
Abstract
The roles of individual bacteria and their relationship in the development of colorectal cancer (CRC) remain unclear. We aimed to determine the prevalence of CRC-associated bacteria using quantitative real-time PCR (qPCR) or 16S rRNA analysis and the statistical correlations of patient demographics and clinical characteristics comprising alcohol consumption with CRC-associated bacteria. We determined the prevalence of five CRC-associated bacterial species in 38 CRC patients (39 samples) and 21 normal individuals using qPCR, and the relative abundance of bacterial taxa in the gut microbiome was assessed using 16S rRNA analysis. Fusobacterium nucleatum was the only bacterium that was significantly (P < 0.0001) more prevalent in the cancer tissue (82.1%) than in the normal tissue (0%) by qPCR. 16S rRNA analysis showed a significant correlation between six operational taxonomic units (OTUs), namely, the genera Fusobacterium, Peptostreptococcus, Collinsella, Prevotella, Parvimonas, and Gemella, in patients with CRC. An integrated analysis using 16S rRNA data and epidemiological characteristics showed that alcohol consumption was significantly correlated with the abundance of Fusobacterium OTUs. The correlation of alcohol consumption with the abundance of Fusobacterium OTUs in cancer tissue discovered using 16S rRNA analysis suggests a possible link between alcohol metabolism and subsequent tumorigenesis caused by F. nucleatum.
Similar content being viewed by others

Introduction
Colorectal cancer (CRC) is the third most common cancer type in men and the second most common in women worldwide, and it is the fourth leading cause of cancer-related deaths in the world1. The mechanisms underlying the development of CRC are not comprehensively understood; however, various risk factors are known to contribute to carcinogenesis in CRC, including age, presence of colon polyps, high consumption of red meat, obesity, smoking, and alcohol consumption2. Furthermore, the response to these risk factors seems to differ between ethnicities and geographical regions, which may affect the prevalence and prognosis of CRC.
Recently, the gut microbiota has been added to the list of CRC risk factors, as it has been implicated in the development of CRC and might contribute to CRC progression, as suggested by the dynamic “driver-passenger” model3. In the past few decades, it has been shown that among the diverse bacterial species that are a part of the gut microbiome, the presence of specific bacteria can play a role in colorectal carcinogenesis4. Bacteria-driven oncogenic mechanisms in CRC have been proposed to include the activation of Wnt signaling (enterotoxigenic Bacteroides fragilis (ETBF)5 and Fusobacterium species6), proinflammatory signaling (Enterococcus faecalis7 and Streptococcus gallolyticus8), and genotoxicity (colibactin-producing Escherichia coli, colB + E. coli9). These carcinogenic effects can occur from very early stages and over the multistep processes of CRC carcinogenesis and can be accompanied by shifts in the gut microbiome and metabolome10. Furthermore, a recent study showed that the gut microbiome may promote the progression of CRC11. Therefore, it is crucial to identify bacteria that may be associated with the development of CRC and to assess which other factors can elicit transformation from a healthy gut microbiome to a tumorigenic microenvironment.
Diet is the most well-known factor that influences the gut microbiome, and differences in diet can substantially affect the entire gut microenvironment12. In South Korea, CRC is one of the most prevalent types of cancer; however, few metagenomic studies have been conducted to determine the association between the gut microbiome and CRC development13. The composition of the gut microbiome may be influenced by factors other than diet, including alcohol consumption14. Numerous epidemiological studies have shown that alcohol consumption is strongly associated with the incidence of CRC15. Retrospective propensity score matching analysis (adjusted hazard ratio: 1.86) revealed that high alcohol consumption increased the development of CRC16. Alcohol consumption is considerably high in South Korea17 and may be a causative factor underlying the high prevalence of CRC in Korea18. However, to our knowledge, the effect of alcohol consumption on the gut microbiome has not been investigated thoroughly in South Korea.
In this study, we aimed to determine the prevalence of five CRC-associated bacteria (ETBF, E. faecalis, colB + E. coli, F. nucleatum, and S. gallolyticus) in the biopsied tissues of patients with CRC using quantitative real-time PCR (qPCR) and the relative abundance of operational taxonomic units (OTUs) by 16S rRNA analysis. Furthermore, we determined the associations between the epidemiological characteristics of CRC and the gut microbiome and alcohol consumption.
Results
Comparison of demographics and clinical characteristics of cases and controls
A total of 39 CRC samples and 21 control samples were examined (Table 1). No significant differences between the groups were observed with respect to epidemiological characteristics, apart from BMI and hypertension. BMI was lower in CRC patients (22.9 ± 2.9) than in controls (24.8 ± 2.7), and hypertension was more prevalent in controls (76.2%, n = 16/21) than in CRC patients (46.2%, n = 18/39). The CRC cases consisted of 71.8% colon cancers and 28.2% rectal cancers, with right- and left-sided cancers accounted for 28.2% and 71.8% of the cases, respectively.
Prevalence of CRC-associated bacteria as assessed by qPCR
The prevalence of the five CRC-associated bacteria was examined in the carcinoma tissues (CTs) of CRC patients and in normal tissues (NTs) of controls using qPCR (Table 2). F. nucleatum was most frequently detected in CRC patients and was not detected in the NTs of controls (82.1% and 0%, respectively; P < 0.0001). However, no significant difference in the prevalence of colB + E. coli, ETBF, E. faecalis, or S. gallolyticus was observed between CRC patients and controls.
Prevalence of F. nucleatum by tumor stage and tissue type
We analyzed the association of F. nucleatum positivity in CRC patients with that in controls based on clinicopathological features. Patients with CRC were classified by tumor stage (early stage [I/II] or late stage [III/IV]). F. nucleatum was detected very frequently in both early (92.3%) and late (76.9%) stages; however, this difference was not significant (Fig. 1A). With respect to the tissue type, the prevalence of F. nucleatum was significantly higher in CTs (82.1%) than in adjacent normal tissues (ATs) (39.9%) as well as distal normal tissues (DTs) (33.3%) in CRC patients; no significant difference was observed between ATs and DTs (Fig. 1B). When the cycle threshold (Ct) values obtained from qPCR were compared among tissue types in F. nucleatum-positive cases, they were found to be significantly lower in CTs (Mann–Whitney U test, P < 0.0001). F. nucleatum was markedly enriched in CTs compared to ATs (Supplementary Fig. S1). The fadA gene, which encodes a virulence factor implicated in adhesion and invasion, was statistically more frequent in the CTs of CRC patients (69.2%) than in the NTs of controls (9.5%) (Fig. 1C).

F. nucleatum statuses of CRC patients and controls. (A) Patients with CRC were categorized according to tumor stage (I/II: early stage, III/IV: late stage). F. nucleatum prevalence was significantly higher in CRC patients at early and late stages than in controls (92% and 76.9% vs. 0%, respectively; Fisher’s exact test, P < 0.0001, each). (B) Tissues of CRC patients were collected from carcinoma tissues (CTs), adjacent normal tissues (ATs) and/or normal tissues (NTs) from non-CRC sites. F. nucleatum was significantly higher in CTs (82.1%) than in ATs (38.9%) and NTs (33.3%) of CRC patients (CT vs. AT, CT vs. NT, AT vs. T, and NT vs. T; Fisher’s exact test, P = 0.0002, < 0.0001, 0.0008, and 0.003, respectively) and was significantly higher in CTs than in tissues (Ts) of controls (P < 0.0001). (C) Comparison of the prevalence of the fadA gene revealed significant differences between the CTs of patients and tissues (Ts) of controls (69.2% vs. 9.5%; Fisher’s exact test, P < 0.0001). Four asterisks (****) indicate P < 0.0001, three asterisks (***) indicate P < 0.001, and two asterisks (**) indicate P < 0.001; Fisher’s exact test was performed using GraphPad Prism version 5.0 for Windows.
16S rRNA analysis
Sequencing of the V3 and V4 regions in the 16S rRNA gene produced 171,988 ± 81,412 sequencing reads, on average. After quality filtration, clustering and taxonomy assignment were performed at the genus level by QIIME software19. A total of 698 OTUs were generated and used for further statistical analysis. In a diversity analysis, no significant difference in α- or β-diversity was observed between CRC cases and controls (Supplementary Fig. S2).
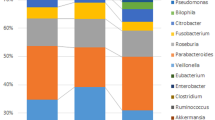
When comparing CRC cases with controls, the proportions of six OTUs, namely, Peptostreptococcus, Collinsella, Prevotella, Parvimonas, Fusobacterium, and Gemella, were significantly different after false discovery rate correction for multiple testing (Fig. 2). Among the epidemiological characteristics with continuous values, Fusobacterium-positive CRC patients were significantly younger than Fusobacterium-negative patients (64.0 ± 10.5 vs. 73.1 ± 9.3; P = 0.034), which was also true for Parvimonas-positive patients (60.4 ± 8.5 vs. 69.0 ± 10.9; P = 0.011; Table 3). With respect to epidemiological characteristics with binomial values, Fusobacterium was associated with alcohol consumption and KRAS mutation (P = 0.088 and P = 0.094), and Parvimonas was associated with tumor location and KRAS mutation, although the difference was only marginally significant (P = 0.070, Table 4). Fusobacterium was observed in 14 of the 15 heavy drinkers (93.3%) but not in 11 of the 17 non/light drinkers (64.7%). The proportion of Fusobacterium OTUs was significantly higher in heavy drinkers than in non/light drinkers (P = 0.003); no corresponding pattern was observed in controls (Fig. 3). With respect to tumor location, the proportion of the Parvimonas OTU was significantly higher in the descending colon and in the rectum (P < 0.001; Supplementary Fig. S3) than in other parts of the colon. Tumor stage and metastasis were not significantly correlated with the presence of specific bacteria. When the Fusobacterium OTU proportions were compared between cases and controls using Ct values, qPCR-positive cases showed higher OTU abundance values than qPCR-negative cases, and the significant positive correlation between these values is shown in Fig. 4.

Relative abundance of six OTUs that were significantly different between cases (CRC patients) and controls were compared using R software.

Differences in the proportion of Fusobacterium between heavy drinkers and non/light drinkers by cases (CRC patients) and controls. The proportion of Fusobacterium OTUs was significantly higher in heavy drinkers than in non/light drinkers (P = 0.003); no corresponding pattern was observed in controls. We used R software to perform the comparison and generate the figure.

Box and whisker plots of the relative abundance of Fusobacterium by 16S rRNA analysis based on F. nucleatum qPCR positivity. Relative abundance (expressed as OTU percentages) was log-transformed for plotting on the Y-axis. Analysis was conducted with R software.
Discussion
CRC is a major health threat in many countries; however, its etiology and underlying mechanisms are still not comprehensively understood. The determination of a cancer’s etiology can result in the development of preventive or therapeutic measures. Several studies have demonstrated enrichment of fecal or tissue samples of CRC patients with specific bacterial pathogens.
ETBF is a well-known pathogen of the gastrointestinal tract that can trigger a carcinogenic multistep process through B. fragilis toxin3,4,5,20. However, we observed no significant difference in the prevalence of ETBF between CRC cases and controls, and its prevalence (35.9%) was significantly lower than that in a previous study (88.5%)20. In a recent study21, it was shown by qPCR that only 36.4% of CRC cases were positive for ETBF, and an equal distribution of B. fragilis was found in tumors, paired normal tissue and diverticula. Thus, the prevalence and distribution of ETBF should be determined by follow-up investigations. It has been shown that pathogenic E. coli can synthesize colibactin, which is genotoxic22. The prevalence of colB + E. coli in CRC patients might be the link between E. coli and CRC. In our study, the prevalence of colB + E. coli in CRC cases was twice as high as that in controls, but the difference was not statistically significant, probably due to an insufficient sample size. S. gallolyticus and E. faecalis were also regarded as CRC-associated bacteria in previous reports23,24; however, recent studies showed that the roles of S. gallolyticus and E. faecalis in CRC carcinogenesis were controversial due to their low prevalence21,25,26. In our study, neither species was frequently detected in cancer patients (Table 2).
In recent decades, F. nucleatum has attracted attention as a potential cause of CRC4,6,27. The role of F. nucleatum in CRC pathogenesis has not been comprehensively understood, but at least four mechanisms have been suggested to describe the same28: (1) cell proliferation through Wnt signaling by an interaction between FadA and E-cadherin, (2) antitumor immune evasion via Fap2 and T cell immunoreceptors having Ig and immunoreceptor tyrosine-based inhibitory motif domains, (3) tumor binding and enrichment of Fap2 and Gal-GalNAc, and (4) chemoresistance by lipopolysaccharide and Toll-like receptor 4.
Fusobacterium species are strictly anaerobic and are difficult to isolate by culture. In this study, the prevalence of F. nucleatum in the tissues of CRC patients as tested by qPCR (82.1%) was notably higher than that obtained using anaerobic culture (7.7%; data not shown). Therefore, non-culture-dependent detection methods such as qPCR or 16S rRNA analysis may be essential for screening Fusobacterium species or for studying its epidemiology in a population during the development of CRC.
The presence of F. nucleatum was significantly correlated with both the early and late stages of CRC (Fig. 1A). This finding suggests that F. nucleatum may be involved in CRC carcinogenesis from an early stage, and one review indicated an association of F. nucleatum with carcinomas throughout the different stages of CRC progression29. Analysis of the abundance of F. nucleatum by tissue type showed that the bacteria seemed to be more prevalent in CTs than in ATs and NTs (Fig. 1B). This finding is consistent with previous studies showing that a high level of Fusobacterium colonization is associated with CRC27,30.
FadA is a well-known oncogenic virulence factor that can induce oncogenic gene expression and promote the growth of CRC cells6. Therefore, FadA is regarded as a major virulence factor of F. nucleatum in CRC, and 69.2% of CT samples were positive for the fadA gene. However, some discrepancies between F. nucleatum presence and fadA gene frequency were observed in our study (Fig. 1C). Thus, it may be necessary to evaluate both F. nucleatum presence and fadA gene frequency to accurately determine the presence and virulence of F. nucleatum31. Interestingly, a recent study suggested that the fadA gene is not specific to F. nucleatum and that it is also present in other Fusobacterium species32. This finding indicates the need for further research regarding the role of other Fusobacterium species in CRC carcinogenesis.
The gut microbiome comprises numerous bacterial species that interact with each other, and it is possible that other bacteria in addition to the five known CRC-associated bacteria that were examined in the present study may affect the carcinogenesis of CRC. Thus, CRC metagenomic analysis is now commonly used to analyze this complex bacterial community. According to our 16S rRNA analysis, six genera, namely, Peptostreptococcus, Collinsella, Prevotella, Parvimonas, Gemella, and Fusobacterium were significantly abundant in CRC patients. Kwong et al. reported that P. anaerobius promoted CRC carcinogenesis in ApcMin/+ mouse models, and a correlation between Peptostreptococcus species and human CRC was reported previously33. The role of Collinsella species in CRC has rarely been investigated; however, Ai et al. reported that Collinsella species were specifically correlated with CRC in the Chinese population using metagenomic analysis34. The association of Prevotella species with CRC was reported in a previous study35, and our results are compatible with this report. Parvimonas and Gemella species were also reported as potential causative agents that may be responsible for CRC carcinogenesis36. However, it is difficult to determine which bacteria are the primary cause of CRC, as the gut microbiome is too complex and can vary by population37.
Based on an integrated analysis of 16S rRNA data and epidemiologic characteristics, we found a marginally significant correlation between Fusobacterium occurrence and a history of alcohol consumption. It has been shown that alcohol consumption is one of the major contributors to CRC carcinogenesis38. The generation of acetaldehyde and other metabolites from alcohol activates cancer-promoting signals39. Moreover, it was suggested that ethanol oxidation by intestinal anaerobes, including Fusobacterium species, under aerobic conditions in the colon and rectum may also play an important role in the pathogenesis of ethanol-related CRC40. To our knowledge, the present study was the first to determine the relationship between alcohol consumption and the presence of Fusobacterium using 16S rRNA analysis. In the integrated statistical analysis, a cutoff of 1% was used to determine the positivity of each OTU41. It is interesting to note that 87.5% (28/32) of qPCR-positive cases and 76.2% (16/21) of qPCR-negative controls showed the same 16S rRNA analysis results when a cutoff of 1% was used. This cutoff seems to be the appropriate threshold to determine abundance in 16S rRNA analysis in CRC tissues, though more investigations are required to confirm the cutoff.
The limitations of this study were the relatively small sample size, which was associated with low statistical significance, and the fact that tubular adenoma could not be analyzed as an independent group due to the small sample size. However, the simultaneous analysis of the presence of specific bacterial pathogens, 16S rRNA analysis, patient demographics and clinical characteristics may be an advantage of this study. The detection of F. nucleatum in stool can be helpful for diagnosing CRC in patients as a biological tumor marker if used concurrently with other tumor markers, and an appropriate testing method should be developed and validated.
In conclusion, F. nucleatum was significantly prevalent in the CTs of patients with CRC and frequently present in both early and late stages. These data support that this bacterium is strongly associated with the development of CRC. The correlation between alcohol consumption and the abundance of Fusobacterium OTUs in cancer tissue discovered using 16S rRNA analysis suggests a possible link between alcohol metabolism and changes in the gut microbiome and subsequent tumorigenesis caused by F. nucleatum.
Methods
Study population
A total of 38 CRC patients and 21 controls who were diagnosed using colonoscopy at a tertiary-care hospital between June 2015 and November 2016 were enrolled. Patients who received preoperative radiation, chemotherapy, and/or antibiotic treatment within 4 weeks were excluded. One patient had two primary adenocarcinoma lesions; thus, 39 CRC samples were analyzed in this study. Among the 21 controls, 12 individuals had tubular adenomas removed during colonoscopy, and 9 individuals showed no specific findings in colonoscopy. Patients with tubular adenomas were analyzed as controls because most of the polyps could be regarded as low risk due to their pathologic findings and small size (< 10 mm), and the resection margin was clear in all samples. G* power 3.1.9.7 analysis was conducted using the chi-square test to justify the appropriate sample size, and the test showed a significance level of 0.05 and a power of 0.81. Patient demographics and clinical characteristics were investigated by reviewing the medical records and interviews. Total alcohol consumption was calculated based on the history of alcohol intake—determined based on factors such as beverage type, amount, and frequency of consumption—and patients were classified as non/light drinkers (0‒1 drink/day) or heavy drinkers (≥ 2 drinks/day). All patients were classified as smokers (persons who are/were smokers) or nonsmokers (persons who never smoked) based on their smoking history.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board at Severance Hospital, Yonsei University, Republic of Korea (approval number: 2014-2768-004). All methods were performed following the relevant guidelines and regulations. Documented informed consent was obtained from each study participant.
Colon tissue collection
Colonoscopy biopsies were obtained from the carcinoma tissues (CTs), adjacent normal tissues (ATs), and/or distant normal tissues (DTs) of CRC patients and from the right (from the cecum to transverse) and left (from descending to the rectum) colons of controls. Tissue samples were categorized as 'normal tissue' (NT) if no macroscopic or pathological evidence of carcinoma was observed. Tissue biopsies were collected in Transystem tubes (Copan, Brescia, Italy) containing anaerobic transport medium. Culture and DNA extraction for qPCR were performed within 30 min after tissue collection, and the remaining tissues were stored in a deep freezer (− 70 ℃) for 16S rRNA analysis. Detailed characteristics of CRC patients and controls are summarized in Supplementary Tables S1A,B.
Detection of CRC-associated bacteria using qPCR
DNA was directly extracted from colon tissue using the QIAamp DNA Micro Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. DNA concentration and purity were recorded using a NanoDrop spectrophotometer (NanoDrop Technologies, Montchanin, U.S.A.). The specific 16S ribosomal RNA (rRNA) genes were amplified by qPCR using the CFX96 Real-Time PCR System (Bio-Rad, Hercules, CA) to detect five CRC-associated bacteria: ETBF42, colB + E. coli43, E. faecalis44, F. nucleatum27, and S. gallolyticus45. The reaction mixture consisted of 10 µL of Ex Taq (TaKaRa, Kusatsu, Japan), 0.4 µL of each specific primer pair (10 µM), and 25 ng/µL DNA template in a total reaction volume of 20 µL. E. faecalis ATCC 29212, F. nucleatum subsp. nucleatum ATCC 25586 and three clinical isolates (ETBF, colB + E. coli and S. gallolyticus) were used for qPCR quality control. Identification of the clinical isolate was performed using MALDI-TOF mass spectrometry and 16S rRNA sequencing. For S. gallolyticus, the rpoB gene was also sequenced. The presence of the fadA gene in F. nucleatum was determined using the conventional PCR method in accordance with a previous study31, and the 211 bp size of the amplicon was confirmed by sequencing.
16S rRNA analysis
The V3 and V4 regions of the 16S rRNA gene (400–450 bp) were PCR amplified using target-specific primers and adapters for Illumina sequencing. Next-generation sequencing was performed on a MiSeq platform (Illumina, San Diego, USA) using the MiSeq Reagent Kit V2 with 500 cycles. Paired-end sequences were merged using PANDASEQ software46, and clustering of operational taxonomic units (OTUs) and taxonomy assignment were performed using QIIME software19. The observed diversity and Chao1 and Shannon indexes for α-diversity were calculated using QIIME. Principal components were calculated and plotted for β-diversity estimation. The relative proportion of each OTU was compared between cases and controls as well as with respect to epidemiological characteristics. OTUs present in > 1% of all OTUs were regarded as positive16. The mock community was analyzed before clinical specimen testing to validate the 16S rRNA analysis results.
Statistical analyses
For 16S rRNA analysis, correction for multiple testing was performed using the false discovery rate (FDR) method. All statistical analyses were performed using GraphPad Prism version 5.0 (https://www.graphpad.com/scientific-software/prism/, GraphPad Software, La Jolla, CA, USA; Tables 1, 2, Fig. 1) and R software (https://www.R-project.org, The R Foundation, version 3.5.3; Tables 3, 4, Figs. 2, 3, 4)47. A value of P < 0.05 was defined as statistically significant, and a P-value between 0.05 and 0.10 was considered marginally significant.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Arnold, M. et al. Global patterns and trends in colorectal cancer incidence and mortality. Gut 66, 683–691 (2017).
Strum, W. B. Colorectal adenomas. N. Engl. J. Med. 374, 1065–1075 (2016).
Tjalsma, H., Boleij, A., Marchesi, J. R. & Dutilh, B. E. A bacterial driver-passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 10, 575–582 (2012).
Sears, C. L. & Garrett, W. S. Microbes, micriobiota, and colon cancer. Cell. Host. Microbe 15, 317–328 (2014).
Wu, S., Morin, P. J., Maouyo, D. & Sears, C. L. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology 124, 392–400 (2003).
Rubinstein, M. R. et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell. Host. Microbe 14, 195–206 (2013).
Balish, E. & Warner, T. Enterococcus faecalis induces inflammatory bowel disease in interleukin-10 knockout mice. Am. J. Pathol. 160, 2253–2257 (2002).
Pasquereau-Kotula, E., Martins, M., Aymeric, L. & Dramsi, S. Significance of Streptococcus gallolyticus subsp. gallolyticus association with colorectal cancer. Front. Microbiol. 9, 614 (2018).
Cuevas-Ramos, G. et al. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 107, 11537–11542 (2010).
Yachida, S. et al. Metagenomic and metabolomic analysis reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 25, 968–976 (2019).
Candela, M. et al. Inflammation and colorectal cancer, when microbiota-host mutualism breaks. World J. Gastroenterol. 20, 908–922 (2014).
Singh, R. K. et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 15, 73 (2017).
Yang, J. et al. Development of a colorectal cancer diagnostic model and dietary risk assessment through gut microbiome analysis. Exp. Mol. Med. 51, 117 (2019).
Engen, P. A., Green, S. J., Voigt, R. M., Forsyth, C. B. & Keshavarzian, A. The gastrointestinal microbiome: Alcohol effects on the composition of intestinal microbiota. Alcohol Res. 37, 223–236 (2015).
McNabb, S. et al. Meta-analysis of 16 studies of the association of alcohol with colorectal cancer. Int. J. Cancer. 146, 861–873 (2020).
Yang, Y. J. et al. Alcohol consumption is associated with the risk of developing colorectal neoplasia: Propensity score matching analysis. Sci. Rep. 9, 8253 (2019).
Ministry of Health and Welfare. Status of alcohol consumption and cigarette smoking. https://www.index.go.kr/potal/main/PotalMain.do (2018). Accessed 12 October 2020.
Lee, S. et al. Cigarette smoking, alcohol consumption, and risk of colorectal cancer in South Korea: A case-control study. Alcohol 76, 15–21 (2019).
Kuczynski, J. et al. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Bioinform. https://doi.org/10.1002/0471250953.bi1007s36 (2012).
Boleij, A. et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin. Infect. Dis. 60, 208–215 (2015).
Bundgaard-Nielsen, C., Baandrup, U. T., Nielsen, L. P. & Sørensen, S. The presence of bacteria varies between colorectal adenocarcinomas, precursor lesions and non-malignant tissue. BMC Cancer 19, 399 (2019).
Buc, E. et al. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS ONE 8, e56964 (2013).
Abdulamir, A. S., Hafidh, R. R. & Bakar, F. A. Molecular detection, quantification, and isolation of Streptococcus gallolyticus bacteria colonizing colorectal tumors: Inflammation-driven potential of carcinogenesis via IL-1, COX-2, and IL-8. Mol. cancer 9, 249 (2010).
Huycke, M. M., Abrams, V. & Moore, D. R. Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis 23, 529–536 (2002).
Andres-Franch, M. et al. Streptococcus gallolyticus infection in colorectal cancer and association with biological and clinical factors. PLoS ONE 12, e0174305 (2017).
de Almeida, C. V., Taddei, A. & Amedei, A. The controversial role of Enterococcus faecalis in colorectal cancer. Therap. Adv. Gastroenterol. 11, 1756284818783606 (2018).
Castellarin, M. et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22, 299–306 (2012).
Garrett, W. S. The gut microbiota and colon cancer. Science 364, 1133–1135 (2019).
Brennan, C. A. & Garrett, W. S. Fusobacterium nucleatum—Symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 17, 156–166 (2019).
Viljoen, K. S., Dakshinamurthy, A., Goldberg, P. & Blackburn, J. M. Quantitative profiling of colorectal cancer associated bacteria reveals associations between Fusobacterium spp., enterotoxigenic Bacteroides fragilis (ETBF) and clinicopathological features of colorectal cancer. PLoS ONE 10, e0119462 (2015).
Huang, S. et al. Rapid detection of nusG and fadA in Fusobacterium nucleatum by loop-mediated isothermal amplification. J. Med. Microbiol. 65, 760–769 (2016).
Manson, M. A. et al. Evolution of invasion in a diverse set of Fusobacterium species. mBio. 5, e01864 (2014).
Kwong, T. N. Y. et al. Association between bacteremia from specific microbes and subsequent diagnosis of colorectal cancer. Gastroenterology 155, 383–90.e8 (2018).
Ai, D. et al. Identifying gut microbiota associated with colorectal cancer using a zero-inflated lognormal model. Front. Microbiol. 10, 826 (2019).
Sobhani, I. et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS ONE 6, e16393 (2011).
Allali, I. et al. Gut microbiome compositional and functional differences between tumor and non-tumor adjacent tissues from cohorts from the US and Spain. Gut Microbes 6, 161–172 (2015).
Dai, Z. et al. Multi-cohort analysis of colorectal cancer metagenome identified altered bacteria across populations and universal bacterial markers. Microbiome 6, 70 (2018).
Bishehsari, F. et al. Alcohol and gut-derived inflammation. Alcohol Res. 38, 163–171 (2017).
Rossi, M., Jahanzaib Anwar, M., Usman, A., Keshavarzian, A. & Bishehsari, F. Colorectal cancer and alcohol consumption-populations to molecules. Cancers (Basel) 10, 38 (2018).
Tsuruya, A. et al. Major anaerobic bacteria responsible for the production of carcinogenic acetaldehyde from ethanol in the colon and rectum. Alcohol Alcohol 51, 395–401 (2016).
Purcell, R. V., Visnovska, M., Biggs, P. J., Schmeier, S. & Frizelle, F. A. Distinct gut microbiome patterns associate with consensus molecular subtypes of colorectal cancer. Sci. Rep. 7, 11590 (2017).
Purcell, R. V. et al. Colonization with enterotoxigenic Bacteroides fragilis is associated with early-stage colorectal Neoplasia. PLoS ONE 12, e0171602 (2017).
Shimpoh, T. et al. Prevalence of pks-positive Escherichia coli in Japanese patients with or without colorectal cancer. Gut. Pathog. 9, 35 (2017).
Ryu, H. et al. Development of quantitative PCR assays targeting the 16S rRNA genes of Enterococcusspp. and their application to the identification of Enterococcusspecies in environmental samples. Applied. Environ. Microbiol. 79, 196–204 (2013).
Kumar, R. et al. Streptococcus gallolyticussubsp. gallolyticuspromotes colorectal tumor development. PLoS Pathog. 13, e1006440 (2017).
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G. & Neufeld, J. D. PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinform. 13, 31 (2012).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2013). Accessed 12 October 2020.
Funding
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning [grant number 2017R1A2B4003785].
Author information
Authors and Affiliations
Contributions
M.K. and S-T.L. designed this study and contributed equally to the manuscript; S.C., S.S.K. and J.H.B. conducted the experiments; Y.A.K. and H.L. analyzed the data; K.-J.R. and J.R.C. gave advice regarding data analysis; T.I.K. and K.L. designed the study and managed the project. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, M., Lee, ST., Choi, S. et al. Fusobacterium nucleatum in biopsied tissues from colorectal cancer patients and alcohol consumption in Korea. Sci Rep 10, 19915 (2020). https://doi.org/10.1038/s41598-020-76467-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-76467-7
This article is cited by
-
Alterations in colorectal cancer virome and its persistence after surgery
Scientific Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
