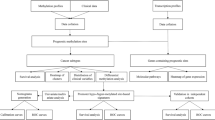

Introduction
Despite the progress of civilization, colon cancer is still a serious health problem in the world, especially in highly developed countries. According to the statistics of the Global Cancer Observatory in 2020 there were almost two million new cases of colorectal cancer, which makes it the third most commonly diagnosed cancer in the world1. The huge scale of the problem prompts many researchers to explore the basis of carcinogenesis.
A small percentage of colon cancers are inherited like Lynch's syndrome or familial adenomatous polyposis (FAP). However, a vast majority of cases are sporadic colorectal cancers associated with older age, low physical activity, a high saturated-fatty acids diet and low fiber intake, as well as spontaneous changes at the molecular level2,3. Disturbances in the TGF-β signaling pathway are believed to be one of the factors associated with the development of colorectal cancer4,5,6,7.
The TGF-β signaling pathway plays a very important role in embryogenesis and cellular homeostasis4,8. It is involved in many biological processes such as control of cell growth, differentiation or migration, and even regulation of apoptosis8,9. The superfamily of the TGF-β signaling pathway consists of 40 different proteins. This group includes, among others, nodal growth differentiation factor (nodal), activin, inhibin, bone morphogenetic proteins (BMPs), growth and differentiation factors (GDFs), anti-Müllerian hormone (AMH) or TGF-β proteins9,10,11. These cytokines, with the participation of specific transmembrane receptors and cytoplasmic proteins, transmit a signal to the cell nucleus and regulate the expression of various genes8,12.
In the canonical TGF-β signaling pathway dependent on SMAD proteins, signal transduction begins with the binding of a dimeric ligand to a type II receptor (TβR-II) binding domain which initiates activation of its kinase domain. As a result of increasing the affinity to the type I receptor (TβR-I), a heterotetramer is created. It consists of two TβR-II and two TβR-I molecules10,13. This results in phosphorylation of the glycine and serine-rich GS domain of the type I receptor by TβR-II, which involves the cytoplasmic proteins of the SMAD family. These proteins enter the cell nucleus where they interact with DNA-binding proteins, transcriptional coactivators and corepressors to regulate the expression of target genes4,8,10,14.
Apart from the canonical pathway, the ligands of the TGF-β may also involve proteins of other signaling pathways in which the regulation of gene expression occurs without SMAD proteins. The pathways of mitogen-activated protein kinases (MAPKs) and phosphoinositide 3-kinases (PI3Ks) are recruited via TGF-β and bone morphogenetic protein receptors8. After binding to the TRAF6 (TNF receptor-associated factor 6), the TGF-β receptors activate the TAK1 protein (TGF-β-associated kinase 1) which activates the signaling cascades of the MKK4-JNK, the MKK3/6-p38 and the IKK-NFκB ones. Through them, the signal is also transmitted to other signaling molecules, such as PAR6, RhoA, Cdc42 or PAK2, which play a significant role in the process of the epithelial-mesenchymal transition12,15,16.
The TGF-β is one of the most important cytokines in the regulation of tissue development and homeostasis. It is released mainly by thrombocytes and, to a lesser degree, by activated macrophages, lymphocytes and neutrophils. Particularly high concentrations of the TGF-β are found at the site of an ongoing inflammatory reaction. In addition, the protein also participates in bone formation, angiogenesis, development of muscle and adipose tissue, inflammation and wound healing. Many reports indicate that dysfunctions of the protein are associated with fibrotic diseases, connective tissue disorders and cancers11,17,18,19.
The TGFB1 gene encoding this cytokine is located on the long arm of chromosome 19 and is composed of seven exons and six introns with a total length of 52.3 kb20,21. The expression product of the gene is the pro-TGF-β protein with a molecular weight of 44.3 kDa and composed of 390 amino acids. The pro-TGF-β is a precursor of the LAP protein (latency associated peptide) and TGF-β (transforming growth factor β) belonging to the TGF-β superfamily ligands22,23. There are three isoforms of transforming growth factor β, i.e., TGF-β1, TGF-β2 and TGF-β3, which in the extracellular space exist in an inactive form, non-covalently associated with other proteins. As a result of the formation of the so-called small latency complex (SLC) and large latency complex (LLC), the ability to bind the cytokine to receptor proteins is lost. Due to the presence of complexes, there is often an excess of inactive TGF-β in the extracellular space which may be quickly used in the case of increased demand, e.g., during embryogenesis, wound healing, fibrosis or carcinogenesis12.
The involvement of the TGFB1 gene in the progression of colorectal cancer has been extensively described. Under physiological conditions, the TGFB1 gene acts as a tumor suppressor that activates apoptosis and lowers the expression of the gene encoding vascular endothelial growth factor24. In turn, it has been observed in many studies that overexpression of this gene is associated with the formation of neoplastic stem cells in the tumor stroma and a worse response to treatment25. Additionally, it promotes the epithelial-mesenchymal transition (EMT) and thus the formation of metastases26,27. It has also been documented that overexpression of the TGFB1 reduces the immune response against cancer cells, which promotes the process of carcinogenesis28.
Evidence available in many publications indicates a connection of the TGFB1 gene with the pathogenesis of neoplastic diseases (among others, melanoma, lung or breast cancers), and therefore the authors of the study decided to assess the relative expression level of the gene in tissues of patients with colorectal cancer29. In the analysis, the relative levels of the TGFB1 gene expression were compared with age, sex and family history of the patients as well as other clinical and pathological features. The most important aim of the study was to understand the mechanism of regulation of the TGFB1 gene expression. To achieve this objective, the authors assessed the presence of methylation within the promoter region of this gene between − 235 and + 22 nucleotides from the start of transcription and then compared the data with demographic and clinico-pathological features.
Materials and methods
The biological material for the research consisted of 64 tissue sections collected from patients with colorectal cancer during cancer removal surgery. All the analyzed tumor tissue fragments were confirmed by histopathological examination and obtained from the Department of Pathology, Medical University of Lodz, Poland. The material was frozen in liquid nitrogen immediately after collection and stored at − 80 °C until the analysis. Patients gave their informed consent to participate in the experiment and the research was conducted with the approvals of the Bioethics Committee of the Medical University of Lodz (RNN/214/00, RNN/8/08/KE, RNN/83/20/KE). All methods used in this study were performed in accordance with the relevant guidelines and regulations.
RNA and DNA isolation
Total RNA and DNA were isolated from frozen tissue fragments of colorectal cancer using the column method according to the Total RNA Mini and Genomic Mini protocols (A&A Biotechnology, Poland). The concentration and purity of the RNA samples after isolation were measured spectrophotometrically using the NanoPhotometer™ (IMPLEN, Germany). RNA samples with a calculated A 260/280 absorbance value ranging from 1.8 to 2.0 were selected to perform the reverse transcription reaction. Isolated DNA from tissues showing adequate purity was stored for further analysis.
RNA analysis—assessment of the level of gene expression
Reverse transcription reaction
At the next stage, the information contained in the isolated RNA was transcribed into complementary DNA. The reverse transcription reaction was performed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, USA). A final RNA concentration of 0.05 µg/µl was used in each sample prepared for the reaction. The composition of the reaction mixture and the temperature conditions for the reaction were provided in accordance with the manufacturer's recommendations.
Qualitative analysis of TGFB1 gene expression
To establish whether cDNA was obtained as a result of RT-PCR, the fragment of GAPDH gene was amplified by PCR. After confirming the correctness of the RT-PCR reaction, a qualitative evaluation of the TGFB1 gene expression was performed using the PCR method. All PCR reactions were carried out on an MJ Mini™ Personal Thermal Cycler (Bio-Rad, USA). The sequence of the primers used in the PCR for GAPDH were 5′-TGGTATCGTGGAAGGACTCATGAC-3′ and 5′-ATGCCAGTGAGCTTCCCGTTCAGC-3′ and 5′-AACCCACAACGAAATCTATG-3′ and 5′-CTTTTAACTTGAGCCTCAGC-3′ for TGFB1. The composition of reaction mixtures and PCR reaction conditions for the analyzed genes are presented in Tables 1 and 2. For each PCR reaction, a negative control, containing the reaction mixture and nuclease-free water without the cDNA template, was run. The presence of the PCR products was confirmed by electrophoresis on a 2% agarose gel using a non-specific ethidium bromide dye, which allowed visualization of the PCR products under UV light. The product size for the GAPDH gene was 189 bp and the size was 146 bp for the TGFB1 gene. Only samples which qualitatively showed presence of the tested gene were subject to further quantitative analysis.
Quantitative analysis of TGFB1 gene expression—real-time PCR
The real-time PCR technique was applied to quantify the level of mRNA expression of the TGFB1 gene. The amplification reaction was performed on a Mx3005P QPCR SYSTEM (Stratagene, USA) using the SYBR® Green JumpStart Taq Ready Mix reagent kit (Sigma Aldrich, Germany). For the analyzed samples, the TGFB1 gene and the GAPDH reference gene were amplified parallelly, in separate tubes. Each reaction was performed in triplicate. The negative control, devoid of template cDNA, was amplified in each cycle of real-time PCR reactions. The reaction mixture for one sample was composed of 7.5 µl of the ready-made reagent mix containing buffer, MgCl2 (7 mM), dNTPs (0.4 mM) and Taq polymerase (0.05 U/μl); 0.7 µl of each primer for the TGFB1 gene or GAPDH gene at a concentration of 10 µM and 1 µl of cDNA and nuclease-free water to a final volume of 16 µl. The same primers were used for both real-time PCR and the qualitative analysis. The time–temperature profile of the real-time PCR reaction included an initial denaturation step at 95 °C for 10 min, then 40 cycles, each of which consisted of three steps, i.e. a 30-s denaturation at 95 °C, 1-min annealing of primers to the template at 59 °C and 1-min extension of the template at 72 °C. After all the reaction cycles were completed, there was a final primer annealing step for 30 s at 55 °C and a final denaturation for 30 s at 95 °C.
Due to the use of a nonspecific fluorescent dye, SYBR Green, which has the ability to intercalate between each double-stranded DNA molecule as a marker, it was necessary to carry out a reaction specificity control. To this end, after each real-time PCR reaction, the process of denaturation of the obtained amplification products was performed and the specificity of these products was assessed based on the obtained melting curves. The amplified products had a characteristic melting temperature (Tm for GAPDH gene = 88 °C, Tm for TGFB1 gene = 81 °C) resulting from their nucleotide sequences. An example of a melting curve plot for the TGFB1 gene was shown in Fig. 1.

The melting curves of the real-time PCR amplification products of the TGFB1 gene.
Based on the obtained amplification diagrams, the Ct point was determined for each sample, which corresponded to the moment when the reaction entered the phase of the logarithmic increase in the amount of the product. The kinetic efficiency of real-time PCR was estimated based on the analysis of the standard curves of the GAPDH and TGFB1 genes. For this purpose, from one of the tests following the PCR reaction, for which the concentration of the obtained product was also determined, a series of dilutions ranging from 10 to 107 were prepared. Then, real-time PCR was performed for each of the dilutions and the Ct values were read. They were used to determine the reaction efficiency. It was calculated from the slopes of the standard curves according to the equation E = 10 [− 1/slope] − 1. PCR efficiency differed between the compared genes significantly (111% for GAPDH and 101% for TGFB1), therefore the Pfaffl's method was used to calculate the relative expression ratio, taking into account the level of reaction efficiency for each of the genes.
DNA analysis—assessment of methylation of the CpG sequence of the TGFB1 gene
Methylation-specific PCR
The assessment of the CpG island methylation of the promoter region of the TGFB1 gene required a stage of DNA purification and preparation for further studies. For this purpose, the conversion of unmethylated cytosines to uracil was performed according to the CiTi Converter DNA Methylation Kit protocol (A&A Biotechnology, Poland). The MS-PCR reaction was subsequently performed using the modified Taq-CiTi HotStart DNA polymerase, which prevents the extension of the template containing the modified nucleotide. The reaction was carried out with the use of the CiTi Converter MSP PCR Kit (A&A Biotechnology, Poland). The composition of the reaction mixture for MS-PCR amplification for one sample was the following: a ready-to-use 2xCiTi MSP PCR Mix (A&A Biotechnology, Poland) in the amount of 25 µl, forward and reverse primers in the volume of 1.75 µl each, DNA after conversion in the amount of 2.5 µl and sterile water to a final volume of 50 µl. The MS-PCR reaction was carried out in a thermocycler in controlled temperature–time conditions, with an initial denaturation of the template and activation of CiTi HotStart DNA polymerase for 5 min at 95 °C, followed by 35 cycles consisting of three repeated steps, i.e., denaturation for 15 s at 95 °C, primer annealing for 30 s at 59 °C and extending the template for 30 s at 72 °C. In the final stage sample was incubated in 10 °C for 10 min. The evaluation of the CpG sequence of the TGFB1 promoter region gene was performed based on electrophoretic separation of the MS-PCR reaction products (Fig. 2). The product size was 257 bp.

Image of gel electrophoresis of MS-PCR products. 1, 4, 5—methylated samples, 2, 3, 6—unmethylated sample, 7—negative control (unmethylated), 8—positive control (methylated), 9—molecular-weight size marker. Original gel is presented in Supplementary Fig. 1.
Bioinformatic analysis of promoter region methylation
TCGA data available in the UALCAN database (http://ualcan.path.uab.edu/index.html) was used to compare the promoter methylation level in the colon adenocarcinoma and healthy colon tissue. The beta value is the ratio of the methylated probe intensity and the sum of methylated and unmethylated probe intensity. Beta value ranges from 0 (unmethylated) to 1 (fully methylated). The boxplot represents beta values of CpG probes located up to 1500 bp upstream of gene's start site.
Results
The STATISTICA 13.1 software (StatSoft Inc., USA) was used to statistically evaluate the obtained results.
The first assessment focused on a comparison between the age of the patients in the study group at the time of developing colorectal cancer and the relative mRNA level of the TGFB1 gene. The median age in the studied group of patients was 63 years (57 years for women and 66 years for men), the youngest patient was 34 years old and the oldest subject was 82 years old. The analysis of dependence did not show any correlation between the compared features (p = 0.1380).
The second step of the statistical evaluation was to compare the relative expression level of the studied gene with respect to sex. Men are statistically more likely to develop colorectal cancer than women. For this reason, it becomes justified to search for molecular bases that may contribute to the emergence of differences in the incidence of this type of cancer. The study compared the relative level of TGFB1 gene expression in the group of 30 women and 20 men, and no statistically significant differences were observed (p = 0.9763).
Some cases of colorectal cancer are hereditary, therefore, in the next stage of the statistical analysis, an association between the relative level of TGFB1 gene expression and presence of cancer in a patient's immediate family was determined. The percentage of patients confirming presence of colorectal cancer in the family was 33%, while a majority of the patients included in the study came from families in which cases of colorectal cancer had not been previously diagnosed. Moreover, no statistically significant correlation was found between family history and the relative level of TGFB1 gene expression (p = 0.8149).
Due to different therapeutic procedures and for prognostic reasons, the location of neoplastic lesions is important. Therefore, another assessed feature was the correlation of the relative expression level of the studied gene with the location of colorectal cancer. Forty nine patients included in this analysis were divided into two groups, i.e., those with cancer of the rectum (20 patients) and those with cancer located in the upper sections of the large intestine (29 patients). However, no statistically significant correlation was found between the relative level of TGFB1 gene expression and cancer location (p = 0.1246).
Another assessed feature was the histopathological subtype of colorectal cancer. Based on histopathological examination, three different types of adenocarcinomas were distinguished in the studied population, i.e., adenocarcinoma tubulare (32 patients), adenocarcinoma mucinosum (9 patients) and adenocarcinoma tubulare/mucinosum (6 patients). In each subgroup, the expression level of the analyzed gene was compared. The comparative analysis showed no differences between the relative level of TGFB1 gene expression and the histopathological type of cancer (p = 0.9561).
Based on the microscopic evaluation, each tumor tissue collected from the study group patients was classified into one of three categories, G1, G2 or G3, depending on the degree of histological differentiation of cells. Most of the observed cases were tumors with moderately (G2) and poorly differentiated cells (G3) (92%). Due to the small number of well-differentiated adenocarcinomas (G1), groups G1 and G2 were combined for statistical purposes. The analysis showed that the relative expression level of the TGFB1 gene did not differ between the groups (p = 0.1102). Despite the lack of a statistically significant association between the transcript level of the TGFB1 gene and the degree of histological malignancy, a tendency was noticed to achieve higher relative expression levels of this gene by G1 and G2 adenocarcinomas as compared to G3.
In order to conduct another comparative analysis, the study group was divided into four subgroups based on the clinical stage of cancer according to the TNM classification (I—16 cases, II—12 cases, III—13 cases, IV—9 cases). The statistical analysis showed that the relative level of TGFB1 gene expression differed depending on the stage of clinical advancement, and that association was statistically significant (p = 0.0113, Fig. 3). It may indicate a potential influence of this gene on the course of carcinogenesis in the large intestine. Moreover, a statistically significant difference in the relative levels of TGFB1 gene expression was demonstrated between TNM grades I and IV (p = 0.0090). Higher relative transcript levels of the TGFB1 gene were observed in stage I patients as compared to stage IV patients. The obtained results indicate that lowering the level of TGFB1 gene expression may be one of the factors contributing to the progression of colorectal cancer.

Relative expression levels of the TGFB1 gene and clinical stage according to the TNM classification.
In the next stage, an association between the individual TNM classification parameters and the expression of the TGFB1 gene was assessed. The first analyzed parameter was the T feature, indicating the size of the primary lesion and tumor penetration of the intestinal wall. A correlation was observed between the relative mRNA levels of the TGFB1 gene and the T feature of the TNM classification, and the association was statistically significant (p = 0.0206). The highest relative expression levels were found in the T1 and T2 stages, which may indicate the inhibitory effect of this gene expression on the size and depth of tumor infiltration. Another assessed parameter of TNM classification was the feature N, which indicates presence of neoplastic cells in the regional lymph nodes. The applied test showed no statistically significant differences in the level of TGFB1 gene expression in different N stages of TNM classification (p = 0.4884). The last element of TNM classification was the feature M indicating presence or absence of distant metastases. It was found that the relative transcript levels of the TGFB1 gene differed in the subjects depending on presence of distant metastases (p = 0.0120). The patients without diagnosed metastases were characterized by higher relative expression levels of the analyzed gene, which may indicate a protective role of TGFB1 in the process of metastasis formation.
Histopathologically confirmed presence of neoplastic cells in blood vessels is an unfavorable prognostic factor in the course of colorectal cancer. The statistical analysis showed an association between the relative levels of TGFB1 gene expression and tumor invasion of blood vessels (p = 0.0125) which is presented in Fig. 4. Higher relative transcript levels of the TGFB1 gene were found in cancer cases where blood vessels were not affected by neoplastic cells, which may indicate that the studied gene plays a role in preventing the formation of secondary neoplastic lesions in distant organs.

Relative expression levels of the TGFB1 gene and tumor invasion of blood vessels.
The presence of lymphocytic infiltration in the tumor mass is a positive prognostic indicator of advanced colorectal cancer. For this reason, another analysis was made to check an association between the expression of the studied gene and presence of lymphocytic infiltration in the neoplastic tissue of the colon. The analysis shows that there is a statistically significant association between the compared parameters (p = 0.0087, Fig. 5).

Relative expression levels of the TGFB1 gene and presence of lymphocytic infiltration in the tumor tissue.
It has been shown that higher relative levels of TGFB1 gene expression are observed more frequently in patients with colon cancer accompanied by lymphocytic infiltration. It may indicate an important role of the TGFB1 gene in regulating the immune response against cancer.
The survival analysis of the patients did not reveal any statistically significant differences in survival times depending on the relative expression level of the studied gene (p = 0.5257).
All results concerning the evaluation of the TGFB1 gene expression in colorectal cancer tissues were collected and presented in Table 3.
The observed differences in the expression levels of the TGFB1 gene in the study group meant that the next goal of the study was to understand the mechanism of regulation of this gene expression. Since DNA methylation is one of them, the frequency of occurrence of methyl groups within the promoter region of the TGFB1 gene in normal and neoplastic tissues was compared using the UALCAN bioinformatics tool (http://ualcan.path.uab.edu/index.html). The analysis showed that the promoter methylation level was substantially higher in colorectal cancer in comparison with normal tissues (p < 0.001, Fig. 6).

The promoter methylation level of the TGFB1 gene in colon adenocarcinoma tissue in comparison with normal colon tissue by the UALCAN database.
Therefore, the next step of the analysis was to check the methylation status in the selected area of the promoter region and refer it to the demographic and pathological parameters of the patients as well as the expression level of the studied gene. In the investigated group, methylation of the promoter region of the TGFB1 gene was confirmed in 26 cases (40.6%), whereas 38 of them (59.4%) were devoid of 5-methylcytosines in the observed area. The mean age of the patients with confirmed methylation within CpG islets preceding the TGFB1 gene was 59 years and it was lower than the mean age of the patients without methylation (62 years), however, that difference was not statistically significant (p = 0.2894). The study population included 32 women and the same number of men. In both groups, an advantage was observed in the patients without methylation of the promoter region of the TGFB1 gene. The statistical analysis did not confirm any statistically significant association between the compared features (p = 0.6107). Out of the 63 patients, 40 had cancer located in the caecum or colon and 23 had cancer of the last section of the large intestine. The comparison did not show any statistically significant association between the frequency of methylation in the promoter region of the TGFB1 gene and the location of colorectal cancer (p = 0.1860). Within the study group, most of the 61 patients had tubular adenocarcinomas (N = 44), while mucinous and tubulo-mucinous adenocarcinomas were sparse (N = 8 and N = 9, respectively). There was no significant correlation between the frequency of methylation within the promoter region of the TGFB1 gene and the histopathological subtype of colorectal cancer (p = 0.1606). Comparing the frequency of methylation with the degree of histological malignancy of cancer cells, a higher frequency of methylated cytosines in the promoter region of the TGFB1 gene was observed in poorly differentiated neoplasms as opposed to cancers with a good and moderate degree of cellular differentiation. The performed statistical analysis did not confirm the association between G1 and G2 groups and G3 group (p = 0.0806). The evaluation, based on a comparison of the frequency of methylation of the TGFB1 gene promoter region with the clinical advancement according to the TNM classification, showed a more frequent methylation of the region regulating TGFB1 gene expression in the most advanced stage III and IV cancers (56% and 53%, respectively) as compared to cancers in earlier stages Io and IIo (33% and 19%, respectively). However, the association turned out to be statistically insignificant (χ2 test, p = 0.1026). Another analyzed feature was the invasion of blood vessels by the neoplasm. Despite the noticeable tendency to a higher frequency of methylation of the area under study in neoplasms with vascular invasion, the performed statistical analysis did not confirm any association between the compared features (p = 0.0505). However, it may be assumed that methylation of the promoter region silencing TGFB1 expression promotes the invasion of blood vessels by the tumor. Comparison of the frequency of cytosine methylation within the promoter region of the TGFB1 gene with the presence of lymphocytic infiltration in the neoplastic tissue also showed no statistically significant correlation (p = 0.3624). When comparing the survival of the patients depending on the methylation status of the CpG islets of the analyzed area, the survival time of the patients was not dependent on the presence of methylation within the promoter region of the TGFB1 gene (p = 0.8000). The final step in the statistical evaluation was to compare the methylation frequency of the promoter region of the TGFB1 gene with the relative expression level of the gene. The performed analysis showed no differences in the levels of relative expression of the TGFB1 gene and methylation (p = 0.2825).
Discussion
The transforming growth factor β signaling pathway is involved in many physiological cellular processes. Disturbances in genes expression within the TGF-β pathway are one of the factors involved in development of many cancers where they can act as both a suppressor and a promoter of carcinogenesis29.
The results obtained in the study are consistent with the phenomenon described as the TGF-β paradox. Numerous reports indicate that in the initial stages of tumorigenesis the TGF-β pathway acts as a tumor suppressor, however, as the disease progresses, it begins to play an oncogenic role28,30. It may be connected with the ability of the TGF-β to arrest the cell cycle in the G1 phase by inhibiting the activity of cyclin-dependent kinases such as p15, p21 and p57. Additionally, the activity of kinases is attenuated by the action of the TGF-β which reduces the synthesis of c-MYC9,12. The suppressor function may be less effective with an increase in the number of mutations in the genes encoding the proteins of the TGF-β pathway as well as in the target genes. As the number of mutations increases, the ability to remodel the extracellular matrix, synthesize chemokines, angiogenesis or avoid immune system control mechanisms is also acquired. At the same time, the oncogenic activity of the TGF-β cytokine, e.g., signalling of the epithelial-mesenchymal transition, remains preserved30.
In the early stages of carcinogenesis, the high relative expression level of the TGFB1 gene may be an element of the suppressor response preventing the fixation of genetic changes in the cell. On the other hand, decreased level of the TGFB1 gene expression during the course of the disease observed in this study, may be an important result in the pathogenesis of colorectal cancer allowing for its further development. When studying the colon cancer cell line, Zhao et al. observed that TGF-β1 decreased the expression of the gene encoding VEGF and blocked the growth of enterocytes. It was also confirmed that TGF-β1 increased the apoptosis by inhibiting the COX-2 and PGE2 production24. This may suggest that the reduction of the TGFB1 expression observed in the most advanced neoplasms is associated with loss of control over this process.
The suppressive effect of the TGF-β on normal cells has been documented in many studies, however, in neoplastic cells its role in promoting carcinogenesis is becoming apparent. The high relative expression levels of the TGFB1 gene observed in the study in the least clinically advanced colorectal cancers may prove these findings. As reported by Suzuki et al., it may mean that inhibition of the TGF-β signaling is one of the most important factors for neoplastic cell proliferation31. The experiment with the breast cancer cell line shows that the TGF-β reduces the expression of the ATM, MSH2 and BRCA1 genes involved in the response to DNA damage. As a consequence, lowering the expression levels of these genes enables cells to escape from control mechanisms and allows their unlimited proliferation9,32. Moreover, studies conducted by Qin et al. on the prostate cancer cell line show that the TGF-β stimulates the process of carcinogenesis already in the initial stages of tumorigenesis33. For this reason, the obtained high expression of the TGFB1 gene in patients with low-advanced colorectal cancer may be a negative prognostic indicator of the disease course. Furthermore, selective inhibition of the TGF-β transmission in its early stages could prove to be a new therapeutic strategy.
In a study conducted in women diagnosed with breast cancer, an increased amount of the TGFB1 transcript promoted development of cancer stem cells and additionally improved their resistance to chemotherapy25. An increase in the cells mobility and a higher likelihood of metastasis were noted after radiotherapy in another group of breast cancer patients. It may be explained by sensitivity of cancer cells to radiation, as a result of which the TGF-β1 is released from LLC complexes. However, it is also very important that blocking the function of this protein eliminated such effects34. There is evidence proving that a dysfunction or loss of TGF-β signaling promotes tumor progression and that enhancement of its transmission has a positive effect on prognosis and prevents progression of colorectal cancer in its early stages35,36. The discrepancy in the research results may be explained by the presence of cytokines in the neoplastic microenvironment which take the form of large protein conglomerates with an activity that is difficult to predict from the level of the gene transcript. Thus, the presence of inactive LLC complexes appears to be one of the ways modulating the TGF-β pathway transmission. An unquestionable limitation of the presented studies is a lack of information on the TGF-β1 protein activity. Moreover, when interpreting the results, it should be noted that increased TGFB1 expression did not have to be equivalent to an increase in cytokine-initiated signaling.
An interesting observation in our study was proving the impact of the TGFB1 gene expression on the presence of liver metastases. The obtained results are consistent with other reports on the involvement of the TGF-β pathway in the formation of metastases in neoplasms of the lung, thyroid, prostate, bladder, esophagus and colon33,35,37,38,39,40. Among the molecular bases influencing progression and the ability to form metastases, the action of TGF-β is believed to stimulate formation of new blood vessels by increasing the expression of vascular endothelial growth factor (VEGF) and connective tissue growth factor (CTGF)31,41,42.
There are many reports that the TGF-β1 cytokine is responsible for acquiring the features typical for mesenchymal cells which they acquire in the process of epithelial-mesenchymal transition. It is related to the increased expression of such transcription factors as SNAIL1/2 (human snail homolog 1/2), HMGA1 (high mobility group AT-hook 1), ZEB1/2 (zinc finger E-box binding homeobox 1/2) and TWIST1 (twist basic helix-loop-helix transcription factor 1)10,28,43. As a result, cells lose the function of E-cadherin and γ-catenin adhesion proteins in favor of increased expression of mesenchymal markers, i.e., vimentin and N-cadherin40,44. It promotes the loss of cells polarity and breaking the connections between them. Consequently, they acquire features typical of mesenchymal cells with greater mobility and ability to migrate26,27.
What is more, equally important observation in the study was related to the high relative expression levels of the TGFB1 gene expression in neoplasms without blood vessel involvement. It is believed that high levels of the gene transcript can initiate vascular invasion during tumor development which is an unfavorable prognostic factor. The obtained results may confirm the contribution of the TGFB1 in increasing the migratory capacity of cells and the formation of secondary cancer foci in distant organs. These actions are explained by the ability of the TGF-β1 to regulate the functions of integrins, e.g., αV, β1 and β3 and promoting the EMT process26. These reports indicate that the TGFB1 affects intensification of proliferative processes and acquisition of the ability to form metastases. In the case of the studied patients, these processes could be initiated at the early stages of tumor development and precede the silencing of this gene transcription45.
Another interesting finding was also higher relative expression levels of the studied gene in the patients with lymphocytic infiltration in the tumor tissue. There are studies showing that overexpression of the TGFB1 gene in neoplastic diseases blocks the anti-tumor immune response by suppressing lymphocytes T and B, NK cells and macrophages28. This is confirmed by a study conducted in patients with advanced colorectal cancer in whom the TGFB1 overexpression was associated with a reduced number of T cells and a blockage of the acquisition of the Th1 phenotype by them46.
Other sources report that high expression of this cytokine inhibits the Th lymphocyte response and induces Treg lymphocytes expressing the Foxp3 transcription factor which suppress the immune response and promote tumorigenesis47,48. Increased infiltration of Treg lymphocytes within the tumor microenvironment may thus create a favorable niche for cancer development49,50. This would also explain the higher relative expression levels of the TGFB1 in tissues with lymphocytic infiltration, which has been observed in this study. Our results do not contradict the current reports on the immunosuppressive role of the TGF-β1 since a large part of the lymphocytes present in the neoplastic tissue may be the Treg cells fraction49. However, to verify the hypothesis about the increased share of Treg cells in relation to Th cells, additional immunohistochemical tests of tissues are required to determine the phenotype of lymphocytes.
The mechanisms regulating gene expression include epigenetic factors, such as modifications of histone proteins or methylation of cytosines within the promoter and regulatory regions of genes. To investigate the regulation of the expression of the studied gene, a qualitative evaluation of the CpG island methylation of the TGFB1 promoter region between − 235 and + 22 nucleotides from the transcription start was performed. The presence of methyl groups within the analyzed region was confirmed in 40.6% of the patients, which was similar to the values obtained by another team that found methylation in 44.0% of lung cancer cases. However, these results were significantly different from those observed in prostate cancer patients with a methylation rate of 82.0%. Moreover, Shah et al. noted that a higher frequency of the CpG islands methylation is characteristic for invasive prostate cancers and metastatic lung tumors51.
It should be emphasized that methylation did not have to occur only in the analyzed area, as it could have taken place simultaneously in other regulatory sites. Therefore, the next stages of the research should exclude the possibility of adding methyl groups within remaining regions influencing the expression of the analyzed gene (e.g. at positions + 91, + 115, + 145 and + 151 from the transcription start site)52. It is worth considering the role of other epigenetic factors that may affect the expression of the TGFB1 gene, including post-translational modifications of histone proteins and the participation of non-coding RNA molecules, such as siRNA and microRNA.
The available scientific data indicate the association of the TGF-β cytokine with the development of many diseases, including cancers. The results presented in the study prove a potential involvement of the TGFB1 gene in the development of colorectal cancer and its progression, however, they should be confirmed in a larger group of patients. Since this experiment did not include an analysis of the immunophenotype of lymphocytes in tumor tissue, assessment of methylation in the remaining regulatory regions of the TGFB1 gene or an impact of other epigenetic factors affecting the level of expression of this gene, such analyzes should be included in subsequent studies. Additional experiments will ensure a better understanding of the mechanisms regulating expression of this gene in the future. These outcomes can be further used to identify epigenetic markers for colorectal cancer or become the target of personalized therapy.
Conclusions
The decreased expression of the TGFB1 gene found in colorectal cancer with high clinical advancement stages indicates that the loss of the physiological function of the gene and the protein it encodes is conducive to disease progression. The protective role of the TGFB1 gene is confirmed by higher expression levels obtained in patients whose blood vessels are not affected by tumor cells and in presence of lymphocytic infiltration in cancer tissue, which are positive prognostic factors for colorectal cancer. The results presented in the study did not confirm an association between methylation of the assessed area and the clinical or pathological features of the patients. The most important conclusion was that the methylation of the studied area had no effect on the expression level of the TGFB1 gene.
References
International Agency for Research on Cancer, https://gco.iarc.fr/today/data/factsheets/cancers/10_8_9-Colorectum-fact-sheet.pdf.
Thanikachalam, K. & Khan, G. Colorectal cancer and nutrition. Nutrients 11, 164 (2019).
Vernia, F., Longo, S., Stefanelli, G., Viscido, A. & Latella, G. Dietary factors modulating colorectal carcinogenesis. Nutrients 13, 143 (2021).
Itatani, Y., Kawada, K. & Sakai, Y. Transforming growth factor-β signaling pathway in colorectal cancer and its tumor microenvironment. Int. J. Mol. Sci. 20, 5822. https://doi.org/10.3390/ijms20235822 (2019).
Jung, B., Staudacher, J. J. & Beauchamp, D. Transforming growth factor β superfamily signaling in development of colorectal cancer. Gastroenterology 152, 36–52 (2017).
Pellatt, A. J. et al. The TGFβ-signaling pathway and colorectal cancer: Associations between dysregulated genes and miRNAs. J. Transl. Med. 16, 1–22 (2018).
Soleimani, A. et al. Role of the transforming growth factor-β signaling pathway in the pathogenesis of colorectal cancer. J. Cell. Biochem. 120, 8899–8907 (2019).
Hata, A. & Chen, Y.-G. TGF-β signaling from receptors to Smads. Cold Spring Harb. Perspect. Biol. 8, a022061 (2016).
Zhao, M., Mishra, L. & Deng, C.-X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 14, 111 (2018).
Liu, S., Chen, S. & Zeng, J. TGF-β signaling: A complex role in tumorigenesis. Mol. Med. Rep. 17, 699–704 (2018).
Walton, K. L., Johnson, K. E. & Harrison, C. A. Targeting TGF-β mediated SMAD signaling for the prevention of fibrosis. Front. Pharmacol. 8, 461 (2017).
Yan, X., Xiong, X. & Chen, Y.-G. Feedback regulation of TGF-β signaling. Acta Biochim. Biophys. Sin. 50, 37–50 (2018).
Clayton, S. W., Ban, G. I., Liu, C. & Serra, R. Canonical and noncanonical TGF-β signaling regulate fibrous tissue differentiation in the axial skeleton. Sci. Rep. 10, 1–11 (2020).
Wrana, J. L., Attisano, L., Wieser, R., Ventura, F. & Massagué, J. Mechanism of activation of the TGF-β receptor. Nature 370, 341–347 (1994).
Burton, J. C., Antoniades, W., Okalova, J., Roos, M. M. & Grimsey, N. J. Atypical p38 signaling, activation, and implications for disease. Int. J. Mol. Sci. 22, 4183 (2021).
Sinha, A., Iyengar, P. V. & Ten Dijke, P. E3 Ubiquitin ligases: Key regulators of TGFβ signaling in cancer progression. Int. J. Mol. Sci. 22, 476 (2021).
Zi, Z. Molecular engineering of the TGF-β signaling pathway. J. Mol. Biol. 431, 2644–2654 (2019).
Xu, X. et al. Transforming growth factor-β in stem cells and tissue homeostasis. Bone research 6, 1–31 (2018).
U.S. National Library of Medicine, https://medlineplus.gov/genetics/gene/tgfb1/#location.
Derynck, R., Rhee, L., Chen, E. Y. & Van Tilburg, A. Intron-exon structure of the human transforming growth factor-beta precursor gene. Nucleic Acids Res. 15, 3188 (1987).
GeneCards The human gene database, https://www.genecards.org/cgi-bin/carddisp.pl?gene=TGFB1.
Robertson, I. B. & Rifkin, D. B. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb. Perspect. Biol. 8, a021907 (2016).
UniProt, https://www.uniprot.org/uniprot/P01137.
Zhao, Y., Xia, S., Cao, C. & Du, X. Effect of TGF-β1 on apoptosis of colon cancer cells via the ERK signaling pathway. J. Buon Off. J. Balk. Union Oncol 24, 449–455 (2019).
Huang, C.-K. et al. Expression of transforming growth factor β1 promotes cholangiocarcinoma development and progression. Cancer Lett. 380, 153–162 (2016).
Asiri, A., Raposo, T. P., Alfahed, A. & Ilyas, M. TGF β1-induced cell motility but not cell proliferation is mediated through Cten in colorectal cancer. Int. J. Exp. Pathol. 99, 323–330 (2018).
Yi, R. et al. Transforming growth factor (TGF) β1 acted through miR-130b to increase integrin α5 to promote migration of colorectal cancer cells. Tumor Biol. 37, 10763–10773 (2016).
Chen, Y. et al. Transforming growth factor β signaling pathway: A promising therapeutic target for cancer. J. Cell. Physiol. 235, 1903–1914 (2020).
Coates, R. F. et al. Significance of positive and inhibitory regulators in the TGF-β signaling pathway in colorectal cancers. Hum. Pathol. 66, 34–39 (2017).
Ramachandran, A. et al. TGF-β uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial-to-mesenchymal transition. Elife 7, e31756 (2018).
Parada, C., Li, J., Iwata, J., Suzuki, A. & Chai, Y. CTGF mediates Smad-dependent transforming growth factor β signaling to regulate mesenchymal cell proliferation during palate development. Mol. Cell. Biol. 33, 3482–3493 (2013).
Liu, L. et al. TGFβ induces “BRCAness” and sensitivity to PARP inhibition in breast cancer by regulating DNA-repair genes. Mol. Cancer Res. 12, 1597–1609 (2014).
Qin, T. et al. Correction: A novel highly potent trivalent TGF-β receptor trap inhibits early-stage tumorigenesis and tumor cell invasion in murine Pten-deficient prostate glands. Oncotarget 8, 57905 (2017).
Bouquet, F. et al. TGFβ1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin. Cancer Res. 17, 6754–6765 (2011).
Chen, W. et al. Silencing Trim59 inhibits invasion/migration and epithelial-to-mesenchymal transition via TGF-β/Smad2/3 signaling pathway in bladder cancer cells. Onco. Targets. Ther. 10, 1503 (2017).
Yang, L., Liu, Z., Tan, J., Dong, H. & Zhang, X. Multispectral imaging reveals hyper active TGF-β signaling in colorectal cancer. Cancer Biol. Ther. 19, 105–112 (2018).
Hao, Y., Yang, X., Zhang, D., Luo, J. & Chen, R. Long noncoding RNA LINC01186, regulated by TGF-β/SMAD3, inhibits migration and invasion through epithelial-mesenchymal-transition in lung cancer. Gene 608, 1–12 (2017).
Wan, F. et al. Knockdown of latent transforming growth factor-β (TGF-β)-binding protein 2 (LTBP2) inhibits invasion and tumorigenesis in thyroid carcinoma cells. Oncol. Res. 25, 503 (2017).
Jing, C. et al. MicroRNA-17/20a impedes migration and invasion via TGF-β/ITGB6 pathway in esophageal squamous cell carcinoma. Am. J. Cancer Res. 6, 1549 (2016).
Wang, P. Suppression of DACH1 promotes migration and invasion of colorectal cancer via activating TGF-β-mediated epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 460, 314–319 (2015).
Hua, Y., Zhang, W., Xie, Z., Xu, N. & Lu, Y. MMP-2 is mainly expressed in arterioles and contributes to cerebral vascular remodeling associated with TGF-β1 signaling. J. Mol. Neurosci. 59, 317–325 (2016).
Ferrari, G., Cook, B. D., Terushkin, V., Pintucci, G. & Mignatti, P. Transforming growth factor-beta 1 (TGF-β1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J. Cell. Physiol. 219, 449–458 (2009).
Zhong, J. et al. TGF-β1 induces HMGA1 expression: The role of HMGA1 in thyroid cancer proliferation and invasion. Int. J. Oncol. 50, 1567–1578 (2017).
Gurzu, S., Kobori, L., Fodor, D. & Jung, I. Epithelial mesenchymal and endothelial mesenchymal transitions in hepatocellular carcinoma: A review. BioMed Res. Int. 2019, 1–12 (2019).
Wang, S. et al. MicroRNA-133b targets TGFβ receptor I to inhibit TGF-β-induced epithelial-to-mesenchymal transition and metastasis by suppressing the TGF-β/SMAD pathway in breast cancer. Int. J. Oncol. 55, 1097–1109 (2019).
Tauriello, D. V. F. et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554, 538–543 (2018).
Stanilova, S., Stanilov, N., Julianov, A., Manolova, I. & Miteva, L. Transforming growth factor-β1 gene promoter -509C/T polymorphism in association with expression affects colorectal cancer development and depends on gender. PLoS ONE 13, e0201775. https://doi.org/10.1371/journal.pone.0201775 (2018).
Li, M. O., Wan, Y. Y., Sanjabi, S., Robertson, A.-K.L. & Flavell, R. A. Transforming growth factor-β regulation of immune responses. Annu. Rev. Immunol. 24, 99–146 (2006).
Toor, S. M. et al. Immune checkpoints in circulating and tumor-infiltrating CD4+ T cell subsets in colorectal cancer patients. Front. Immunol. 10, 2936 (2019).
Gyori, D. et al. Compensation between CSF1R+ macrophages and Foxp3+ Treg cells drives resistance to tumor immunotherapy. JCI Insight 3, 1–12 (2018).
Shah, J. N., Shao, G., Hei, T. K. & Zhao, Y. Methylation screening of the TGFBI promoter in human lung and prostate cancer by methylation-specific PCR. BMC Cancer 8, 1–11 (2008).
Zhang, Q. et al. Effect of TET2 on the pathogenesis of diabetic nephropathy through activation of transforming growth factor β1 expression via DNA demethylation. Life Sci. 207, 127–137 (2018).
Funding
This work was supported by statutory funds of the Department of Pharmaceutical Biochemistry and Molecular Diagnostics, Medical University of Lodz, 503/3-015-02/503-31-001.
Author information
Authors and Affiliations
Contributions
D.W.: investigation, writing—original draft, formal analysis. A.W.: investigation, validation, methodology. J.W.: data curation, statistical analysis. R.S.: data curation, writing—review and editing. R.K.: conceptualization, visualization. E.B.: project administration, supervision, writing—review and editing. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit movehttp://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wodziński, D., Wosiak, A., Pietrzak, J. et al. Assessment of the TGFB1 gene expression and methylation status of the promoter region in patients with colorectal cancer. Sci Rep 12, 11488 (2022). https://doi.org/10.1038/s41598-022-15599-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-15599-4
This article is cited by
-
Virtual Screening, Molecular Docking, and Dynamic Simulations Revealed TGF-β1 Potential Inhibitors to Curtail Cervical Cancer Progression
Applied Biochemistry and Biotechnology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
