
Abstract
Clostridium perfringens (Cp) is a ubiquitous opportunistic pathogen of humans and animals in the natural environment and animal intestines. The pathogenicity of Cp depends on the production of toxins encoded by genes on the chromosomes or plasmids. In contemporary literature, there is no clear consensus about the pathogenicity of CpA β2 toxin. To analyze the homology of the genome of piglet source CpA and its β2 toxin, we sequenced the whole genome of strain JXJA17 isolated from diarrhea piglets using the Illumina Miseq and Pacbio Sequel platforms. The genome was composed of a circular chromosome with 3,324,072 bp (G + C content: 28.51%) and nine plasmids. Genome and 16S rDNA homology analysis revealed a close relation of the JXJA17 strain with the JGS1495, Cp-06, Cp-16, and FORC_003 strains. These strains were isolated from different samples and belonged to different toxin-types. JXJA17 strain was found to carry two toxin genes (plc and cpb2). In contrast to other Cp strains, the cpb2 of JXJA17 was located on a large plasmid (58 kb) with no co-localization of other toxin genes or antibiotic resistance genes. Analysis of JXJA17 genome homology and its cpb2 would facilitate our further study the relationship between β2 toxin and piglet diarrhea.
Similar content being viewed by others

Introduction
Clostridium perfringens is a ubiquitous Gram-positive, rod-shaped anaerobic bacterium1. Based on the four secreted toxins (α, β, ε, and ι), this bacterium is categorized into five classical types (A to E); in addition, two other new types (F and G) were recently proposed based on the presence of C. perfringens enterotoxin (CPE) and necrotic enteritis toxin B (NetB)2. C. perfringens is a common pathogen in humans and animals and causes various diseases, including gas gangrene, diarrhea, and food poisoning3. The relationship between piglet diarrhea and toxins produced by Clostridium perfringens type A (CpA) is not well characterized4,5.
As an opportunistic pathogen, Cp produces many toxins and virulence factors closely related to the diseases it causes6. More than 18 toxins are produced by Cp types A-F. CpA predominantly produces α toxin and β2 toxin which is encoded by two cpb2 alleles. β2 toxin is categorized into two types: consensus and atypical. The consensus cpb2 is preferentially associated with C. perfringens isolated from pigs7. However, there is no clear consensus on the pathogenesis of CpA β2 toxin3. Farzan et al. found no significant difference in the presence of β2 toxin in the intestinal contents of normal and diarrheic piglets8. On the contrary, in 2003 Bueschel found that most porcine enteritis isolates were cpb2 positive (> 85%), while only 11.1% of normal porcine isolates contained cpb29. There is a paucity of data related to the complete genome sequences of CpA isolated from diarrheal piglets. Moreover, there is an increasing interest in the toxin genes carried by the CpA plasmids for their impact on animal disease10,11.
In this study, strain JXJA17 was one of 229 Cp isolated from the rectal contents of neonatal piglets with diarrhea and was identified as CpA by toxin typing. This strain carries cpb2, which is more pathogenic than the CpA isolated from healthy piglets not carrying cpb2, based on animal experiments of mouse toxicity12 and rabbit ileal loop model. For better characterization β2 toxin of CpA isolated from piglet with diarrhea, we sequenced the JXJA17 complete genome and analyzed the cpb2.
Results
General features of JXJA17 genome
The sequencing coverage was 357 × with 5,250,394 high-quality reads, and 1,283,480,560 high-quality sequences (bp) were obtained on the Illumina MiSeq platform. The percentage of high-quality reads was 99.17%, reflecting the actual nucleotide composition. A total of 163,553 sequences were obtained on the PacBio Sequel platform. The total sequence length was 1,302,938,004 bp, and the GC content was 32.67%. The complete genome of JXJA17 was composed of a circular chromosome and nine extrachromosomal elements or plasmids. The genome had a chromosome with 3,324,072 bp, G + C content of 28.51%, 2967 ORFs (chromosome), 30 rRNA genes (10 5S, 10 16S, and 10 23S), and 95 tRNA genes. The identified extrachromosomal elements and their respective size (bp) were JXJA17_p1 (58,796), JXJA17_p2 (38,284), JXJA17_p3 (62,027), JXJA17_p4 (40,125), JXJA17_p5 (36,275), JXJA17_p6 (12,326), JXJA17_p7 (12,133), JXJA17_p8 (3,550), and JXJA17_p9 (11,801) (Table S1). rRNA genes, tRNA genes, or other non-coding RNA genes were not found in these plasmids, with the exception of JXJA17_p2 which had one tRNA gene and one non-coding RNA gene.
CRISPR is a unique family of DNA repeat sequences that are widely found in prokaryotic genomes. In the JXJA 17 genome, one CRISPR array was predicted. This array contained 2,865 bp (nucleotide positions 1,723,039 to 1,725,903), 44 repeats, and 43 spacer sequences. The length of each repeat was 36 bp, and the average length of the spacer sequences was 29 bp.
Among the Clusters of Orthologous Groups (COG) categories in JXJA17, seven had the largest proportions (each with ≥ 5% of the total COG classifications): R (general function prediction only, 268 ORFs, 9.03%), G (carbohydrate transport and metabolism, 199 ORFs, 6.71%), S (function unknown, 192 ORFs, 6.47%), E (amino acid transport and metabolism, 180 ORFs, 6.07%), L (replication, recombination, and repair, 169 ORFs, 5.70%), J (translation, ribosomal structure, and biogenesis, 160 ORFs, 5.39%), and K (transcription, 160 ORFs, 5.39%). The detailed numbers of COG functional categories are shown in Fig. S1 and Table S2. The physical chromosome map with the location of functional genes, transcription in both directions from a specific site, and origins of replication (oriC) are shown in Fig. 1. The CDSs on the forward or reverse strand were identified by the GC skew value. The former was more inclined towards positive values, whereas the latter showed the opposite trend. The information that each CDS belonged to the specific COG category was analyzed.

Circular map of the Clostridium perfringens JXJA17 strain chromosome. Summary of gene annotation and GC skew analysis of the genome of the JXJA17 strain. Circles (from inner to outer): circle 1 represents the scale; circle 2 shows the GC skew; circle 3 shows the GC content; circles 4 and 7 show the COG of each coding sequences (CDSs); and circles 5 and 6 show the positions of CDS. The genome atlas was drawn using CGView 1.0 (http://stothard.afns.ualberta.ca/cgview_server/).
Homology analyses of JXJA17 genome
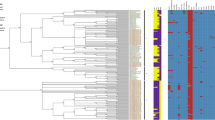
The phylogenetic tree based on genome between JXJA17 and all 205 Cp strains included in the NCBI genome database was constructed (Fig. 2). The dendrogram showed that the JXJA17 strain was more closely related to the JGS1495 (NZ_ABDU01000042.2, 84 contigs) strain and clustered with Cp-06(NZ_JAALNH010000001.1, 100 contigs) and Cp-16 (NZ_JAALMX010000001.1, 77 contigs) strains. However, the full genome sequences of these three C. perfringens were not de novo sequenced, and just spliced by some scaffolds and contigs. Therefore, the phylogenetic tree analysis is based on 16SrDNA genes and house-keeping genes between JXJA17 and 19 Cp strains complete genome using the i-sang platform (https://www.i-sanger.com/) 13 (Fig. S2 and S3). The dendrogram showed that JXJA17 strain was more closely related to the Cp FORC_003 (NZ_CP009557.1). Therefore, we compared and analyzed the general features of four C. perfringens genomes, and the results are shown in Table 1. Surprisingly, strain JXJA17 and its four genetically related strains were isolated from different hosts and environments, and their toxinotype were different as well. If a comparison were based on de novo sequencing, toxin genotyping of JXJA17 and Cp FORC_003 would be type A.

Phylogenetic tree depicting the relationship between JXJA17 and 205 reference Cp strains, constructed based on the genomic BLAST in NCBI (https://www.ncbi.nlm.nih.gov/genome/tree/158).
Collinearity analyses of plasmid carrying cpb2 of JXJA17
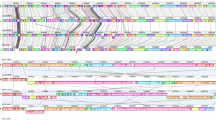
The sequencing results showed consensus cpb2 (798 bp) in JXJA17. It (from 41,169 to 41,961 bp) was individually located on a large plasmid p1 (58,000 bp) and there was no co-carriage with other toxin (such as enterotoxin or ε toxin) genes and antibiotic resistance genes. In contrast to other plasmids carrying cpb2, no Tcp conjugation locus or insertion sequence (IS) was found in plasmid p1 of JXJA17 (Table 2 and Fig. 3). Collinearity analysis of plasmid carrying cpb2 was conducted between JXJA17 and eight reference plasmids which were all cpb2 encoding plasmids among the 50 Cp plasmids included in the NCBI. The results (Fig. 4) showed sharing of eight distinct locally collinear blocks (LCBs) by these plasmids, and cpb2 was located in the blue LCB.

Comparative alignment of the sequenced C. perfringens plasmids carrying the cpb2. Arrows represent ORFs, and are colored as follows: red arrows—cpb2; yellow arrows—cpe; fuchsia arrows—etx; dark blue arrows—Pcp conjugation locus; green arrows—Tcp conjugation locus; gray arrows—antibiotic resistance; light blue arrows—insertion sequence. ORFs open reading frames.

Collinearity analyses of plasmids JXJA17_p1, pFORC3, pJFP55G, pJFP838D, pJIR3844, pCpb2-CP1, pCP8533etx, pCP13, and pCPF5603. Results aligned by using Mauve 2.4.0. (http://darlinglab.org/mauve).
Pathogenesis and virulence factors
Some important pathogenic and virulence related genes were identified in the chromosome and plasmids of the JXJA17 strain. JXJA17 contains ten known virulence related genes, including the alpha toxin phospholipase C (PLC, CPE_RS00500) gene on the genome and the β2 toxin on JXJA17_p1. Other virulence factors identified in the genome were perfringolysin O (theta toxin, pfoA, CPE_RS01165), thiol-activated cytolysin (BAS3109), collagenase (kappa-toxin, colA, CPE_RS01210), hyaluronidase (mu-toxin, CPE_RS07880), exo-alpha-sialidase (CPE_RS04035), sialidase (nanH, CPF_RS07165), alpha-clostripain (cloSI, CPR_0833), and an ATP-dependent Clp protease proteolytic subunit (clpP, lmo2468) (Table S3). However, no other toxin-related gene was carried with cpb2 in plasmid JXJA17_p1.
Antibiotic resistance genes
Screening of the genome sequences against the Comprehensive Antibiotic Resistance Database (CARD) identified 21 antibiotic resistance genes and 18 antibiotic targets in the JXJA17 strain. This strain was most likely to develop resistance to fluoroquinolone, daptomycin, tetracycline, streptomycin, and erythromycin. The 21 antibiotic resistance genes (Table S4) included one aminocoumarin gene, three fluoroquinolone genes, four daptomycin genes, two tetracycline genes, one transmembrane protein gene, one dihydropteroate synthase gene, two elfamycin genes, one streptomycin aminoglycoside adenylyl transferase gene, one UDP-glucose-6-dehydrogenase gene, one Van response regulator gene, one two-component response regulator (vanRF) gene, and three unknown product genes. In addition, the gene encoding erythromycin resistance was found in the JXJA17_p4 plasmid. However, no antibiotic resistance gene was carried with cpb2 in plasmid JXJA17_p1.
Discussion
The genomic B LAST in NCBI and homology analysis of 16S rDNA revealed high homology of JXJA17 with CP-06, CP-16, JGS1495, and FORC_003 obtained from different hosts and geographical regions. The results indicated no significant correlation of the strains with the host sources, toxinotypes, or geographical distribution. Homology analysis based on 16S rDNA sequencing is a common method for C. perfringens taxonomy14,15. Among the 19 de novo sequenced C. perfringens included in the i-sang platform, the JXJA17 strain was most closely related to the Cp FORC_003 based on 16S rDNA homology analysis. The toxin genotyping of JXJA17 and Cp FORC_003 were type A, notwithstanding the fact that Cp FORC_003 was isolated from aquarium water in South Korea. Based on the above aspects, there is no significant correlation of the strains with the host sources, toxinotypes, or geographical distribution. The location of the pathogenic toxin on plasmids may be a key characteristic that facilitates the spread of C. perfringens.
The complete genome sequences of strain JXJA17 was found to contain nine extrachromosomal elements or plasmids. One of the plasmids contains the cpb2, and another plasmid carries the erythromycin resistance gene. Many plasmid-containing strains have recently been reported. Profeta et al. reported two plasmids in the C. perfringens netB positive strain 2016TE7641_69 isolated from the intestine of a diseased turkey in Italy16. In C. perfringens CBA7123 isolated from humans, Kim et al. identified a 46,640-bp plasmid (with a G + C content of 27.1%)17. Li. et al. identified four plasmids in C. perfringens Del1 isolated from chicken18. The strain JP55 isolated from foal was found to contain five plasmids, and their G + C content was lower than that in the chromosome19. The G + C content of chromosomes from the above reported strains was higher than that in their respective plasmids. The JXJA17 strain had nine plasmids, it was also consistent with the rule that the G + C content of each plasmid is lower than that of its chromosome.
In this study, ten known virulence related genes were found in JXJA17. α toxin is the most toxic extracellular enzyme generated by C. perfringens type A20. This toxin is produced by all C. perfringens types and is essential for virulence. Furthermore, its core protein, phospholipase C, hydrolyzes phosphatidylcholine, and sphingomyelin, which are key constituents of eukaryotic cell membranes21. This toxin causes diseases such as myonecrosis22. The soluble toxins perfringolysin O and phospholipase C can cause host cell lysis and exhibit synergistic effects in C. perfringens-mediated gas gangrene23. β2 toxin is a very interesting compound. Some studies have indicated its lethal toxicity. Other reports indicate that β2 is an α auxiliary toxin24,25. Therefore, we are interested in the characteristics of β2 toxin from piglets CpA. In the JXJA17 strain, the size of plasmid carrying cpb2 was 58 kb. This is consistent with previous studies in which the size of the plasmids encoding β2 toxin was 45–97 kb. Among the fifty plasmids of C. perfringens included in the NCBI, only eight plasmids contain cpb2, including one recombinant plasmid (pCpb-CP1)26. Of the seven natural plasmids, four carried cpb2 along with another toxin (enterotoxin or ε toxin) and one plasmid was conjugated with antibiotic resistance gene. The plasmids pJFP55G and pJFP838D carrying the enterotoxin gene were from Cp JP55 and JP838 strains isolated from horses and dogs with necrotizing enteritis, respectively19. The plasmid pCPF5603 was from the Cp F5603 isolated from sporadic diarrhea (SPOR)27. The plasmid pCP8533etx carrying ε toxin was from CpB NCTC8533B4D. However, in JXJA17_p1, there were no other toxin genes, insertion sequence, antibiotic resistance gene, or Tcp conjugation locus conjugated with cpb2. By comparing these plasmids, JXJA17_p1 was most similar to plasmid pCP13, which was isolated from soil bacteria that cause gas gangrene. Plasmid pCP13 is a new family of conjugative toxin plasmids of C. perfringens strain 1328. CpCna, which encodes a putative collagen binding protein in the plasmid pCP13, is a potential virulence factor of porcine enteritis caused by C. perfringens7. The plasmid JXJA17_p1 contains the Pcp conjugation locus and CpCna. In addition, CRISPR is a prokaryotic immune system that recognizes foreign genes and silences their expression29. One CRISPR array predicted in the genome of JXJA 17 may lead to the sustained expression of toxin genes and their products. Further studies should investigate whether CRISPR in the genome would affect the expression of the toxin on JXJA17 plasmids.
In summary, we sequenced the complete genome and nine associated plasmids of the C. perfringens strain JXJA17. On 16SrDNA and whole-genome sequence analysis, the strain JXJA17 exhibited high homology with JGS1495, Cp-06, Cp-16, and Cp FORC_003. These strains were isolated from different hosts and environments, and their toxinotype were different as well. Public health authorities must be vigilant of the fact that C. perfringens carry multiple plasmids that harbor pathogenic toxin genes and are easily transmitted. Moreover, cpb2 in JXJA17 was located on plasmid p1, and was independent of other toxins, antibiotic resistance genes, or insertion sequences. Plasmid JXJA17_p1 contains no Tcp but Pcp conjugation locus, which is a new family of conjugative toxin plasmids in C. perfringens28. Analysis of JXJA17 genome homology and its cpb2 gene would facilitate our further study concerning the relationship between β2 toxin and piglet diarrhea.
Methods
Isolation and DNA extraction of Clostridium perfringens JXJA17
The study was approved by the Institution Animal Care and Use Committee of Jiangxi Agricultural University and performed according to its guidelines. All the piglets involved in the study were obtained after informed consent of the pig farm owner. We obtained the rectal contents by dipping rectal contents directly with sterilized cotton swabs through the anus of piglets. JXJA17 is a CpA strain isolated from the rectal contents of diarrhea piglets in the Jiangxi region of China and identified by biochemical tests, 16S rDNA sequencing, and toxin subtyping. A pure culture of the C. perfringens JXJA17 strain was obtained utilizing tryptose-sulfite-cycloserine agar (Haibo Biological Engineering Co. Ltd., Qingdao, China). Genomic DNA was extracted and sequenced by the Shanghai Personal Biotechnology Co. Ltd (Shanghai, China). The whole-genome sequence data was deposited in GenBank (Accession number CP028149).
Whole-genome sequencing, assembly, and annotation
The genomic DNA was sequenced using the PacBio Sequel platform (SMRTbell Template Prep kit 1.0-SPv3, Pacific Biosciences, Menlo Park, CA, USA) and Illumina MiSeq platform (Rapid Plus DNA Lib Prep Kit, Illumina, USA). MiSeq reads were trimmed using AdapterRemoval 1.5.4 (https://adapterremoval.readthedocs.io/en/latest/) and SOAPec 2.0 (https://github.com/aquaskyline/SOAPdenovo2). Next-generation sequence data were assembled using the A5-MiSeq pipeline version 20160825, and contig and scaffold sequences were obtained. Data from the PacBio Sequel platform were assembled utilizing Canu 1.3 (https://canu.readthedocs.io/en/latest/), and scaffold sequences were identified. The locations and gaps among contigs from both sequencing results were fixed using MUMmer 3.2.3 (http://mummer.sourceforge.net/) 30. Assembly errors were corrected using Pilon version 1.22 (https://github.com/broadinstitute/pilon/releases/download/v1.22/pilon-1.22.jar), and the complete genome was obtained. The hypothesized coding DNA sequences (CDSs) were identified using Glimmer version 3.0 (http://cbcb.umd.edu/software/glimmer/), and the length of the open reading frame (ORF) was set to be no less than 110 bp. tRNA genes and ribosomal RNA genes were predicted using the tRNAscan-SE 2.0 (http://lowelab.ucsc.edu/tRNAscan-SE/index.html) and RNAmmer 1.2 Server (http://www.cbs.dtu.dk/services/RNAmmer/), respectively 31. Other non-coding RNA genes were obtained by comparing the genome sequences to the Rfam database (http://rfam.sanger.ac.uk). The clustered regularly interspaced short palindromic repeats (CRISPRs) recognition tool (CRT, http://crispr.u-psud.fr/crispr/) was used to predict CRISPR regions. Functional annotation of CDSs was achieved by searching a protein database and a non-redundant protein database of the National Center for Biotechnology Information (NCBI). The predicted proteins were functionally annotated using the evolutionary genealogy of genes: Non-supervised Orthologous Groups (eggNOG) database version 3 (http://eggnog.embl.de/version_3.0). Orthologs and paralogs were defined as the same protein when the degree of similarity of sequences was > 30%. The genome atlas was drawn using CGView 1.0 (http://stothard.afns.ualberta.ca/cgview_server/).
Homology analysis
The complete genome sequence and annotation of 205 Cp strains were obtained from the GenBank database based on the whole-genome sequence of the JXJA17 strain. These strains were isolated from different hosts and the surroundings. To construct a dendrogram, we performed a homology analysis of 206 Cp in the NCBI database based on approximately 2967 genes of the JXJA17 using the genomic BLAST; the phylogenetic tree of 16S rRNA genes or house-keeping gene of 19 Cp which de novo sequencing for complete genome using the i-sang platform (https://www.i-sanger.com/). A list of the general features of the C. perfringens genomes which exhibit homology with JXJA17 was built.
Comparative study of beta2 toxin-encoding plasmids
The key JXJA17-derived toxin genes are alpha and beta2. Alpha toxin gene, which is carried by C. perfringens types A-E, is known to be carried on genome; however, the cpb2 is carried on plasmids. In order to explore the toxin genes of JXJA17, eight beta2 toxin-encoding plasmids were obtained from the GenBank using de novo assembly-graphs (https://www.ncbi.nlm.nih.gov/); subsequently, collinearity analysis was performed using the Mauve software. The sequences of eight plasmids were pFORC3 (NZ_CP009558.1/CP009558.1); pJFP55G (NZ_CP013042.1/CP013042.1); pJFP838D (NZ_CP013039.1/CP013039.1); pJIR3844 (NC_019257.1/JN689217.1); pCP8533etx (NC_011412.1/AB444205.1; pCpb2-CP1 (NC_019687.1/JQ655732.1); pCP13 (NC_003042.1/AP003515.1), and pCPF5603 (NC_007773.1/AB236337.1). The purpose of this analysis was to identify cpb2 and align the plasmid 1 of JXJA17 with the eight reference beta2 toxin-encoding plasmids complete sequences.
Prediction of virulence and antibiotic resistance genes
Potential virulence and antibiotic resistance genes in the complete genome were predicted using the virulence factors database (VFDB, http://www.mgc.ac.cn/VFs/main.htm) and the comprehensive antibiotic resistance database (CARD, http://arpcard.mcmaster.card) 32, respectively. Using BLAST (ftp://ftp.ncbi.nlm.nih.gov/blast/) to predict genes in the genome that are associated with virulence factors and antibiotic resistance, E-value threshold were set to 1e-6, more than 45% amino acid sequence consistency, and the ratio of the length of the sequence alignment to the length of the sequence not less than 70%.
References
Kazanavičiūtė, V. et al. Plant-expressed bacteriophage lysins control pathogenic strains of Clostridium perfringens. Sci. Rep. 8, 10589. https://doi.org/10.1038/s41598-018-28838-4 (2018).
Rood, J. I. et al. Expansion of the Clostridium perfringens toxin-based typing scheme. Anaerobe 53, 5–10. https://doi.org/10.1016/j.anaerobe.2018.04.011 (2018).
Uzal, F. A. et al. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 9, 361–377. https://doi.org/10.2217/fmb.13.168 (2014).
Chan, G. et al. The epidemiology of Clostridium perfringens type A on Ontario swine farms, with special reference to cpb2-positive isolates. BMC Vet. Res. 8, 156. https://doi.org/10.1186/1746-6148-8-156 (2012).
Lee, K. E. et al. Distribution of Clostridium perfringens isolates from piglets in South Korea. J. Vet. Med. Sci 76, 745–749. https://doi.org/10.1292/jvms.13-0430 (2014).
Moreira, G. M. et al. Immunogenicity of a trivalent recombinant vaccine against Clostridium perfringens alpha, beta, and epsilon toxins in farm ruminants. Sci. Rep. 6, 22816. https://doi.org/10.1038/srep22816 (2016).
Jost, B. H., Billington, S. J., Trinh, H. T. & Songer, J. G. Association of genes encoding beta2 toxin and a collagen binding protein in Clostridium perfringens isolates of porcine origin. Vet. Microbiol. 115, 173–182. https://doi.org/10.1016/j.vetmic.2006.01.012 (2006).
Farzan, A. et al. An investigation into the association between cpb2-encoding Clostridium perfringens type A and diarrhea in neonatal piglets. Can. J. Vet. Res 77, 45–53 (2013).
Bueschel, D.M., Jost, B.H., Billington, S.J., Trinh, H.T., & Songer, J.G. Prevalence of cpb2, encoding beta2 toxin, in Clostridium perfringens field isolates: Correlation of genotype with phenotype. Vet. Microbiol 94, 121–129, https://doi.org/10.1016/S0378-1135(03)00081-6 (2003)
Miseon, P. & Fatemeh, R. The prevalence of plasmid-coded cpe enterotoxin, β toxin, tpeL toxin, and tetracycline resistance in Clostridium perfringens strains isolated from different sources. Anaerobe 56, 124–129. https://doi.org/10.1016/j.anaerobe.2019.02.007 (2019).
Teuber, M. Spread of antibiotic resistance with food-borne pathogens. Cell Mol. Life Sci. 56, 755–763. https://doi.org/10.1007/s000180050022 (1999).
Zhou, J. et al. CPA isolation and its β 2 toxin investigation from the intestinal tract of piglets in Jiangxi province. Chin. J. Prevent. Vet. Med. 40, 101–105. https://doi.org/10.3969/j.issn.1008-0589.201708015 (2018).
Gardner, S. N. & Hall, B. G. When whole-genome alignments just won’t work: kSNP v2 software for alignment-free SNP discovery and phylogenetics of hundreds of microbial genomes. PLoS ONE 8, e81760. https://doi.org/10.1371/journal.pone.0081760 (2013).
Ashish, B., Tanmoy, M., Jayadev, J., Pratap, S. & Vipin, C. K. Insights into the origin of Clostridium botulinum strains: Evolution of distinct restriction endonuclease sites in rrs (16S rRNA gene). Indian J. Microbiol. 55, 140–150. https://doi.org/10.1007/s12088-015-0514-z (2015).
Kalia, V. C. et al. Analysis of the unexplored features of rrs (16S rDNA) of the genus Clostridium. BMC Genomics 12, 18. https://doi.org/10.1186/1471-2164-12-18 (2011).
Profeta, F. et al. Draft genome sequence of Clostridium perfringens netB-positive strain 2016TE7641_69, isolated from the intestine of a diseased Turkey in Italy. Microbiol. Resour. Announc. 9, e00065-e120. https://doi.org/10.1128/mra.00065-20 (2020).
Kim, Y. B. et al. Complete genome sequence of CBA7123 isolated from a faecal sample from Korea. Gut Pathog. 9, 32. https://doi.org/10.1186/s13099-017-0181-1 (2017).
Li, C., Yan, X. & Lillehoj, H. S. Complete genome sequences of Del1 strain isolated from chickens affected by necrotic enteritis. Gut Pathog. 9, 69. https://doi.org/10.1186/s13099-017-0217-6 (2017).
Mehdizadeh, G. I. et al. Plasmid characterization and chromosome analysis of two netF+ Clostridium perfringens isolates associated with foal and canine necrotizing enteritis. PLoS ONE 11, e0148344. https://doi.org/10.1371/journal.pone.0148344 (2016).
Gao, X. W. et al. Oral immunization of mice with a probiotic Lactobacillus casei constitutively expressing the α-toxoid induces protective immunity against Clostridium perfringens α-toxin. Virulence 10, 166–179. https://doi.org/10.1080/21505594.2019.1582975 (2019).
Rood, J. I. Virulence genes of Clostridium perfringens. Annu. Rev. Microbiol 52, 333–360. https://doi.org/10.1146/annurev.micro.52.1.333 (1998).
Sakurai, J., Nagahama, M. & Oda M. 2004. Clostridium perfringens alpha-toxin: Characterization and mode of action. J. Biochem. 136, 569–574, https://doi.org/https://doi.org/10.1093/jb/mvh161 (2004).
Awad, M. M., Ellemor, D. M., Boyd, R. L., Emmins, J. J. & Rood, J. I. Synergistic effects of alpha-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect. Immun. 69, 7904–7910. https://doi.org/10.1128/iai.69.12.7904-7910.2001 (2001).
Freedman, J. C. et al. Clostridium perfringens type A-E toxin plasmids. Res. Microbiol. 166, 264–279. https://doi.org/10.1016/j.resmic.2014.09.004 (2015).
Smedley, J. G., Fisher, D. J., Sayeed, S., Chakrabarti, G. & McClane, B. A. The enteric toxins of Clostridium perfringens. Rev. Physiol. Biochem. Pharmacol. 152, 183–204. https://doi.org/10.1007/s10254-004-0036-2 (2004).
Parreira, V. R., Costa, M., Eikmeyer, F., Blom, J., & Prescott, J. F. Sequence of two plasmids from Clostridium perfringens chicken necrotic enteritis isolates and comparison with C. perfringens conjugative plasmids. PLoS ONE 7, e49753, https://doi.org/10.1371/journal.pone.0049753 (2012).
Miyamoto, K. et al. Complete sequencing and diversity analysis of the enterotoxin-encoding plasmids in Clostridium perfringens type A non-food-borne human gastrointestinal disease isolates. J. Bacteriol. 188, 1585–1598. https://doi.org/10.1128/jb.188.4.1585-1598.2006 (2006).
Watts, T. D. et al. pCP13, a representative of a new family of conjugative toxin plasmids in Clostridium perfringens. Plasmid 102, 37–45. https://doi.org/10.1016/j.plasmid.2019.02.002 (2019).
Weissman, J. L., Stoltzfus, A., Westra, E. R. & Johnson, P. L. F. Avoidance of self during CRISPR immunization. Trends Microbiol. 28, 543–553. https://doi.org/10.1016/j.tim.2020.02.005 (2020).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12. https://doi.org/10.1186/gb-2004-5-2-r12 (2004).
Lagesen, K. et al. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. https://doi.org/10.1093/nar/gkm160 (2007).
Chen, L. H., Xiong, Z. H., Sun, L. L., Yang, J. & Jin, Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 40, D641–D645. https://doi.org/10.1093/nar/gkr989 (2012).
Acknowledgements
We are grateful to all the personnel who contributed to this study. This research was funded by the National Natural Science Foundation of China (No. 31860694).
Author information
Authors and Affiliations
Contributions
J.H.Z. conceived the study, J.Z. and X.Z. conducted the experiments. B.L., C.H., and L.W. analyzed the results. B.L., Y.D., and X.Z. aggregated the resulting data and wrote the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zeng, X., Liu, B., Zhou, J. et al. Complete genomic sequence and analysis of β2 toxin gene mapping of Clostridium perfringens JXJA17 isolated from piglets in China. Sci Rep 11, 475 (2021). https://doi.org/10.1038/s41598-020-79333-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-79333-8
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
