
Abstract
The genomic diversity of Plasmodium malariae malaria parasites is understudied, partly because infected individuals tend to present with low parasite densities, leading to difficulties in obtaining sufficient parasite DNA for genome analysis. Selective whole genome amplification (SWGA) increases the relative levels of pathogen DNA in a clinical sample, but has not been adapted for P. malariae parasites. Here we design customized SWGA primers which successfully amplify P. malariae DNA extracted directly from unprocessed clinical blood samples obtained from patients with P. malariae-mono-infections from six countries, and further test the efficacy of SWGA on mixed infections with other Plasmodium spp. SWGA enables the successful whole genome sequencing of samples with low parasite density (i.e. one sample with a parasitaemia of 0.0064% resulted in 44% of the genome covered by ≥ 5 reads), leading to an average 14-fold increase in genome coverage when compared to unamplified samples. We identify a total of 868,476 genome-wide SNPs, of which 194,709 are unique across 18 high-quality isolates. After exclusion of the hypervariable subtelomeric regions, a high-quality core subset of 29,899 unique SNPs is defined. Population genetic analysis suggests that P. malariae parasites display clear geographical separation by continent. Further, SWGA successfully amplifies genetic regions of interest such as orthologs of P. falciparum drug resistance-associated loci (Pfdhfr, Pfdhps, Pfcrt, Pfk13 and Pfmdr1), and several non-synonymous SNPs were detected in these genes. In conclusion, we have established a robust SWGA approach that can assist whole genome sequencing of P. malariae, and thereby facilitate the implementation of much-needed large-scale multi-population genomic studies of this neglected malaria parasite. As demonstrated in other Plasmodia, such genetic diversity studies can provide insights into the biology underlying the disease and inform malaria surveillance and control measures.
Similar content being viewed by others

Introduction
Malaria, a mosquito-borne disease caused by Plasmodium parasites, is a continuing threat to global health. There were an estimated 228 million cases and 405,000 deaths in 20181. The majority of mortality events are due to P. falciparum malaria and therefore disease control and elimination efforts have primarily targeted this species. Molecular surveillance has demonstrated that non-falciparum malaria has been underestimated by microscopy diagnosis2,3,4,5, and rapid diagnostic tests (RDT), which are unable to diagnose non-falciparum malaria to the species level6,7. Molecular studies are beginning to demonstrate alarmingly high levels (4–24%) of P. malariae mono- and co-infections across continents2,8,9,10,11,12.
P. malariae infections commonly present with mild or no symptoms, however, severe disease, including anaemia, renal pathologies, and splenomegaly13,14,15,16,17 can occur, complications which can prove fatal16. P. malariae infections present with quartan fevers with parasites that can remain in the host for decades13,18,19 . This persistence is a threat to disease elimination strategies19. Severe P. malariae infections are commonly treated with an Artemisinin Combination Therapy (ACT), similar to P. falciparum infections in the same region18. The high prevalence of mixed infections with P. falciparum and P. vivax means that populations of P. malariae may have been experiencing substantial drug pressure. Several reports have described P. malariae parasites that have not been cleared after treatment with standard antimalarials8,17,20 or have initiated successful infections despite effective chemoprophylaxis21, leading to fears of reduced drug efficacy.
Advances in whole genome sequencing (WGS) technologies now allow for large scale genome diversity studies. Such studies in P. falciparum and P. vivax have provided significant new understanding of the structure of parasite populations, intra- and inter-population genomic diversity, and identified genomic regions under selective pressure, such as drug resistance associated genes22,23,24,25. However, to date only a few complete genomes have been assembled for P. malariae (n = 5; genome size 31.9 Mb)26,27, which have led to insights into genome structure including species-specific gene expansions, causing the characteristically large genome of P. malariae27. One expansion of note is a family encoding transmembrane domain proteins, known as Pm-fam, containing fam-m and fam-l genes, which are hypothesised to be involved in host–pathogen interactions and are unique to P. malariae parasites26,27. To date, investigations of P. malariae genetic diversity have used microsatellite data and demonstrated considerable levels of genetic diversity and differences between and within populations from different countries28,29. However, microsatellite markers reflect only a minority of the genome (< 0.1%), and further investigation using WGS data is needed to explore genetic diversity and population structure across endemic regions.
A major challenge in performing WGS studies using clinical parasite isolates is the difficulty in obtaining sufficient Plasmodium DNA from infected individuals. This is due to low parasite densities and the presence of human DNA from host lymphocytes and other circulating nucleated cells. For P. malariae, genome studies are further complicated by the lack of an in vitro culture method for this parasite species. Until now, WGS data for Plasmodium parasites has been obtained using DNA extracted from venous blood of clinical cases that were pre-filtered to remove human leukocytes, in order to reduce the amount of co-extracted human DNA30. This methodology is efficient when parasite densities are high, however, this is not the case for the majority of P. malariae infections, particularly asymptomatic individuals, where this approach would not yield sufficient parasite DNA for WGS. Recently, a selective whole genome amplification (SWGA) strategy has been used to successfully sequence P. falciparum, P. vivax and P. knowlesi genomes from non-filtered blood and from dried blood spots of clinical samples31,32,33. The SWGA method uses oligonucleotide primers that preferentially bind with high frequency to the pathogen DNA, and rarely bind to the host genome34. The high fidelity Phi29 polymerase, which works through multiple displacement amplification (MDA), is used to amplify large segments (~ 70 kb) of DNA, primed by the SWGA oligonucleotides.
The unique but poorly understood characteristics of the P. malariae parasite, and the threat of unpredictable drug resistance, indicate a need for better understanding of the biological features of this neglected species. Knowledge of the complexity and variability of the P. malariae genome, and comparative studies with the well characterised P. falciparum and P. vivax genomes23,24,25, could provide insights into the biology of this human parasite species. Here, we adapt and validate the SWGA approach for amplification of the P. malariae genome, successfully processing and sequencing 19 clinical samples. After selecting 18 high quality samples, we demonstrate that the resulting WGS data can be used to assess genetic diversity in P. malariae genes orthologous to known drug resistance markers in other species, and to inform population structure. In doing so, we provide proof-of-principle for large-scale WGS studies using blood samples collected from malaria endemic regions to inform malaria control efforts, and provide new molecular information for development of diagnostics, vaccines and drugs.
Results
SWGA enriches P. malariae DNA and increases WGS data coverage
We performed SWGA using a designed primer set (denoted as Pmset1) consisting of five primers (see S1 Table) that preferentially bind the P. malariae genome (average binding sites located once every 2.9 kb within the P. malariae genome, compared to once every 45.1 kb in the human genome). For successful selective amplification it is essential that the binding sites are in close proximity in the parasite genome and spaced further apart in the human genome35. Using two test samples (PM_THA_001 and PM_THA_002), we demonstrate that Pmset1 successfully amplifies the P. malariae genome, allowing for higher quality WGS data in comparison to non-amplification (S1 Fig.). Whilst all four samples were sequenced at a similar depth, we observed that amplified samples have a significant increase in coverage, with a mean 18.6-fold increase in the percentage of the genome covered with ≥ 5 reads when compared to non-amplification (S2 Table). The increase in genome coverage seen with SWGA allows for greater detection of SNPs which can be used for downstream population genetics analysis. As a result, there was an 800- to 13,000-fold increase in the number of callable SNPs detected in samples amplified using Pmset1 (S2 Table).
After validation of Pmset1, 17 additional clinical samples were amplified using Pmset1 and underwent WGS. One sample (PM_THA_009), with a low parasitaemia of 0.0016% presented with low coverage after the first sequencing run (27% genome covered ≥ 5 reads), this sample was re-sequenced, and the second run had better results (44% genome covered ≥ 5 reads) (S2 Table). The two sequencing runs were combined to generate PM_THA_009com (52% genome covered ≥ 5 reads). Excluding the separate runs for PM_THA_009, and one sample with low genome coverage (PM_LBR_003), the remaining samples had an average of 67.4% (± 15%) of the genome covered by ≥ 5 reads (S2 Table). The coverage profile after amplification was uneven, as reported for other Plasmodia32, but generally, across all chromosomes, reaching coverage above the recommended cut off point for SNP calling (five reads or above) (Fig. 1). Coverage of the mitochondria was variable but consistently high in comparison to other chromosomes (mean: 26-fold coverage). The average chromosomal coverage of the two unamplified samples was much lower, with only 0.82% of the genome with a coverage ≥ 5 reads (S2 Fig).

Sequencing coverage by chromosome after SWGA. The distribution of coverage for each position within the genome between 18 high quality samples, among the 14 nuclear chromosomes and the organellar apicoplast (the mitochondrial coverage plot is excluded due to high coverage). The blue horizontal line represents the recommended coverage cut-off point for SNP calling (≥ 5 reads).
SWGA is dependent on the initial parasitaemia of a sample
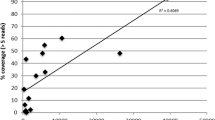
To determine a potential limit of parasitaemia for WGS, a measure of genomic coverage was assessed in nine Thailand samples for which parasitaemia data was available (range of parasitaemia: 0.0004% to 0.2024%). We determine a parasitaemia limit using both microscopy estimates and cycle threshold (CT) values calculated using the qPCR method36. We plotted the CT values of each sample alongside the percentage of the genome that was covered by ≥ 5 reads. We determined that a CT value of 30 will lead to an estimate of 50% of the genome covered by ≥ 5 reads (Fig. 2a). Coverage results are unpredictable below this limit, however, as with PM_THA_001, sequence data may be usable below this limit. When using percentage parasitaemia, we verified that all sequence data from parasite densities higher than 0.01% (400 parasites/ul) led to > 50% of the genome covered by five or more reads; this is a lower limit than previously defined for P. falciparum, and P. knowlesi32,37 (Fig. 2b). For difficult samples with lower parasitaemia it is possible to improve genome coverage by performing independent SWGA reactions and by increasing sequence data, as observed previously for P. vivax31, and also demonstrated here for PM_THA_009, for which merging data lead to > 50% of genome covered with at least 5 reads (S2 Table).

Correlation between parasite density and genome coverage. (a) Nine samples (amplified with SWGA approach) were used to assess the effect of parasite density (as measured by CT values obtained from qPCR) on the proportion of the genome covered by five or more reads. Each point demonstrates a single sample, with a lowess line of best fit. The dashed horizontal line represents a cut off of 50% of the genome covered by 5 or above, and the dashed vertical line indicates the suggested CT cut-off of 30. (b) The same plot is shown using parasitaemia as the measure of parasite density (percentage of RBCs parasitized). Parasitaemias range from 0.0004% to 0.2024%, and the vertical dashed line represents the suggested parasitaemia cut-off of 0.01%.
Determining and excluding hypervariable regions
Many Plasmodium species are known to contain large regions of repetitive sequences within the subtelomeres, which is exaggerated in the case of P. malariae, leading to an enlarged genome in comparison to other species27. We defined the core genome by both excluding regions with > 2.25 SNPs on average per 5 kb window (S3 Fig.) or containing Pm-fam genes (Fig. 3, S4 Fig., core genome coordinates are listed in S3 Table), to leave a total core genome size of 23,960,057 bases (81% of the total PmUG01 reference genome).

Defining and excluding subtelomeric regions, an example with chromosome 1. The average coverage (pink) and number of SNPs (blue) between all 18 samples for chromosome 1. The black dashed horizontal line demonstrates the previously chosen SNP limit per 5 kb window (as defined in S2 Fig.). Black dashed lines are placed at the suggested hypervariable region cut-off points, where clusters of windows demonstrating > 2.25 SNPs are seen. The midpoints of Pm-fam gene families are annotated; Pm-fam-l gene positions are denoted by a black plus, whilst Pm-fam-m gene positions are denoted with a grey circle. (S3 Fig. for all chromosomes, S3 Table for coordinates).
Genetic diversity and population structure
We investigated the multiplicity of infection (MOI) in all samples using the core genome, initially through determining the proportion of SNPs that were heterozygous, alongside running estMOI38 for each sample which calculates the percentage of the genome that supports a MOI of 1 (S2 Table, S5 Fig.). The samples were P. malariae mono-infections, that is, where no other Plasmodium species were detected by qPCR. However, it is possible that > 1 clone of P malariae is present in a sample i.e. polyclonal. Using this sample set, three isolates displayed evidence of polyclonal infections (PM_LBR_002, PM_UGA_007 and PM_THA_012). This observation was confirmed by assessing the minor allele frequency (MAF) distribution of these isolates, where they presented with a higher proportion of SNPs with a non-reference MAF in the range 0.2 to 0.8 (S6 Fig.). For these three isolates only the major allele strain in each isolate was used in further population genetics analysis.
A total of 868,476 genome-wide SNPs were found within the 18 high quality samples (average of 48,249 SNPs per sample), of which 194,709 were unique. However, as with other Plasmodium spp., the subtelomeric region of the P. malariae genome contains large sections of repetitive DNA sequence27. These regions are problematic when interpreting WGS data from short-read technologies such as Illumina as short reads are likely to be aligned to incorrect regions along the reference genome, leading to deceptively high coverage and number of SNPs.
After removing hypervariable regions, we analysed the core genome (see S3 Table for coordinates) of 18 samples (≥ 40% of the genome covered by ≥ 5 reads) and identified 29,899 unique SNPs (mean: 5,810 ± 2,229 SNPs per sample) for downstream population genetic analysis. We found that geographically proximal samples displayed less pairwise diversity than geographically separated samples, with parasites from Thailand appearing more closely related to each other than to parasites obtained from Africa. Nucleotide diversities (π) > 3 × 10−4 nucleotide differences per site are only seen when comparing samples between Thailand and Africa, and π < 2 × 10−4 was only seen when comparing samples within Thailand or Africa (S4 Table).
A maximum-likelihood tree was constructed using core genome SNP data and demonstrates clear regional separation of P. malariae parasites, with samples from the African continent clustering together, and independently from samples originating in Thailand (Fig. 4).

Population structure of P. malariae parasites. A maximum likelihood (ML) tree was generated using 29,899 unique SNPs from 18 amplified and sequenced samples (> 40% genome with at least fivefold coverage). The ML tree is unrooted and was generated using Iqtree39 with Modelfinder software used to select the best model of substitution40. Horizontal branch lengths are drawn to scale demonstrating the number of substitutions per position, and branch bootstrap values (determined using UFBoot241) above 50 are denoted with a grey circle at the start of each branch. The tree was visualised in iTOL42, and branches were coloured by country (country codes: KEN = Kenya, SLE = Sierra Leone, LBR = Liberia, UGA = Uganda, THA = Thailand, SDN = Sudan).
Genetic variation in in orthologs of known P. falciparum genes associated with drug resistance
P. malariae parasites are commonly subject to antimalarial treatments, therefore we investigated the coverage and prevalence of mutations in orthologs of known P. falciparum genes associated with drug resistance (Pfcrt, Pfdhfr, Pfdhps, Pfk13 and Pfmdr1; gene IDs are in S5 Table). SNPs were only found in Pmdhfr (n = 3; 2 non-synonymous), Pmdhps (n = 5; 1 non-synonymous) and Pmmdr1 (n = 4, 2 non-synonymous) (Fig. 5, Table 1). SNPs within Pmdhfr at positions 1,292,026 and 1,292,193 in chromosome 5 appear to be more common globally than other SNPs, whereas SNPs within Pmdhps and Pmmdr1 appear to be more prevalent in Thailand than Africa (Table 2). All of the non-synonymous mutations found within Pmdhfr led to amino acid alterations (F57L, R58S and N114S) at positions that align with known drug-resistance associated positions within the Pfdhfr ortholog (C59R and S108N respectively) upon amino acid allignment (Table 1, S7 Fig.)43. In addition, the mutation at position 527,528 within Pmmdr1 (chromosome 10), which leads to the amino acid substitution L1063F, aligns in close proximity to N1042D in the Pfmdr1 ortholog that is associated with quinine resistance, and increased mefloquine and artemisinin susceptibility (Table 1, S7 Fig.)44.

Average sequencing coverage and SNPs found within P. malariae orthologues of P. falciparum drug resistance associated genes. Average sequencing coverage for 18 samples across 5 genes is represented. The blue horizontal line indicates the coverage of 5 reads and black vertical lines are locations of SNPs (Pmdhfr: 1,292,023, 1,292,026 and 1,292,193; Pmdhps: 1,657,704, 1,657,993, 1,658,014, 1,658,019 and 1,658,049; Pmmdr1: 525,728, 527,072, 527,449 and 527,528).
Mixed infections
P. malariae parasites are commonly found in mixed infections with other Plasmodium spp.911,12. This provides a further obstacle for WGS, as not only is the human genome a potential contaminant, but also the other Plasmodium species present. We used four further unprocessed clinical blood samples from Thailand which were found to be mixed infections after qPCR36 and underwent SWGA to determine whether Pmset1 was specific to only the P. malariae genome. Each sample contained varying mixtures of other parasite species present and our results suggest that SWGA is likely to work if P. malariae is initially the most prevalent parasite in the mixed infection (i.e. has the lowest CT value) (S6 Table). However, when DNA from other species is present at high concentrations, SWGA may not be effective for amplification of P. malariae (S6 Table).
Discussion
P. malariae is a neglected malaria parasite with unique features, such as a longer quartan cycle and the ability to persist in the human host for years or decades13. Genetic investigation of this parasite may allow us to understand how P. malariae is able to cause chronic infections, why there are accounts of P. malariae parasites persisting after treatment with ACT, and why some P. malariae infections lead to severe outcomes whilst others remain asymptomatic. Malaria parasite genomics can provide important biological insights to understand this disease, but the difficulty of obtaining sufficient parasite DNA for WGS has been a challenge for genomic studies of P. malariae. Here we present the first application of SWGA for this species. We have customized the SWGA approach to successfully amplify P. malariae DNA extracted directly from unprocessed blood from clinical samples which were obtained from six different countries. In agreement with others31,32, we have demonstrated that the parasitaemia affects the efficiency of SWGA, and recommend using samples with a percentage parasitaemia > 0.01%, which is a lower threshold than reported for other species31,32,37. The WGS data generated from SWGA-treated samples is of high quality with good overall coverage, leading to an average of 67.4% (± 15%) of the genome covered by ≥ 5 reads between the 18 samples assessed in this study. Using these samples, we were able to identify 868,476 total SNPs (average 48,249 SNPs per sample), filtered to 104,583 total SNPs after exclusion of hypervariable regions (average of 5,810 SNPs per sample). This is lower than SNP prevalence documented in P. knowlesi (115,995 SNPs per sample including hypervariable regions)37, yet higher than SNPs found in P. vivax (14,463 SNPs per sample before filtering for core genome) after SWGA31.
It is important to note that differences in the number of SNPs per sample reported could also be due to differences in the method used for variant calling.
A maximum likelihood tree based on SNP data revealed geographic clusters, with clear separation of African and Asian samples. This geographical clustering is consistent with data for P. falciparum45 and P. vivax parasites24,45,46. Similar geographic clustering was observed in the phylogenetic analysis of SNPs in the circumsporozoite gene from P. malariae isolates from Africa and Asia47. To improve geographical clustering resolution (i.e. by country), the number of samples investigated needs to be increased. Our data suggests that parasites display isolation by distance, therefore country or multi-country regional analysis of P. malariae populations could be used in future studies to identify regions under selection in different populations.
We further demonstrate that SWGA successfully amplifies genes orthologous to those associated with drug resistance in P. falciparum, and identify SNPs in Pmdhfr, Pmdhps and Pmmdr1. The effects of these SNPs are unknown, and to date, there are no characterised molecular markers of drug resistance in P. malariae parasites, even though treatment failures have been reported19,48. Despite this, potential mutations of interest were found, particularly at positions 1,292,023, 1,292,026 and 1,292,193 in chromosome 5 in the Pmdhfr gene. These mutations lead to amino acid substitutions F57L, R58S and N114S respectively, and align almost perfectly with P. falciparum amino acid substitutions C59R and S108N which are associated with reduced susceptibility to sulfadoxine/pyrimethamine49. The nonsynonymous mutation N114S has been previously reported in two P. malariae samples from Thailand and the F57L and R58L mutations have been reported in P. vivax samples from several geographical regions50,51. In addition, one mutation within Pmmdr1 at position 525,728 in chromosome 10 leads to amino acid substitution L1063F, which aligns with close proximity to N1042 in the Pfmdr1 ortholog, associated with reduced susceptibility to quinine and increased susceptibility to mefloquine, halofantrine and artemisinin44. It is important to note that whilst treatment failures are seen with P. malariae infections, it is not clear whether this is due to mutations within the parasite genome leading to reduced drug efficacy, or perhaps a specific phenotype of this species due to the longer parasite life cycle which may reduce drug absorption48; therefore further functional studies are required to determine the effect, if any, of these substitutions.
The subtelomeres, containing the fam-l and fam-m gene families are of great interest when studying P. malariae, as they are unique to this species and are thought to be involved in host-parasite interactions26. Unfortunately, sequence analysis of these regions is notoriously difficult using short-read technologies, therefore longer-read sequencing will be needed to further investigate these regions.
In conclusion, the SWGA approach offers a fast, cost effective way to explore the genome diversity of P. malariae from unprocessed blood of infected individuals. Further studies should consider the analysis of a larger number of samples from a greater geographical range and different clinical outcomes, in addition to studies investigating the subtelomeric regions with long read technologies. Such studies are necessary to characterize the epidemiology and genetic diversity of P. malariae populations, with the potential to provide biological insights for disease control.
Methods
Ethics statement
Isolated from Thailand were collected with ethical approval from the Mahidol Faculty of Tropical Medicine Ethics Committee (Ref: 2015-001.01); PHE-MRL samples are analysed under NHS Ethics approval (#18/LO/0738). In both instances, samples were collected according to relevant guidelines and regulations in both Thailand and the UK, and informed consent was obtained for all subjects over the age of 18 (for subjects under 18 years old, consent was obtained from the appropriate legal guardian.
Sample collection and processing
This project used nine P. malariae DNA samples extracted from unprocessed venous blood from infected individuals in Thailand. Parasite density (parasites/µl) determined by microscopy was available for these isolates. Genomic DNA was extracted from frozen unprocessed blood using the QIAamp DNA Blood Mini Kit (Qiagen) or the QIAsymphony DSP DNA Kit in combination with a QIAsymphony SP instrument (Qiagen), according to manufacturer’s instructions. As microscopy is prone to human errors, all extracted DNA samples were subject to qPCR as outlined by Shokoples et al.36 to ensure that only P. malariae single species infections were used.
A further ten DNA samples were provided by the Public Health England-Malaria Reference Laboratory (PHE-MRL) at the London School of Hygiene and Tropical Medicine (LSHTM). These samples were sourced from individuals who had reported recent travel to only one country with malaria transmission, including: Kenya (n = 2), Liberia (n = 2), Sierra Leone (n = 2), Sudan (n = 1) and Uganda (n = 3) between 2010 and 2017. PHE-MRL samples are commonly sourced from individuals returning to visit relatives in their original native country. For species identification, PHE-MRL samples perform both a nested PCR52 and qPCR36 and are archived according to the species present.
Total DNA concentration for all samples was quantified using a Qubit v2.0 fluorometer (Thermo Fisher Scientific).
Selective whole genome amplification
The swga program (www.github.com/eclarke/swga) was used to identify primers that preferentially amplify the P. malariae genome35, using its reference genome (PmUG01, https://plasmodb.org) as the target (foreground), and the human genome (GRCh37; https://grch37.ensembl.org/) as the background. The swga program ranks primers dependant on the ratio of foreground genome binding to the background genome binding, combined with the evenness of primer binding along the target genome and generates multiple potential primer sets. The five highest-ranked sets consist of combinations of 4 to 6 oligonucleotides each, with overlapping primers. The set that ranked highest (Pmset1) consisted of five primers: TATGTATA*T*T, TTATTC*G*T, TTCGTT*A*T, TTTTTA*C*G, TATTTC*G*T, that were ordered with a phosphorothioate bond (represented by *) modifications to prevent primer degradation by the exonuclease activity of the Phi29 polymerase. To evaluate the efficacy of Pmset1 for SWGA of the P. malariae genome, we tested two samples (PM_THA_001 and PM_THA_002) and sequenced both before and after SWGA.
DNA samples were subject to SWGA following previously published protocols31,32,37. All SWGA reactions were carried out in a UV Cabinet for PCR Operations (UV-B-AR, Grant-Bio) to eliminate potential contamination. Briefly, a maximum of 60 ng of gDNA (minimum of 5 ng) was added to a total 50 µl reaction alongside 5 µl of 10 × Phi29 DNA Polymerase Reaction Buffer (New England BioLabs), 0.5 µl of Purified 100 × BSA (New England BioLabs), 0.5 µl of 250 µM Primer mix, 5 µl 10 mM dNTP (Roche), 30 units Phi29 DNA Polymerase (New England BioLabs) and Nuclease-Free Water (Ambion, The RNA Company) to reach a final reaction volume of 50 µl. The reaction was carried out on a thermocycler with the following step-down program: 5 min at 35 °C, 10 min at 34 °C, 15 min at 33 °C, 20 min at 32 °C, 25 min 31 °C, 16 h at 30 °C and 10 min at 65 °C. After successful validation of Pmset1, the remaining samples underwent SWGA as described above. After SWGA, samples were purified using a 1:1 ratio of AMPure XP beads (Beckman-Coulter), following manufacturer’s instructions.
Library preparation and WGS
SWGA samples and the unamplified negative controls were sequenced on either an Illumina MiSeq or HiSeq4000 platform. For the MiSeq runs, the QIAseq FX DNA Library Kit (QIAGEN) was used for library preparation according to the manufacturer’s protocol, with a 20-min fragmentation step. For the HiSeq4000 runs, samples were prepared using the NEB Next Ultra DNA Library Prep Kit for Illumina (from New England BioLabs Inc., E7370). Library DNA concentration was analysed using a Qubit 2.0 fluorometer. All sequencing reactions were performed using paired (2×) 150 bp reads.
Sequence data analysis
Raw fastq files were trimmed using trimmomatic set to default parameters53, and aligned to the P. malariae UG01 reference genome (PlasmoDB) using bwa-mem software54. SNPs were identified using the samtools software suite (samtools.sourceforge.net)55 and filtered for quality based on previously described methods56. The coverage of each nucleotide position was analysed using sambamba57, which was set to include only SNPs with coverage levels of at least fivefold. Poor quality samples were removed (< 40% of the genome covered by 5 reads) to leave 18 high quality samples. We used estMOI38 to determine MOI for samples, and the major allele was used when heterozygous SNP calls were found.
Determining and excluding subtelomeric regions
To exclude hypervariable subtelomeric regions the P. malariae genome was split into 5 kb segments and the average number of SNPs was calculated. We defined an upper limit for the number of SNPs within each window in order to identify highly polymorphic windows. This SNP limit was used in conjunction with the positions of the Pm-fam gene families to define the subtelomeric regions of each chromosome and exclude these from downstream analysis.
Population genetics
To investigate the population structure of P. malariae parasites, a distance matrix was created which was based on a matrix of pairwise identity calculated from the SNPs present in each sample. Using the distance matrix, a maximum likelihood tree was produced using Iqtree39 with Modelfinder40 to select the best model of substitution and ultrafast bootstrap analysis41. The resulting Newick tree was visualised in iTOL42. The nucleotide diversity (π) metric was used to investigate the genetic variability between samples, and was calculated using the pegas (v0.10) package58, which defines nucleotide diversity as the average number of SNPs per position between two sequences.
Drug resistance orthologs
Orthologs of known genes involved in drug resistance in P. falciparum were analysed. The SNPs were described using the snpEff software59 which annotates the genes affected, the type of mutation, and if non-synonymous, the amino-acid change that has occurred. The coverage of genes of interest was also analysed using the output file from applying sambamba software57. The genes investigated and their respective IDs are summarised in S5 Table.
Data availability
All raw sequence data is listed in the European Nucleotide Archive (study accession number PRJEB33837).
References
WHO. World malaria report 2018 (World Health Organization, Geneva, 2018).
Scopel, K. K. G., Fontes, C. J. F., Nunes, ÁC., Horta, M. F. & Braga, ÉM. High prevalence of Plamodium malariae infections in a Brazilian Amazon endemic area (Apiacás—Mato Grosso State) as detected by polymerase chain reaction. Acta Trop. 90, 61–64 (2004).
Fançony, C. et al. Various pfcrt and pfmdr1 genotypes of plasmodium falciparum cocirculate with P. malariae, P. ovale spp., and P. vivax in Northern Angola. Antimicrob. Agents Chemother. 56, 5271–5277 (2012).
Mayxay, M., Pukrittayakamee, S., Newton, P. N. & White, N. J. Mixed-species malaria infections in humans. Trends Parasitol. 20, 233–240 (2004).
Diversity, G. et al. Plasmodium malariae Prevalence. 23, (2017).
Mathison, B. A. & Pritt, B. S. Update on Malaria Diagnostics and Test Utilization. J. Clin. Microbiol. 55, 2009–2017 (2017).
Daniels, R. F. et al. Evidence of non-Plasmodium falciparum malaria infection in Kédougou, Sénégal. Malar. J. 16, 9 (2017).
Betson, M. et al. Detection of persistent Plasmodium spp. infections in Ugandan children after artemether-lumefantrine treatment. Parasitology 141, 1880–1890 (2014).
Niño, C. H. et al. Plasmodium malariae in the Colombian Amazon region: you don’t diagnose what you don’t suspect. Malar. J. 15, 576 (2016).
Doritchamou, J. Y. A. et al. Submicroscopic placental infection by non-falciparum Plasmodium spp. PLoS Negl. Trop. Dis. 12, e0006279 (2018).
Camargo-Ayala, P. A. et al. High Plasmodium malariae prevalence in an endemic area of the Colombian Amazon region. PLoS ONE 11, e0159968 (2016).
Zhou, M. et al. High prevalence of Plasmodium malariae and Plasmodium ovale in malaria patients along the Thai–Myanmar border, as revealed by acridine orange staining and PCR-based diagnoses. Trop. Med. Int. Health 3, 304–312 (1998).
Collins, W. E. & Jeffery, G. M. Plasmodium malariae: parasite and disease. Clin Miscrobiol Rev 20, 579–592 (2007).
Hendrickse, R. G. & Adeniyi, A. Quartan malarial nephrotic syndrome in children. Kidney Int. 16, 64–74 (1979).
Badiane, A. S. et al. Acute kidney injury associated with Plasmodium malariae infection. Malar. J. 13, 1–5 (2014).
Langford, S. et al. Plasmodium malariae infection associated with a high burden of anemia: a hospital-based surveillance study. PLoS Negl. Trop. Dis. 9, e0004195 (2015).
Betson, M., Clifford, S., Stanton, M., Kabatereine, N. B. & Stothard, J. R. Emergence of nonfalciparum plasmodium infection despite regular artemisinin combination therapy in an 18-month longitudinal study of Ugandan children and their mothers. J. Infect. Dis. 217, 1099–1109 (2018).
Sutherland, C. J. Persistent parasitism: the adaptive biology of malariae and ovale malaria. Trends Parasitol. 32, 808–819 (2016).
Dinko, B., Oguike, M. C., Larbi, J. A., Bousema, T. & Sutherland, C. J. Persistent detection of Plasmodium falciparum, P. malariae, P. ovale curtisi and P. ovale wallikeri after ACT treatment of asymptomatic Ghanaian school-children. Int. J. Parasitol. Drugs Drug Resist. 3, 45–50 (2013).
Maguire, J. D. et al. Chloroquine-resistant Plasmodium malariae in south Sumatra, Indonesia. Lancet 360, 58–60 (2002).
Teo, B.H.-Y. et al. Delayed onset of symptoms and atovaquone-proguanil chemoprophylaxis breakthrough by Plasmodium malariae in the absence of mutation at Codon 268 of pmcytb. PLoS Negl. Trop. Dis. 9, e0004068 (2015).
Miotto, O. et al. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat. Genet. 47, 226–234 (2015).
Manske, M. et al. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature 487, 375–379 (2012).
Pearson, R. D. et al. Genomic analysis of local variation and recent evolution in Plasmodium vivax. Nat. Genet. 48, 959–964 (2016).
Hupalo, D. N. et al. Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nat. Genet. 48, 953–958 (2016).
Rutledge, G. G. et al. Plasmodium malariae and P. ovale genomes provide insights into malaria parasite evolution. Nature 542, 101–104 (2017).
Ansari, H. R. et al. Genome-scale comparison of expanded gene families in Plasmodium ovale wallikeri and Plasmodium ovale curtisi with Plasmodium malariae and with other Plasmodium species. Int. J. Parasitol. 46, 685–696 (2016).
Bruce, M. C., Macheso, A., Galinski, M. R. & Barnwell, J. W. Characterization and application of multiple genetic markers for Plasmodium malariae. Parasitology 134, 637–650 (2006).
Bruce, M. C., Macheso, A., McConnachie, A. & Molyneux, M. E. Comparative population structure of Plasmodium malariae and Plasmodium falciparum under different transmission settings in Malawi. Malar. J. 10, 38 (2011).
Auburn, S. et al. An effective method to purify plasmodium falciparum DNA directly from clinical blood samples for whole genome high-throughput sequencing. PLoS ONE 6, 4–11 (2011).
Cowell, A. N. et al. Selective whole-genome amplification is a robust method that enables scalable whole-genome sequencing of Plasmodium vivax from unprocessed clinical samples. MBio 8, e02257-e2316 (2017).
Oyola, S. O. et al. Whole genome sequencing of Plasmodium falciparum from dried blood spots using selective whole genome amplification. Malar. J. 15, 597 (2016).
Sundararaman, S. A. et al. Genomes of cryptic chimpanzee Plasmodium species reveal key evolutionary events leading. Nat. Commun. 7, 1–14 (2016).
Leichty, A. R. & Brisson, D. Selective whole genome amplification for resequencing target microbial species from complex natural samples. Genetics 198, 473–481 (2014).
Clarke, E. L. et al. SWGA: a primer design toolkit for selective whole genome amplification. Bioinformatics 33, 2071–2077 (2017).
Shokoples, S. E., Ndao, M., Kowalewska-Grochowska, K. & Yanow, S. K. Multiplexed real-time PCR assay for discrimination of plasmodium species with improved sensitivity for mixed infections. J. Clin. Microbiol. 47, 975–980 (2009).
Benavente, E. D. et al. Whole genome sequencing of amplified Plasmodium knowlesi DNA from unprocessed blood reveals genetic exchange events between Malaysian Peninsular and Borneo subpopulations. Sci. Rep. 9, 9873 (2019).
Assefa, S. A. et al. estMOI: estimating multiplicity of infection using parasite deep sequencing data. Bioinformatics 30, 1292–1294 (2014).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016).
Cowman, A. F., Morry, M. J., Biggs, B. A., Cross, G. A. M. & Foote, S. J. Amino acid changes linked to pyrimethamine resistance in the dihydrofolate reductase-thymidylate synthase gene of Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 85, 9109–9113 (1988).
Sidhu, A. B. S., Valderramos, S. G. & Fidock, D. A. pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol. Microbiol. 57, 913–926 (2005).
Volkman, S. K. et al. A genome-wide map of diversity in Plasmodium falciparum. Nat. Genet. 39, 113–119 (2007).
Neafsey, D. E. et al. The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat. Genet. 44, 1046–1050 (2013).
Saralamba, N. et al. Genetic polymorphisms in the circumsporozoite protein of Plasmodium malariae show a geographical bias. Malar. J. 17, 269 (2018).
Rutledge, G. G. et al. Genomic characterization of recrudescent Plasmodium malariae after treatment with Artemether/Lumefantrine. Emerg. Infect. Dis. 23, 1300–1307 (2017).
McCollum, A. M. et al. Antifolate resistance in Plasmodium falciparum: multiple origins and identification of novel DHFR alleles. J. Infect. Dis. 194, 189–197 (2006).
Auliff, A. et al. Amino acid mutations in Plasmodium vivax DHFR and DHPS from several geographical regions and susceptibility to antifolate drugs. Am. J. Trop. Med. Hyg. 75, 617–621 (2006).
Tanomsing, N. et al. Genetic analysis of the dihydrofolate reductase-thymidylate synthase gene from geographically diverse isolates of Plasmodium malariae. Antimicrob. Agents Chemother. 51, 3523–3530 (2007).
Singh, B. et al. A genus- and species-specific nested polymerase chain reaction malaria detection assay for epidemiologic studies. Am. J. Trop. Med. Hyg. 60, 687–692 (1999).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Campino, S. et al. Genomic variation in two gametocyte non-producing Plasmodium falciparum clonal lines. Malar. J. 15, 229 (2016).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015).
Paradis, E. Pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26, 419–420 (2010).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms. SnpEff. Fly (Austin) 6, 80–92 (2012).
Acknowledgements
Sequence analysis was performed on the MRC UK funded eMedlab computing resource. AI and PJGG are supported by an MRC LID PhD studentship. TGC is funded by the Medical Research Council UK (Grant no. MR/M01360X/1, MR/N010469/1, MR/R025576/1, and MR/R020973/1) and BBSRC (Grant no. BB/R013063/1). SC is funded by Medical Research Council UK grants (MR/M01360X/1, MR/R025576/1, and MR/R020973/1). The project was funded by a Research England Bloomsbury SET Project Grant (award reference CCF17-7779). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
S.C. and T.G.C. conceived and directed the project. D.N., S.P., H.P.F. and F.N. organised sample collection and processing. D.N. and J.M. undertook sample processing and DNA extraction for samples from the PHE-MRL. A.I. and S.C. undertook laboratory work including amplification and sequencing. A.I. performed bioinformatic analysis with guidance and training from E.D.B. and under the supervision of S.C. and T.G.C., and together they interpreted the results. Additional advice from M.H., P.J.G.G., C.S., H.P.F. and C.R. was sought during analysis. A.I. wrote the first draft of the manuscript with guidance from E.D.B., T.G.C. and S.C. All authors commented on versions of the manuscript and approved the final manuscript. A.I., E.D.B., S.C. and T.G.C. compiled the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ibrahim, A., Diez Benavente, E., Nolder, D. et al. Selective whole genome amplification of Plasmodium malariae DNA from clinical samples reveals insights into population structure. Sci Rep 10, 10832 (2020). https://doi.org/10.1038/s41598-020-67568-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-67568-4
This article is cited by
-
New reference genomes to distinguish the sympatric malaria parasites, Plasmodium ovale curtisi and Plasmodium ovale wallikeri
Scientific Reports (2024)
-
Rapid profiling of Plasmodium parasites from genome sequences to assist malaria control
Genome Medicine (2023)
-
The primate malaria parasites Plasmodium malariae, Plasmodium brasilianum and Plasmodium ovale spp.: genomic insights into distribution, dispersal and host transitions
Malaria Journal (2022)
-
Zoonotic origin of the human malaria parasite Plasmodium malariae from African apes
Nature Communications (2022)
-
Single-cell sequencing of the small and AT-skewed genome of malaria parasites
Genome Medicine (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
