
Abstract
Arenaviruses represent a family of viruses that are naturally present in rodents belonging to subfamily Murinae, Neotominae or Sigmodontinae. Except for Lassa virus, little information is available on other Old-World arenaviruses. Here, we describe strain AnRB3214, a virus isolated from a presumed Praomys sp. rodent in the Central African Republic in 1981 and assigned to Ippy virus based on antigenic similarity. The strain was simultaneously sequenced on Illumina NovaSeq 6000 and MinION Mk1B devices and analysed with various bioinformatics tools. We show that the best genome coverage and depth were obtained with the Kaiju and Minimap2 classification and identification tools, on either the MinION or the Illumina reads. The genetic analysis of AnRB3214 fragments showed 68% to 79% similarity with the Mobala and Gairo mammarenaviruses at the nucleic acid level. Strain AnRB3214 had a truncated nucleoprotein smaller than that of other Old World arenaviruses. Molecular clock analysis suggests that this strain diverged from Mobala virus at least 400 years ago. Finally, this study illustrates the importance of genomics in the identification of archived viruses and expands on the diversity of African arenaviruses, because strain AnRB3214 is either a variant or a close relative of Mobala virus, and not Ippy virus.
Similar content being viewed by others

Background
Arenaviruses are enveloped, single-stranded RNA viruses belonging to family Arenaviridae, order Bunyavirales. With approximately 43 species, arenaviruses are subdivided into four genera (Antennavirus, Hartmanivirus, Reptarenavirus and Mammarenavirus) that infect fishes, snakes or mammals1. Arenaviruses typically have ambisense, bisegmented RNA genomes. Each RNA segment encodes two non-overlapping open reading frames (ORFs) of opposite polarity. The small (S) 3500 nt genomic segment encodes the nucleocapsid protein (NP) and a glycoprotein precursor (GPC), which later matures into two membrane glycoproteins, GP1 and GP2. The large (L) ~ 7000 nt genomic segment codes for the RNA-dependent RNA polymerase (RdRp) and exceptionally a zinc-binding protein (Z) in the Mammarenavirus and Reptarenavirus genera2,3. Interestingly, Antennavirus has a trisegmented RNA genome, with NP and GPC being on two distinct segments4.
Mammarenaviruses are geographically segregated into two monophyletic groups based on genomic features and antigenic properties. The New World (NW) arenaviruses, represented by the Tacaribe virus complex, are prevalent in North and Latin America; the Old World (OW) arenaviruses, represented by the Lassa-Lymphocytic choriomeningitis virus (LCMV) complex, are found in Africa3,5. The natural reservoirs of mammarenaviruses are generally rodent subfamilies Murinae, Neotominae and Sigmodontinae, in which they establish persistent infections with no overt pathology for the animal host6,7. These viruses seem to be associated with specific rodent host species, although many host-switching events have been recorded, which implies that virus-host co-evolution may not be as tightly linked as previously thought6,8,9. Most mammalian arenaviruses are non-pathogenic to humans, but seven are recognized as haemorrhagic fever viruses including the Lassa and Lujo viruses in Africa and the Sabia, Machupo, Chapare, Junin and Guaranito viruses in Latin America10,11. LCMV, which has a worldwide distribution, can cause neurological disease in immune-compromised individuals12. Except for Lassa virus, knowledge of OW arenavirus diversity, evolution and involvement in human diseases is sparse. Several arenaviruses of unknown pathogenic potential have been described in African rodents such as Gbagroube, Kodoko and Menekre viruses in West Africa and Mopeia, Morogoro, Luna, Merino Walk, Mariental and Okahandja viruses in southern Africa2. In Central Africa, wild and domestic rodents act as reservoirs of numerous parechoviruses, rhabdoviruses, phleboviruses, flaviviruses, picornaviruses, astroviruses, paramyxoviruses, orthopoxviruses and arenaviruses, some of which are directly responsible for human diseases, including monkeypox, Mokola virus infection or lymphocytic choriomenigitis13,14,15.
Few arenaviruses are known in Central African rodents; however, LCMV, Souris, Mobala and Ippy viruses have been described in Mus, Praomys, Mastomys and Arvicanthis rodents in Gabon, Cameroon and the Central African Republic (CAR)13,16,17,18. Seroconversion has been highlighted in the CAR for Mobala virus infection in humans in locations where the virus has been isolated from Praomys rodents17,19. Interestingly, seroprevalence in humans seems to reflect virus isolation in its reservoir host. Indeed, for a prevalence of 7.7% and 3.2% in Praomys sp. rodents in the Bouboui and Gomoka villages, respectively, using the indirect fluorescent antibody test (IFAT), the prevalence in humans ranges from 3.1 to 3.8%; however, in Botambi, with a prevalence of 2.2% in the reservoir, no antibodies have been detected in the human population19. Seroconversion in humans for Souris and Ippy viruses remains to be determined.
Next-generation sequencing (NGS) platforms have been a breakthrough in many scientific fields. In virology, NGS has facilitated a better understanding of the diversity and evolutionary history of viruses, the discovery of new viruses and the rapid response to outbreaks through diagnosis of the causative agents4,20,21,22. The leading NGS technologies, among which Illumina, are based on the incorporation of terminating nucleotides in a DNA polymerization process. These technologies, which produce very accurate and high-quality results, are widely used. However, they are usually cumbersome and only available in well-equipped, specialized laboratory facilities that can provide all the necessary utilities, such as a stable electric supply and internet connection. On the other hand, MinION uses a different approach in which a specific nanopore protein is used to recognize a k-mer in the sequence of interest via a disturbance in the basal electrical current. This innovation led to the miniaturization of the sequencing device and the ability to perform real-time sequence analysis23,24. This portable tool is gaining interest because it brings more flexibility to sequencing, albeit with a high error rate, and can, therefore, be used in remote regions as a diagnosis tool or in response to outbreaks as seen in the recent Lassa, Ebola and Zika epidemics22,25,26,27,28,29. Despite its shortcomings, MinION sequencers can be implemented in resource-limited settings such as the CAR as a first-line tool to respond instantly to emergencies or to analyse infectious samples subject to transportation restrictions.
Here, we describe a divergent arenavirus, the AnRB3214 strain isolated from wild Praomys rodents in the CAR in 1981. The strain was sequenced using the MinION Mk1B (Oxford Nanopore Technologies, Oxford, UK) and Illumina NovaSeq 6000 (Integragen, Ivry, France) devices and analysed with various bioinformatics pipelines.
Methods
Description of the strain
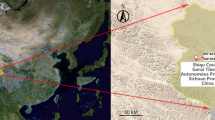
Strain AnRB3214, initially identified as Ippy virus, was isolated from a presumed Praomys sp. rodent caught near Botambi, Ombella-M’poko prefecture, CAR in October 1981 (Fig. 1). The initial isolation procedure was performed by intracerebral infection of two-day-old newborn mice up to the sixth passage. The viral isolate was then freeze-dried (lyophilized) for long-term conservation at room temperature.

Distribution of Old word arenaviruses across Africa. Coloured circles indicate countries in which OW arenaviruses have been isolated. Countries shown in grey are those where Lassa outbreaks have been recorded in humans and the virus detected in various rodents such as Mastomys natalensis. The CAR is highlighted (dot-filled). Ippy virus has been identified in the Ouaka prefecture, and Mobala and AnRB3214 (formerly identified as Ippy) originate from the Ombella-M’poko prefecture. The maps were drawn in QGIS v3.14.16 (https://www.qgis.org/fr/site/) using geopackages provided by GADM v3.6 (https://gadm.org/index.html).
Virus isolation
A volume of 0.2 mL of 1X PBS-diluted lyophilized virus was used to inoculate a litter of eight newborn mice (24–72 h old). Only mice that died after a 7-day follow-up were harvested and their brains were collected, crushed and suspended in a 1X PBS solution, and then filtered. This filtered brain suspension was used to infect fresh confluent Vero E6 cells culture in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, Life Technologies, Carlsbad, California, USA) supplemented with 2% Foetal Calf Serum (FCS, Gibco, Life Technologies) and a 1% v/v antimicrobial solution (10,000 units/mL of penicillin, 10,000 µg/mL of streptomycin and 25 µg/mL of amphotericin B, Gibco, Life Technologies). The cell cultures were monitored for 7 days for cytopathic effects using an inverted optical microscope. The brain suspensions and cell culture supernatant were then stored at -80 °C until further use.
Nucleic acid extraction
Total RNA was extracted from the culture supernatant using the QIAamp Viral RNA Mini kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. These RNAs were then quantified using the Qubit RNA HS Assay kit (Invitrogen, Life Technologies, Carlsbad, California, USA) and stored at -80 °C.
Preparation of libraries for Illumina and MinION sequencing
The first cDNA strand was prepared using SuperScript III reverse transcriptase and random hexamers (Invitrogen, Life Technologies, Carlsbad, California, USA) on total RNA. The second cDNA strand was then prepared using the NEBNext Ultra II Non-Directional RNA Second Strand Synthesis Module kit (New England BioLabs, Hitchin, Hertfordshire, UK) according to the manufacturer's instructions. All final repair and adapter ligation steps were based on the use of the NEBNext Ultra II RNA Library Prep Kit for Illumina (New England BioLabs, Hitchin, Hertfordshire, UK). All DNA purification steps were performed using Agencourt AMPure XP beads (Beckmann Coulter, Woerden, Netherlands). The library quality was checked on the 2100 BioAnalyzer instrument (Agilent Technologies, Santa Clara, California, USA) and then sequenced on Illumina NovaSeq 6000 sequencer (Integragen, Ivry, France) to obtain 2 × 150 bp paired-end reads. In parallel, the double-stranded cDNA obtained after the end repair step was used to prepare the MinION library with the PCR Barcoding kit (SQK-PBK004) (Oxford Nanopore Technologies. Oxford, Oxfordshire, UK) according to the manufacturer's instructions. The library was multiplexed with five other samples and loaded on a R9.4 flow cell (FLO-MIN106) and sequenced on the MinION Mk1B device in 37 h. The raw sequencing data were collected using ONT MinKNOW software (version 19.05.0).
Bioinformatics analysis
The quality of the raw reads from the Illumina sequencing was first assessed, filtered, and then trimmed using CLC Genomics workbench v10.0.1 (Qiagen, Hilden, Germany). All trimmed reads were then de novo assembled using SPAdes v3.10 to obtain two contigs corresponding to the S and L segments of an arenavirus genome. All reads were mapped back on the two contigs previously obtained using Geneious Prime (Biomatters, Auckland, New Zealand). All the reads from the Illumina sequencing that have been mapped were grouped together in a constituted dataset referred to as "AnRB-3214-Illumina". Three subsets, derived from the AnRB-3214-Illumina dataset, were constituted by collecting random reads from the raw fastq files in such a way that the first dataset represented 10% of the initial reads, the second 1%, and the last 0.1%. In parallel, the raw data obtained from MinION sequencing were acquired in real-time in files containing 4000 sequences in FAST5 format. These files were base-called and demultiplexed using Guppy (version 3.4.1). As carried out for the Illumina raw reads, the MinION raw reads were mapped on the obtained sequences of the two segments using Minimap230. All the reads that have been mapped were grouped in a dataset referred to as "AnRB-3214-MinION". From this dataset, three sub-datasets were constituted in the same way as previously. Viral diversity was determined from the two above-described datasets (AnRB-3214-MinION and AnRB-3214-Illumina) and their three sub-datasets using three different taxonomic classification tools: Kaiju31, Centrifuge32, and Kraken233. These tools use reference databases in which the sequence of our AnRB-3214 strain was absent. In parallel with the use of these three taxonomic classification tools, two other traditional approaches were used for the identification of viral reads. The first approach consisted in a homology search from reference sequences using the BLASTN tool whereas the second approach was to map the reads on other reference sequences using Minimap2. To use these approaches, the genomes of viruses closest to our AnRB3214 strain were screened for in GenBank and identified as Mobala virus (DQ328876, AY342390) and Gairo virus (KJ855308, KJ855307). Then, we screened the AnRB-3214-MinION and AnRB-3214-Illumina datasets against these Mobala and Gairo virus sequences using the BLASTN (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and Minimap2 tools. Hits produced by both methods were reformatted in fastq files and remapped to obtain bam files for both BLASTN and Minimap2. Bam files were converted into consensus sequences by applying BCFtools on the results of SAMtools mpileup.
Phylogenetic analysis and molecular clock estimation
S and L segment nucleic-acid sequences as well as the coding sequences of the NP, GPC, RdRp and Z genes were aligned using ClustalO in Unipro Ugene version 34.034 and manually edited. Then, phylogenetic trees were inferred for each segment using the maximum-likelihood (ML) method implemented in MEGA X (version 10.1.1) under the GTR + I + Γ4 nucleotide substitution model as determined by the best model finder in MEGA X35. The node supports were estimated using 1000 bootstrap replicates.
Concurrently, a molecular clock was tested on concatenated S and L segments using Bayesian inference. S + L concatenates of genomic sequences were aligned using MAFFT v736 and manually edited with AliView37. Starting from this alignment, ML and Bayesian analyses were conducted. A global ML evolutionary tree was reconstructed using IQ-TREE38 according to a GTR + F + I + Γ4 substitution model, recommended by the “ModelFinder” model fitter included IQ-TREE. Branch robustness was assessed using the ultrafast bootstrap approximation with 1000 bootstrap replicates39. The resulting ML tree was visualized and edited in FigTree (https://github.com/rambaut/figtree/releases/tag/v1.4.4). TempEst v1.5 was used for investigating the temporal signal of our molecular phylogeny40. Analysis of the extreme values of residuals (tangential deviation from the regression line) permitted us to spot problematic sequences. Those genomes were discarded from the following steps. BEAST v1.10.4 was used for Bayesian Monte Carlo Markov chain (MCMC) analysis to estimate the divergence time of nodes41. We retained the simplest model: strict clock and constant population size with the HKY substitution model and four gamma categories. BEAST runs were completed checking chain convergence and sufficient sampling of the posterior space (ESS > 200). The final chronogram was generated using TreeAnnotator v1.10.441.
Ethical statement
The study was approved by the institutional committee of the Institute Pasteur of Bangui. For the experiments realised out of CAR, material transfer agreements were established with regards to national and international policies. All the experiments carried on mice were done in accordance with relevant guidelines and regulations on the use of laboratory animals including the ARRIVE guidelines.
Results
Bioinformatics analysis
De novo assembly of the raw reads from Illumina sequencing allowed us to obtain two sequences with lengths of 3390 nt and 7390 nt for the S and L fragments, respectively. Then, mapping back all the raw reads identified a total of 9,275,654 reads corresponding to the arenavirus. The mean depth was of 1902 × and 155 × for the S and L fragments, respectively. Mapping also made it possible to identify a total of 77,133 reads from MinION sequencing. After mapping MinION data on these two sequences, the mean coverage was 18 × and 3x. The taxonomic assignment using three different classification tools was performed only on the dataset that contained only reads corresponding to our virus. Taxonomic assignments showed that the percentage of reads from the AnRB-3214-Illumina dataset assigned as belonging to the Arenaviridae family ranged from 0.02 to 0.4% whereas for reads belonging to the AnRB-3214-MinION dataset, taxonomic assignment percentages were 0.01% and 0.88% for Centrifuge32 and Kaiju31, respectively (Table 1).
Moreover, the percentage of reads from the computer-diluted Illumina datasets assigned as belonging to the Arenaviridae family only varied from 0.050 to 0.070% and from 0.020 to 0.024% for Kraken233 and Centrifuge, respectively. The variation was also low with Kaiju (0.45 to 0.46%) except for the 0.1% subset for which no reads were assigned. Excluding the 0.1% subset in which no MinION reads could be classified, the percentages in the other datasets ranged from 0.03 to 1.04% for computer-diluted datasets from MinION sequencing (Table 1). In parallel to these three classification tools, two other tools were used to identify viral reads, a mapping tool (Minimap230) and a homology search tool (BLASTN) using the sequence of the closest variant available in the GenBank (Mobala virus). From the AnRB-3214-Illumina dataset, a total of 26,726 reads (0.29%) were identified with Minimap2, but only 2364 (0.025%) were identified with BLASTN. Finally, although only 0.45% of the reads were classified with Kaiju, almost 100% of both the S and L segments were covered. The second best coverage was obtained with Minimap2 with 95% (573 positions uncovered), but using either BLASTN or the other two classification tools (Kraken2 and Centrifuge), the coverage percentages were lower, varying between 17% (BLASTN) and 45% (Kraken2) and were concentrated on only certain regions of the different fragments (Fig. 2).

Map of the raw reads on the de novo assembled AnRB3214 genome. The figure shows the coverage and depth of arenavirus reads retrieved with different tools on the small (S) and large (L) segments of strain AnRB3214. Minimap2 and Kaiju afforded the most extensive coverage of both segments with a depth of more than × 1000. Blastn, Centrifuge and Kraken 2 map the reads only at certain genomic regions with low coverage of other portion of the genome.
Genome analysis
We obtained two genomic segments, an S segment of 3390 nt and an L segment of 7391 nt, each segment encoding two non-overlapping open reading frames (ORFs) in an ambisense organization typical of arenaviruses. The S segment encodes GPC [position 146 to 1618] and NP [position 3190 to 1646] and the L segment encodes the Z protein [position 153 to 458] and the RdRp protein [position 7256 to 570].
Nucleotide sequences and the deduced amino acid sequence of AnRB3214 strain were compared with those of other OW arenaviruses species. The S and L segments of the AnRB3214 strain are phylogenetically very distant from the other described Ippy virus strain DakAnB188d42,43, with respectively 64% and 56% nucleic acid similarity, evidence that they belong to two distinct virus species. Although the two strains were identified in two locations in the CAR from Arvicanthis sp. (DakAnB188d) and Praomys sp. (AnRB3214) rodents almost 10 years apart, the AnRB3214 strain was classified as an Ippy virus due to cross-reactivity with DakAnB188d. Serum-based assays were widely used for the classification of viruses in the 1970-1980s and viruses were historically assigned to antigenic groups (Group A, Group B) given their antigenic relationships.
Additionally, for the S segment, at the nucleic acid level, the AnRB3214 strain displayed 79%, 72%, 70% and 68% similarity with Mobala virus, Gairo virus, Luna virus and Morogoro virus, respectively (Table 2). Similar trends were observed when comparing the L segment, where these viruses are the closest relatives with however divergences of 22%, 32%, 38% and 39%, respectively. Gairo, Luna and Morogoro viruses have been isolated from Mastomys natalensis in Tanzania and Zambia and the Mobala virus has been identified in Bouboui village in Praomys sp. from the CAR one year prior to the isolation of strain AnRB3214 in Botambi, another village of the same prefecture. When considering each protein in their respective segments, we observed an increase in the level of conservation. At the amino acid level, there was a high level of conservation for GPC with 94%, 83% and 82% of similarity with Mobala, Gairo, Luna, and Morogoro viruses (Table 2). The level of similarity with the other arenaviruses was low for the NP, Z and RdRp proteins. The Z protein of AnRB3214 was less conserved at the protein level than at the nucleic level and showed higher homology with that of Mobala virus, but less than 70% similarity with other close relatives, such as the Gairo and Luna viruses. Furthermore, the AnRB3214 NP appears to have been truncated of 43 to 78 amino acids (aa) at the 5′ end during the evolution of the virus. The AnRB3214 strain NP protein contains 515 aa, but OW arenavirus NP length ranges from 558 to 593 aa44,45. Indeed, when compared to NP of OW arenaviruses like Lassa, Mopeia, Mobala, Gairo and Luna, the NP deletion of AnRB3214 corresponds to the first 53 aa of these viruses. This feature was already observed at the nucleic acid level with the NP gene length of 1545 nt with the other arenavirus NP genes ranging from 1674 to 1779 nt.
Phylogenetic trees were built for each segment and coding region using references available in GenBank. All the tree topologies confirmed the conclusions drawn from the pairwise comparison of the sequences. For example, strain AnRB3214 belongs to the cluster comprising the Mopeia, Morogoro, Luna, Gairo and Mobala viruses (Fig. 3).

Phylogenetic trees built on the large (A) and small (B) segments. The phylogenetic trees were built using the whole genome segment under the maximum-likelihood method and the GTR-I-Γ4 substitution model with the 1000 bootstrap replicates. The trees show two groups of arenaviruses, one from the Old World (OW) and the other from the New World (NW). AnRB3214 (in blue) forms a cluster with Mobala, which is supported by a bootstrap value of 100. The phylogenetic trees were built in MEGA X v10.1.1 (https://www.megasoftware.net/).
The AnRB3214 strain is genetically very divergent from the Ippy virus DakAnB188d strain, because they are found in two distant groups in the phylogenetic trees. Moreover, strain AnRB3214 shares a most recent common ancestor (MRCA) with Mobala virus. Molecular clock analysis shows that the two viruses diverged from one another around the year 1600 AD with a substitution rate of 4.28 × 10–4 subt/site/year (1374–1885, 95% highest posterior density (HPD)) (Fig. 4).

Molecular clock analysis of strain AnRB3214 using BEAST v1.10.4. The clock shows two monophyletic arenavirus groups which seems to have diverged some 5000 years ago. AnRB3214 strain (in blue) diverged from an ancestor in common with Mobala virus around year 1602 [year 1373- year 1884]. BEAST v1.10.4 (https://www.beast2.org/) was used for the Bayesian Monte Carlo Markov chain (MCMC) inference and the chronogram was generated with TreeAnnotator v1.10.4 (https://beast.community/treeannotator).
Discussion
In this study, the AnRB3214 strain was sequenced on two sequencing platforms, Illumina and MinION, and we used a diverse set of taxonomic classification and mapping tools to retrieve arenavirus-specific reads from the obtained datasets. We obtained 77,133 and 9,275,654 reads specific to arenaviruses from the MinION and Illumina datasets, respectively. It is no surprise that we obtained less reads from MinION sequencing as this technology produces longer reads compared to Illumina. Additionally, the high amount of reads obtained during our experiment is representative of the type of sample that was used (here, cell culture supernatant) compared with direct sequencing of biological samples in which the viruses are present at low abundance with high levels of the host nucleic acid background26,46. When using reference viral genome to recover arenaviruses reads in our AnRB3213 dataset, the best read identifications were obtained with Kaiju and Minimap2. Few, if any, studies have compared tools for the classification of reads, either from long reads (MinION) or short reads (Illumina) for the analysis of divergent viruses. Minimap was initially developed for long reads, but has been updated to work with smaller paired-end reads, through a genuine management of gaps30. Furthermore, Minimap2 has been shown to be a good tool for the alignment of nanopore reads, with a higher speed than GraphMap although with comparable accuracy47. Contrary to Kraken2 and Centrifuge which primarily classify nucleotides, Kaiju uses amino acid sequences instead of genomic sequences for metagenomic classification. This feature allows Kaiju to overcome the limitations introduced by the redundancy of the genetic code and therefore make it suitable for investigating divergent organisms31. For more insight on our dataset and sequencing approaches, randomly generated subsets corresponding to 10%, 1%, and 0.1% of the overall initial outputs were generated for subsequent analysis to simulate a situation similar to that of a primary biological sample, i.e. from infected rodent reservoirs with low viral load. We showed that, except for the 0.1% dilution subset using Kaiju, arenavirus reads were detected by all the classification tools, although at varying rates. However, this test also demonstrated the limitations that can arise in the detection of such viruses in their reservoirs because the corresponding reads will be present in limited amounts.
OW mammarenaviruses are zoonotic viruses hosted by rodents of subfamily Murinae, which includes approximately 129 genera and 584 species. Members of about eight rodent genera are recognized as harbouring arenavirus species across the African continent6,7. For example, Praomys rodent species have a very extended geographic range and can be found across Sub-Saharan Africa. Ippy virus, Souris virus and Mobala virus have been isolated from Praomys rodents and Praomys clade rodents host almost all the OW arenaviruses48. Here, we described the AnRB3214 strain which was initially identified as an Ippy virus in 1981 in Botambi, CAR based on serological cross-reactivities. We demonstrated that strain AnRB3214 is distinct from the only other Ippy strain described to date, strain DakAnB188d, which also originates from the CAR. This classification was certainly due to cross-reactivity between the two viruses, because immunological assays were often used for virus identification and taxonomic assignment in the 1980s. Furthermore, “Ippy virus” has already been referred to as a complex comprising at least two viral species. Immunological microcompliment fixation data rapidly corroborated the presence of at least two distinct strains of Ippy virus, with low cross-reaction level, one of which was found in an Arvicanthis sp. rodent and the other in a Praomys sp. rodent7. With the description of the AnRB3214 strain, we confirm the huge arenavirus diversity in CAR rodents and the importance of the molecular characterization of viruses stored in biobanks.
Species delineation among arenaviruses is based on many criteria, including antigenic cross-reactivity, geographic distribution, host species and divergence at the amino acid level49. For OW arenaviruses, the interspecies amino acid cut-off divergences vary according to the protein: 8.6–20.4% (GPC), 10.2–21.5% (NP), 24.2–30.2% (Z) and 20.8–36.6% (L)49. Even so, some authors consider 12% divergence at the amino acid level in the NP enough to distinguish between two OW arenaviruses50,51. However, the latter criterion raises significant limitations, especially when considering the intra-species diversity of Lassa virus and NW arenaviruses48,52. Compared with Mobala virus at the amino acid level, strain AnRB3214 showed 6%, 13%, 17% and 17% genetic distance for the GPC, RdRp, Z and NP genes, respectively. Mobala virus and strain AnRB3214 were both isolated from unspecified Praomys rodents in the CAR in 1980 and 1981, respectively. Given the wide diversity of rodent species found in the forest areas of Central Africa, their morphological identification is often difficult and may sometimes require the use of molecular tools. Unfortunately, the absence of reference sequences for few rodent species mean that they cannot be identified53. Furthermore, Praomys rodents, which are subdivided into at least 20 putative species, are morphologically similar to Mastomys and Hylomyscus rodents54,55. Given that all these rodent species are abundant in CAR and that it is sometimes impossible to distinguish them on morphological criteria alone, it is difficult to confirm that strain AnRB3214 was isolated with certainty from a rodent of the Praomys genus. Mobala virus has also been isolated in Botambi, the same village from where strain AnRB3214 originates. Moreover, field and experimental evidence suggest that co-infections of a rodent species by two different arenaviruses are possible, although it has not yet been reported in the literature6,54. It is therefore difficult to conclude whether strain AnRB3214 is distinct from Mobala virus or not, especially because no further information is available for either virus. Molecular clock analysis under the strict clock model shows that the MRCA of strain AnRB3214 and Mobala virus can be dated back to year 1600 AD, almost 400 years ago. OW arenaviruses are thought to have originated nearly 3000–7000 years ago, as illustrated by LCMV speciation events which are estimated to have occurred around 1000 to 5000 years ago and the spread of Lassa virus from Nigeria to Mali and Ivory Coast some 1000–2000 years ago2,55,56,57. However, molecular clock analysis within Lassa virus lineages shows more contemporary virus speciation events, of less than 100 years up to 700 years58,59, similar to what was observed for strain AnRB3214. Additionally, the substitution rate deduced for strain AnRB3214 matches that of other arenaviruses, ranging from 3.3 × 10–4 to 6.3 × 10–4 subt/site/year, rates similar to those for Gbagroube virus, LCMV or Lassa virus55,59,60. Hence the circumstances that have led to the diversification of Mobala virus and strain AnRB3214 are probably similar to those observed within the Lassa virus lineages.
The AnRB3214 NP protein seems to be shorter than that of Mobala virus or the other OW arenaviruses, as it shows a ~ 53 aa deletion at the beginning of the N-terminal domain. Arenavirus NP proteins are essential for many aspects of the viral life cycle, such as the formation of the ribonucleoprotein complex when associated with RdRp, and the interference with the host immune system45,61. The N- and C-terminal domains have been shown to be the main NP regions directly involved in NP-NP oligomerisation, ribonucleoprotein complex formation and RNA synthesis. In comparison with the results obtained through an amino acid mutagenesis experiments in the N-terminus using the Tacaribe virus, a NW arenavirus44,62, we can conclude that the deletion observed in the AnRB3214 NP protein may not impact its biological function. However, further investigations are needed to understand the origin and consequences of this truncated nucleoprotein on viral particles.
More studies are needed to understand the implications of strain AnRB3214 for public health, especially in the CAR where civil unrest and political instability force the population to resort to feeding on forest-dwelling rodents. Although the prevalence of antibodies against Mobala virus in human population was associated with its prevalence in the rodent reservoirs17,19, cross reactivity with arenavirus strain AnRB3214 cannot be rule out.
Conclusion
OW arenavirus diversity remains to be explored. Here, we revealed that strain AnRB3214, classified as an Ippy virus, is clearly a species close or identical to Mobala virus. This article shows once again the usefulness of molecular tools for taxonomic assignment, especially considering archived viral strains obtained using classical virology tools and classified based on antigenic similarities. In the CAR, 22 viral isolates identified as Ippy virus have been reported from Lemniscomys striatus, Praomys spp., Arvicanthis spp. and Mastomys spp. rodents, of which 16 were isolated from Praomys spp.7,63. As seen in our study, these strains can represent distinct arenaviruses, especially because a specific arenavirus is usually associated with a single host species. Therefore, this study opens new ways to characterize the remaining CAR arenavirus strains, and especially those classified as Ippy virus and improve knowledge on the ecology of OW arenaviruses.
References
Radoshitzky, S. R. et al. ICTV Virus Taxonomy Profile: Arenaviridae. J. Gen. Virol. 100, 1200–1201 (2019).
Pontremoli, C., Forni, D. & Sironi, M. Arenavirus genomics: novel insights into viral diversity, origin, and evolution. Curr. Opin. Virol. 34, 18–28 (2019).
Charrel, R. N., de Lamballerie, X. & Emonet, S. Phylogeny of the genus Arenavirus. Curr. Opin. Microbiol. 11, 362–368 (2008).
Shi, M. et al. Redefining the invertebrate RNA virosphere. Nature 540, 539–543 (2016).
Wolff, H., Lange, J. V. & Webb, P. A. Interrelationships among arenaviruses measured by indirect immunofluorescence. Intervirology 9, 344–350 (1978).
Gonzalez, J. P., Emonet, S., de Lamballerie, X. & Charrel, R. Arenaviruses. Curr. Top. Microbiol. Immunol. 315, 253–288 (2007).
Salazar-Bravo, J., Ruedas, L. A. & Yates, T. L. Mammalian reservoirs of arenaviruses. Curr. Top. Microbiol. Immunol. 262, 25–63 (2002).
Forni, D. et al. Ancient Evolution of Mammarenaviruses: Adaptation via Changes in the L Protein and No Evidence for Host-Virus Codivergence. Genome Biol. Evol. 10, 863–874 (2018).
Bowen, M. D., Peters, C. J. & Nichol, S. T. Phylogenetic analysis of the Arenaviridae: patterns of virus evolution and evidence for cospeciation between arenaviruses and their rodent hosts. Mol. Phylogenet Evol. 8, 301–316 (1997).
Fichet-Calvet, E. & Rogers, D. J. Risk maps of Lassa fever in West Africa. PLoS Negl. Trop. Dis. 3, e388 (2009).
Briese, T. et al. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog. 5, e1000455 (2009).
Bonthius, D. J. Lymphocytic choriomeningitis virus: an underrecognized cause of neurologic disease in the fetus, child, and adult. Semin. Pediatr. Neurol. 19, 89–95 (2012).
Nadine, N. et al. Evidence of lymphocytic choriomeningitis virus (LCMV) in domestic mice in Gabon: risk of emergence of LCMV encephalitis in Central Africa. J. Virol. 89, 1456–1460 (2015).
Doty, J. B. et al. Assessing monkeypox virus prevalence in small mammals at the human-animal interface in the Democratic Republic of the Congo. Viruses 9, 283 (2017).
Nakoune, E. et al. A Nosocomial outbreak of human monkeypox in the Central African Republic. Open Forum Infect. Dis. 4, ofx168 (2017).
Gonzalez, J. P. et al. An arenavirus isolated from wild-caught rodents (Pramys species) in the Central African Republic. Intervirology 19, 105–112 (1983).
Gonzalez, J. P., McGomick, J. B., Saluzzo, J. F. & Georges, A. J. Les fièvres hémorragiques africaines d’origine virale: contribution à leur étude en République Centrafricaine. Cahiers ORSTOM Série Entomologie Médicale et Parasitologie 21, 119–130 (1983).
Olayemi, A. et al. Arenavirus diversity and phylogeography of Mastomys natalensis Rodents, Nigeria. Emerg. Infect. Dis. 22, 694–697 (2016).
Georges, A. J. et al. Antibodies to Lassa and Lassa-like viruses in man and mammals in the Central African Republic. Trans. R. Soc. Trop. Med. Hyg. 79, 78–79 (1985).
Shi, M. et al. The evolutionary history of vertebrate RNA viruses. Nature 556, 197–202 (2018).
Hetzel, U. et al. Isolation, identification, and characterization of novel arenaviruses, the etiological agents of boid inclusion body disease. J. Virol. 87, 10918–10935 (2013).
Quick, J. et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 530, 228–232 (2016).
Loman, N. J. & Watson, M. Successful test launch for nanopore sequencing. Nat. Methods 12, 303–304 (2015).
Loman, N. J. et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 30, 434–439 (2012).
Mongan, A. E., Tuda, J. S. B. & Runtuwene, L. R. Portable sequencer in the fight against infectious disease. J. Hum. Genet. 65, 35–40 (2020).
Faria, N. R. et al. Mobile real-time surveillance of Zika virus in Brazil. Genome Med. 8, 97 (2016).
Kafetzopoulou, L. E. et al. Metagenomic sequencing at the epicenter of the Nigeria 2018 Lassa fever outbreak. Science 363, 74–77 (2019).
Wawina-Bokalanga, T. et al. Complete genome sequence of a new Ebola virus strain isolated during the 2017 Likati outbreak in the Democratic Republic of the Congo. Microbiol. Resour. Announc. 8, e00360 (2019).
Gardy, J. L. & Loman, N. J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 19, 9–20 (2018).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Menzel, P., Ng, K. L. & Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 7, 11257 (2016).
Kim, D., Song, L., Breitwieser, F. P. & Salzberg, S. L. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 26, 1721–1729 (2016).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Okonechnikov, K., Golosova, O. & Fursov, M. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167 (2012).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Nakamura, T., Yamada, K. D., Tomii, K. & Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 34, 2490–2492 (2018).
Larsson, A. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278 (2014).
Minh, B. Q. et al. Corrigendum to: IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 37, 2461 (2020).
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Rambaut, A., Lam, T. T., Max Carvalho, L. & Pybus, O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2, vew007 (2016).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016 (2018).
Swanepoel, R. et al. Identification of Ippy as a Lassa-fever-related virus. Lancet 1, 639 (1985).
Digoutte, J. P. Annual report of Institut Pasteur, Bangui, Central African Republic 59 (1970).
Levingston Macleod, J. M. et al. Identification of two functional domains within the arenavirus nucleoprotein. J. Virol. 85, 2012–2023 (2011).
Pinschewer, D. D., Perez, M. & de la Torre, J. C. Role of the virus nucleoprotein in the regulation of lymphocytic choriomeningitis virus transcription and RNA replication. J. Virol. 77, 3882–3887 (2003).
Quick, J. et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 12, 1261–1276 (2017).
Senol Cali, D., Kim, J. S., Ghose, S., Alkan, C. & Mutlu, O. Nanopore sequencing technology and tools for genome assembly: computational analysis of the current state, bottlenecks and future directions. Brief Bioinform. 20, 1542–1559 (2019).
Emonet, S. F., de la Torre, J. C., Domingo, E. & Sevilla, N. Arenavirus genetic diversity and its biological implications. Infect. Genet. Evol. 9, 417–429 (2009).
Emonet, S., Lemasson, J. J., Gonzalez, J. P., de Lamballerie, X. & Charrel, R. N. Phylogeny and evolution of old world arenaviruses. Virology 350, 251–257 (2006).
Bowen, M. D. et al. Genetic diversity among Lassa virus strains. J. Virol. 74, 6992–7004 (2000).
Radoshitzky, S. R. et al. Past, present, and future of arenavirus taxonomy. Arch. Virol. 160, 1851–1874 (2015).
Weaver, S. C. et al. Guanarito virus (Arenaviridae) isolates from endemic and outlying localities in Venezuela: sequence comparisons among and within strains isolated from Venezuelan hemorrhagic fever patients and rodents. Virology 266, 189–195 (2000).
Gryseels, S. et al. Role of Wildlife in Emergence of Ebola Virus in Kaigbono (Likati), Democratic Republic of the Congo, 2017. Emerg. Infect. Dis. 26, 2205–2209 (2020).
Fulhorst, C. F., Ksiazek, T. G., Peters, C. J. & Tesh, R. B. Experimental infection of the cane mouse Zygodontomys brevicauda (family Muridae) with guanarito virus (Arenaviridae), the etiologic agent of Venezuelan hemorrhagic fever. J. Infect. Dis. 180, 966–969 (1999).
Coulibaly-N’Golo, D. et al. Novel arenavirus sequences in Hylomyscus sp. and Mus (Nannomys) setulosus from Cote d’Ivoire: implications for evolution of arenaviruses in Africa. PLoS ONE 6, e20893 (2011).
Albarino, C. G. et al. High diversity and ancient common ancestry of lymphocytic choriomeningitis virus. Emerg. Infect. Dis. 16, 1093–1100 (2010).
Andersen, K. G. et al. Clinical Sequencing Uncovers Origins and Evolution of Lassa Virus. Cell 162, 738–750 (2015).
Fichet-Calvet, E. et al. Spatial and temporal evolution of Lassa virus in the natural host population in Upper Guinea. Sci. Rep. 6, 21977 (2016).
Ehichioya, D. U. et al. Current molecular epidemiology of Lassa virus in Nigeria. J. Clin. Microbiol. 49, 1157–1161 (2011).
Zhang, L. et al. Isolation and genomic characterization of lymphocytic choriomeningitis virus in ticks from northeastern China. Transbound Emerg. Dis. 65, 1733–1739 (2018).
Martinez-Sobrido, L., Giannakas, P., Cubitt, B., Garcia-Sastre, A. & de la Torre, J. C. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J. Virol. 81, 12696–12703 (2007).
D’Antuono, A., Loureiro, M. E., Foscaldi, S., Marino-Buslje, C. & Lopez, N. Differential contributions of tacaribe arenavirus nucleoprotein N-terminal and C-terminal residues to nucleocapsid functional activity. J. Virol. 88, 6492–6505 (2014).
Meunier, D. Y., McCormick, J. B., Georges, A. J., Georges, M. C. & Gonzalez, J. P. Comparison of Lassa, Mobala, and Ippy virus reactions by immunofluorescence test. Lancet 1, 873–874 (1985).
Acknowledgements
We acknowledge Dr Carolyn Engel-Gautier for editing the English in the manuscript. This study was supported by the World Organisation for Animal health (OIE) through the European Union (capacity building and surveillance for Ebola virus disease; EBO-SURSY, project reference: FOOD/2016/379-660).This project was also supported by the Chinese Academy of Sciences, a Shanghai Municipal Science and Technology Major Project (Grant No. 2019SHZDZX02) and External Cooperation Program of Chinese Academy of Sciences (Grant No. 153211KYSB20160001).
Author information
Authors and Affiliations
Contributions
H.S.T., S.D.D. and N.B.: wrote the manuscript; S.D.D., H.S.T. and M.V.: performed the bioinformatic analyses; B.S. and A.K.: performed the experiments; J.C.M., A.G. and V.C.: reviewed and edited the manuscript; E.N. and N.B.; developed and designed the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Simo Tchetgna, H., Descorps-Declère, S., Selekon, B. et al. Molecular characterization of a new highly divergent Mobala related arenavirus isolated from Praomys sp. rodents. Sci Rep 11, 10188 (2021). https://doi.org/10.1038/s41598-021-88046-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-88046-5
This article is cited by
-
Predicting the evolution of the Lassa virus endemic area and population at risk over the next decades
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
