
Abstract
Gene co-expression network analysis (GCNA) can detect alterations in regulatory activities in case/control comparisons. We propose a framework to detect novel genes and networks for predicting breast cancer recurrence. Thirty-four prognosis candidate genes were selected based on a literature review. Four Gene Expression Omnibus Series (GSE) microarray datasets (n = 920) were used to create gene co-expression networks based on these candidates. We applied the framework to four comparison groups according to node (+/−) and recurrence (+/−). We identified a sub-network containing two candidate genes (LST1 and IGHM) and six novel genes (IGHA1, IGHD, IGHG1, IGHG3, IGLC2, and IGLJ3) related to B cell-specific immunoglobulin. These novel genes were correlated with recurrence under the control of node status and were found to function as tumor suppressors; higher mRNA expression indicated a lower risk of recurrence (hazard ratio, HR = 0.87, p = 0.001). We created an immune index score by performing principle component analysis and divided the genes into low and high groups. This discrete index significantly predicted relapse-free survival (RFS) (high: HR = 0.77, p = 0.019; low: control). Public tool KM Plotter and TCGA-BRCA gene expression data were used to validate. We confirmed these genes are correlated with RFS and distal metastasis-free survival (DMFS) in triple-negative breast cancer (TNBC) and general breast cancer.
Similar content being viewed by others

Introduction
Breast cancer (BC) is perhaps the most well-studied malignancy in the world. Approximately 1.7 million women were diagnosed with the disease in 2012, making it a global priority1. There is an urgent need to identify risk factors associated with recurrence to address this serious problem2.
Microarray analysis has contributed to our understanding of the heterogeneity and complexity of BC3, and it has enabled the identification of gene signatures for diagnosis, molecular characterization, prognosis prediction and treatment recommendation4,5,6. Networks of topological characteristics can potentially serve as predictive biomarkers through network-based classification7,8, and the topology of biological networks has increasingly been used to complement studies of individual genes or gene sets9,10. Several gene network analysis tools based on various methodologies have been developed, including GeneMania11, BisoGenet12, Cytoscape13, and DAVID14.
Gene co-expression network analysis (GCNA) provides insight into novel biological mechanisms and is complementary to standard differential expression (DE) analysis. This method has proven to be an attractive and effective tool for understanding BC10,15,16,17. However, gene co-expression networks (GCN) from single transcriptomic studies are often less informative and generalizable due to cohort bias and a limited sample size, whereas the use of integrated analysis through the combination of multiple transcriptomic studies provides more accurate and comprehensive results18. Therefore, we applied GCNA and integrated microarray analysis, and we considered candidate genes related to BC prognosis to design an analysis procedure and to investigate novel genes and networks related to BC recurrence.
Results
We made comparisons between groups using GCNA with r > 0.9 and edge limit = 1. Comparison networks between cases of recurrence and no recurrence were generated, and UBE2C, MCM6 and IGHG1 were found to be highly differentially co-expressed genes. (Table 1 and Fig. 1) Common genes in the two networks were IGHA1, IGHD, IGHG3, IGLC2, and IGLJ3. Highly co-expressed genes in each of the four comparison groups classified by node (+/−) and recurrence (+/−) are shown in Table 2 and Fig. 1. Regardless of node status, highly co-expressed genes within the network of no recurrence were IGHA1, IGHD, IGHG1, IGHG3, IGLC2, and IGLJ3. Cox proportional hazard ratio regression analysis found these genes to be significantly correlated with the recurrence of BC, regardless of node status (Table 3, Fig. 1). Logistic regression analysis revealed a significant correlation with node status, with an odds ratio (OR) range of 2.3–23 (p < 0.001). These six highly co-expressed genes for LST1 and IGHM belong to a cluster and are related to immune function (Fig. 1).

Gene co-expression networks. Co-expression networks of six subgroups, (a) Recurrence (+), (b) Recurrence (−), (c) Node (+) and Recurrence (+), (d) Node (+) and Recurrence (−), (e) Node (−) and Recurrence (+), (f) Node (−) and Recurrence (−). The width of the gene connection indicates the degree of correlation between genes. Colors of the gene icons and connecting lines denote similar gene expression patterns for genes in the same color, which were analyzed by hierarchical clustering. Connection lines in green denote neighboring genes that do not belong to the same cluster. Size of the gene icon reflects the absolute value of cv of gene expression. The 34 candidate genes are represented by diamonds; co-expressed genes are represented by circles, and significant recurrence associated co-expressed genes are represented by stars. The gene icon frame is shown in red if 0.01 ≤ p < 0.05, and it is shown in yellow if p < 0.01. Up-regulated genes are shown by a dashed line, whereas down-regulated genes are shown by a solid line.
Recently, studies have found that robust levels of tumor-infiltrating lymphocytes (TILs) are associated with increased disease-free survival (DFS) and overall survival (OS) rates in triple-negative breast cancer (TNBC) patients with and without treatment. There have also been efforts to develop a standardized methodology for evaluating TILs19. Their presence at diagnosis is associated with a pathologic response to neoadjuvant therapy as well as increased DFS and OS following adjuvant chemotherapy in certain subtypes20,21.
We speculate that highly co-expressed immune-related genes can be used for the prognosis and treatment of general BC as well as TNBC, and we have established an immune response index to explore the relationships between specific genes and BC recurrence. Because these six genes were highly correlated, they were replaced with a component score by using principle component analysis (PCA). This score had a value range of −1.93~ 1.83 (mean = 0, sd = 1). Cox proportional hazard ratio regression analysis (under the control of node status) was also performed to investigate the impact of the component score on recurrence. It was found that the risk of recurrence was reduced by approximately 13% (HR = 0.87, p = 0.014) for each additional unit of the component score (Table 4). In addition, to divide the samples into high and low immune index groups, we used the 40th percentile (value: −0.5) of the component score as the group’s cut-off point. Using the low group as the control, the immune index effectively distinguished recurrence status, with a hazard ratio (HR) of 0.774 (p = 0.019). (Table 4, Fig. 2).

Cox proportional hazard ratio regression prediction model of a relapse-free survival curve based on the immune index.
Since TNBC is an aggressive subtype and difficult to treat, we wanted to know whether these immune-related genes are correlated by using KM Plotter online cancer survival analysis tool (http://kmplot.com/analysis/)22. The TNBC validation data sets consist of 255 RFS and 43 DMFS cases. All immunoglobulin-related genes were significantly associated with RFS and DMFS with the exception that IGHD was not related to RFS (Table 5).
In order to validate these genes in a larger BC data set, we used TCGA-BRCA gene expression data sets representing 1,215 tumors (Supplementary Fig. 1). DMFS (n = 68) and RFS (TNBC (+): n = 131; TNBC (−): n = 637) samples were selected and stratified by TNBC status (negative/positive) (Supplementary Table 1). All of these BC patients had received initial treatment. Because there are no corresponding gene symbols for our six immunoglobulin-related genes in TCGA-BRCA microarrays, we validated all related immunoglobulin genes: IGLL3, IGLL1, IGSF9B, IGDCC3, IGDCC4, IGBP1, IGSF5, IGSF11, IGSF22, IGSF21, IGHMBP2, IGSF10, IGSF8, IGSF9, IGSF6, IGSF1, IGSF3, IGFN1, and IGJ. Stage, TNM stage, PR status and node status were significantly associated with RFS and DMFS in the univariable Cox proportional-hazards regression models (Supplementary Table 2). The clinical variables were analyzed with the immunoglobulin-related genes by using multivariable Cox proportional-hazards regression. Only node status was found to be significantly related to recurrence with biomarkers in the multivariable Cox proportional-hazards regression models (Table 6). The results showed that IGDCC3, IGJ and IGSF9B were significantly associated with RFS and DMFS; IGSF3 was significantly associated with RFS; and IGSF22, IGSF6 and IGSF9 were significantly associated with DMFS in BCs (Table 6).
Discussion
We showed that without prior information, comparison of co-expression networks between case and control groups can confirm and reveal novel disease mechanisms using a systems approach. In addition, we found that co-expression networks estimated from integrated publicly available genomic studies provide more accurate and robust results than those from a single study18.
We found 34 candidate genes related to BC recurrence from six studies23,24,25,26,27,28 that identified marker genes for BC prognosis. A GCN was established based on these 34 candidates, and eight sub-networks related to immune function were found using GCNA, which consisted of two candidate genes, LST1 and IGHM, and six co-expressed genes, IGHA1, IGHD, IGHG1, IGHG3, IGLC2, and IGLJ3. Studies have found the functional pathways of significant recurrent genes in BC to be associated with the immune response and sensitivity to drugs2 indicating that the immune-related genes identified in this study may also be related to drug sensitivity. Gene function annotation was performed by using DAVID (https://david.ncifcrf.gov/home.jsp). Although a corresponding function for IGLJ3 was not identified, the Gene Ontology (GO) terms of the other five novel genes include “the immunoglobulin complex” and “circulation”. These biological processes are positive regulators of B cell activation, phagocytosis recognition, engulfment, and B cell receptor signaling. B cells infiltrating a patient’s BC and B cells present in the tumor-draining lymph node are clonally and functionally related. Heavy and light chains selected for tumor binding from the BC and tumor-draining lymph node (TDLN) libraries indicate a physiologic relationship that may be important to the tumor-specific immune response29.
Studies have also found that B cell-specific immunoglobulin genes, including both heavy (IGHA1, IGHA2, IGHV1-5, and IGHM) and light (IGLJ3, IGLV6-57, IGKC, IGKV1-5, and IGK@) chain-encoding genes, are up-regulated during the immune response in formalin-fixed paraffin-embedded stroma-rich TNBC tumors30.
Inflammatory cells and their mediators are important constituents of the tumor microenvironment, and they can affect the prognosis of various cancers, including BC31,32. Gene expression of immunoglobulin normally associates with lineage fidelity in B lymphocytes33. Growing evidence indicates that immunoglobulins are produced by mature B lymphocytes, plasma cells and BCs34.
The six immunoglobulin-related genes that we examined have not previously been identified as having roles in BC, but widespread evidence has shown that immunoglobulin-related genes are effective diagnostic and prognostic biomarkers for BC31,34,35,36,37,38,39. Recently, many immunoglobulin superfamily (IgSF) genes were found to serve as effective prognostic biomarkers for BC36. In addition, immunoglobulin free light chains (FLCs) were identified as ligands in the pro-tumorigenic activation of mast cells. FLCs may be helpful in the diagnosis and prognosis of BC31.
The stromal immunoglobulin kappa chain (IGKC) has been validated as an immunological biomarker of prognosis and response to therapy in BC38,39. Immunoglobulin gamma heavy-chain marker and kappa light-chain marker allotypes are associated with humoral immunity to HER-2, a finding with potential implications for BC immunotherapy40. All of the evidence suggests that our six immunoglobulin-related genes have potential for use in prognosis prediction and targeted therapy.
TNBC is an aggressive disease without established targeted treatment options for patients. It represents a major challenge, and there is an urgent need for new therapeutic targets41,42. We sought to determine whether the immunoglobulin-related genes are associated with RFS and distal metastasis-free survival (DMFS) in TNBC samples. After validate in KM Plotter online cancer survival analysis tool22, we found that all immunoglobulin-related genes were significantly associated with RFS and DMFS with the exception that IGHD was not related to RFS (Table 5). Further validation was conducted using TCGA-BRCA gene expression data sets (Supplementary Table 1). Though there are no corresponding gene symbols for our six immunoglobulin-related genes, we still found that related immunoglobulin genes are associated with the RFS and DMFS in BC or TNBC. In summary, the results indicate that immunoglobulin-related genes play significant roles in RFS and DMFS both in general BC and TNBC. This has implications for targeted therapy for TNBC.
TILs are reported to be positively associated with improved survival21, particularly in TNBC, but they can also aid in the prediction of responses to neoadjuvant and adjuvant chemotherapy treatments. There have been increasing efforts to target the immune system as part of BC therapy, primarily in patients with TNBC19. Accordingly, we established an immune index score system with six immune-related genes. This score is a protective indicator for the recurrence of BC: as the score increases, the risk of recurrence decreases. This index may be used as a TIL-related indicator and a TNBC treatment marker in the future.
To the best of our knowledge, this is the first study showing that the immunoglobulin-related genes IGHA1, IGHD, IGHG1, IGHG3, IGLC2, and IGLJ3 serve as suppressor genes in the recurrence of general BC and TNBC patients. The validation results from the public tool KM Plotter and TGCA-BRCA confirmed their significant roles in DMFS and RFS of general BC or TNBC. Our results also show that the analysis workflow of GCNA can effectively and efficiently detect novel prognostic biomarkers of BC. These six immunoglobulin genes are warrant further study of their roles in TNBC and we are working on verifying their function in cell lines.
Materials and Methods
Microarray datasets
The microarray data in this study were collected from the BC datasets generated by Chou et al.23, including GSE 2034 (n = 286)24, GSE 2990 (n = 189)25, GSE 4922 (n = 249)26, and GSE 7390 (n = 198)27 of the NCBI GEO database. These datasets comprise 922 cases and 13,452 genes. In total, 354 cases showed BC recurrence (38%) and 566 cases showed no recurrence (61%); there were two missing cases, 111 cases with node positivity (12%), and 796 negative cases (86%). All of the BC patients had received surgical therapy. The four datasets revealed no difference in determining the distribution of recurrence. The average follow-up time of the four datasets was 6–9 years, and the length of the tracking time differed according to the analysis of variance (ANOVA) results (Table 7). The Desmedt27 dataset was selected as the reference standard. The other three gene expression datasets24,25,26 were log-transformed so that they had similar distributions in terms of central tendency, dispersion tendency, skewness and kurtosis. The detailed conversion formulae and microarray correction method are described by Chou et al.23.
The four datasets in this study were generated by using an Affymetrix GeneChip Human Genome U133 Array (HG-U133A), which included 22,283 probes, of which 21,187 detected human functional genes (noted by the NCBI), and contained 13,452 genes (1–14 probes for each). For genes with multiple probes, the median value was used to represent the performance of the gene.
The 34 candidate genes
We chose the significant gene signatures from the six studies23,24,25,26,27,28 that reported the microarray datasets. In our previous study, we selected the top 100 significant genes (Supplementary Table 3) related to BC recurrence23, but in this study, we screened out the identical genes, of which there were 34 in total. (Supplementary Table 4) These 34 candidate genes influence the recurrence of BC; thus, these candidates were used to plot the GCN.
Co-expression network analysis
This study used R software version 3.2.2 (http://www.r-project.org)43. The co-expression network was developed using the visNetwork kit. The correlation coefficient, hierarchical clustering, coefficient of variation, and Cox proportional hazard ratio regression were computed using the cor, hclust, co.var, and coxph functions.
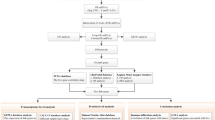
Due to variations in genotype and recurrence, for the analysis, the data were divided into two datasets based on recurrence. The 34 candidate genes were set as the included genes, and Spearman correlation coefficient analysis was performed on the other 13,418 genes with these 34 candidate genes. We identified highly correlated genes with a correlation coefficient over 0.9. The analysis flow chart is shown in Fig. 3.

Study flowchart.
The co-expression networks were established as follows: (1) Width of the gene connection: indicates the degree of correlation between genes by the method of Spearman correlation; a thicker connection line indicates a greater degree of correlation, and the weight of the correlation between two genes is shown by clicking the connection line. (2) The number of gene connections: to simplify the co-expression networks, the number of edges for each gene was limited to one, and the selection started from the gene with the highest correlation coefficient. (3) Colors of the gene icons and connecting lines: These denote similar gene expression patterns for genes in the same color. The R function “hclust” was used with the method set to “spearman” and “complete”; the tangent point was set to be the same cluster when the kinship distance was 1/1.5 of all lengths, and the cluster results are illustrated using the same color connecting line as in the gene networks. Connection lines in green denote neighboring genes that do not belong to the same cluster. (4) Size of the gene icon: the coefficient of variation (cv) of each gene was calculated, and the absolute value of the cv was used to represent the size of the dot; as the cv increases in value, the variation in mRNA gene expression also increases. (5) Shape of the gene icons: the 34 candidate genes are represented by diamonds; co-expressed genes are represented by circles, and significant recurrence associated co-expressed genes are represented by stars (univariable Cox proportional hazard ratio regression test, p < 0.05). (6) Frame color of gene icons: correlation between the mRNA gene expression of each gene and recurrence was analyzed by univariable Cox proportional hazard ratio regression; if 0.01 ≤ p < 0.05, then the icon frame is shown in red, and if p < 0.01, then the icon frame is shown in yellow. (7) Style of the gene icon frame: up-regulated genes are shown with dashed lines, whereas down-regulated genes are shown by solid lines.
Conclusions
We identified and validated six genes related to immune function as potential biomarkers of recurrence for both general breast cancer and TNBC. Our results suggest that GCNA can effectively and efficiently detect novel prognostic biomarkers of breast cancer.
Data Availability
All the GEO dataset are available in NCBI GEO data base (https://www.ncbi.nlm.nih.gov/geo/).
References
Woolston, C. Breast cancer. Nature 527, S101, https://doi.org/10.1038/527S101a (2015).
Chen, X. et al. Identification of breast cancer recurrence risk factors based on functional pathways in tumor and normal tissues. Oncotarget 8, 20679–20694, https://doi.org/10.18632/oncotarget.11557 (2017).
Colombo, P. E., Milanezi, F., Weigelt, B. & Reis-Filho, J. S. Microarrays in the 2010s: The contribution of microarray-based gene expression profiling to breast cancer classification, prognostication and prediction. Breast Cancer Res 13, 212, https://doi.org/10.1186/bcr2890 (2011).
Nannini, M. et al. Gene expression profiling in colorectal cancer using microarray technologies: results and perspectives. Cancer treatment reviews 35, 201–209, https://doi.org/10.1016/j.ctrv.2008.10.006 (2009).
Denkert, C., Pfitzner, B. M., Heppner, B. I. & Dietel, M. Molecular pathology for breast cancer: Importance of the gene expression profile. Der Pathologe 36, 145–153, https://doi.org/10.1007/s00292-015-0009-z (2015).
Zhang, X., Cha, I. H. & Kim, K. Y. Use of a combined gene expression profile in implementing a drug sensitivity predictive model for breast cancer. Cancer research and treatment: official journal of Korean Cancer Association 49, 116–128, https://doi.org/10.4143/crt.2016.085 (2017).
Tuck, D. P., Kluger, H. M. & Kluger, Y. Characterizing disease states from topological properties of transcriptional regulatory networks. BMC bioinformatics 7, 236, https://doi.org/10.1186/1471-2105-7-236 (2006).
Chuang, H. Y., Lee, E., Liu, Y. T., Lee, D. & Ideker, T. Network-based classification of breast cancer metastasis. Molecular systems biology 3, 140, https://doi.org/10.1038/msb4100180 (2007).
Ramadan, E., Alinsaif, S. & Hassan, M. R. Network topology measures for identifying disease-gene association in breast cancer. BMC bioinformatics 17(Suppl 7), 274, https://doi.org/10.1186/s12859-016-1095-5 (2016).
Hallett, R. M. et al. Identification and evaluation of network modules for the prognosis of basal-like breast cancer. Oncotarget 6, 17713–17724, https://doi.org/10.18632/oncotarget.4034 (2015).
Montojo, J., Zuberi, K., Rodriguez, H., Bader, G. D. & Morris, Q. GeneMANIA: Fast gene network construction and function prediction for Cytoscape. F1000Research 3, 153, https://doi.org/10.12688/f1000research.4572.1 (2014).
Martin, A. et al. BisoGenet: A new tool for gene network building, visualization and analysis. BMC bioinformatics 11, 91, https://doi.org/10.1186/1471-2105-11-91 (2010).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome research 13, 2498–2504, https://doi.org/10.1101/gr.1239303 (2003).
Jiao, X. et al. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 28, 1805–1806, https://doi.org/10.1093/bioinformatics/bts251 (2012).
Mao, Y. et al. Network pharmacology-based and clinically relevant prediction of the active ingredients and potential targets of Chinese herbs in metastatic breast cancer patients. Oncotarget 8, 27007–27021, https://doi.org/10.18632/oncotarget.15351 (2017).
Zhang, X. H. et al. A network meta-analysis for toxicity of eight chemotherapy regimens in the treatment of metastatic/advanced breast cancer. Oncotarget 7, 84533–84543, https://doi.org/10.18632/oncotarget.13023 (2016).
Tilli, T. M., Carels, N., Tuszynski, J. A. & Pasdar, M. Validation of a network-based strategy for the optimization of combinatorial target selection in breast cancer therapy: siRNA knockdown of network targets in MDA-MB-231 cells as an in vitro model for inhibition of tumor development. Oncotarget 7, 63189–63203, https://doi.org/10.18632/oncotarget.11055 (2016).
Zhu, L. et al. MetaDCN: Meta-analysis framework for differential co-expression network detection with an application in breast cancer. Bioinformatics 33, 1121–1129, https://doi.org/10.1093/bioinformatics/btw788 (2017).
Garcia-Teijido, P., Cabal, M. L., Fernandez, I. P. & Perez, Y. F. Tumor-infiltrating lymphocytes in triple negative breast cancer: The future of immune targeting. Clinical Medicine Insights. Oncology 10, 31–39, https://doi.org/10.4137/CMO.S34540 (2016).
Demaria, S. et al. Development of tumor-infiltrating lymphocytes in breast cancer after neoadjuvant paclitaxel chemotherapy. Clin Cancer Res 7, 3025–3030 (2001).
Denkert, C. et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 28, 105–113, https://doi.org/10.1200/JCO.2009.23.7370 (2010).
Szasz, A. M. et al. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget 7, 49322–49333, https://doi.org/10.18632/oncotarget.10337 (2016).
Chou, H. L. et al. Gene expression profiling of breast cancer survivability by pooled cDNA microarray analysis using logistic regression, artificial neural networks and decision trees. BMC bioinformatics 14, 100, https://doi.org/10.1186/1471-2105-14-100 (2013).
Wang, Y. et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 365, 671–679, https://doi.org/10.1016/S0140-6736(05)17947-1 (2005).
Sotiriou, C. et al. Gene expression profiling in breast cancer: Understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst 98, 262–272, https://doi.org/10.1093/jnci/djj052 (2006).
Ivshina, A. V. et al. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res 66, 10292–10301, https://doi.org/10.1158/0008-5472.CAN-05-4414 (2006).
Desmedt, C. et al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res 13, 3207–3214, https://doi.org/10.1158/1078-0432.CCR-06-2765 (2007).
Xu, L., Tan, A. C., Winslow, R. L. & Geman, D. Merging microarray data from separate breast cancer studies provides a robust prognostic test. BMC bioinformatics 9, 125, https://doi.org/10.1186/1471-2105-9-125 (2008).
Novinger, L. J., Ashikaga, T. & Krag, D. N. Identification of tumor-binding scFv derived from clonally related B cells in tumor and lymph node of a patient with breast cancer. Cancer Immunol Immunother 64, 29–39, https://doi.org/10.1007/s00262-014-1612-1 (2015).
Winslow, S., Leandersson, K., Edsjo, A. & Larsson, C. Prognostic stromal gene signatures in breast cancer. Breast Cancer Res 17, 23, https://doi.org/10.1186/s13058-015-0530-2 (2015).
Groot Kormelink, T. et al. Immunoglobulin free light chains are biomarkers of poor prognosis in basal-like breast cancer and are potential targets in tumor-associated inflammation. Oncotarget 5, 3159–3167, https://doi.org/10.18632/oncotarget.1868 (2014).
Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature 454, 436–444, https://doi.org/10.1038/nature07205 (2008).
Babbage, G., Ottensmeier, C. H., Blaydes, J., Stevenson, F. K. & Sahota, S. S. Immunoglobulin heavy chain locus events and expression of activation-induced cytidine deaminase in epithelial breast cancer cell lines. Cancer Res 66, 3996–4000, https://doi.org/10.1158/0008-5472.CAN-05-3704 (2006).
Yang, B. et al. Correlation of immunoglobulin G expression and histological subtype and stage in breast cancer. Plos One 8, e58706, https://doi.org/10.1371/journal.pone.0058706 (2013).
Tong, W. L., Tu, Y. N., Samy, M. D., Sexton, W. J. & Blanck, G. Identification of immunoglobulin V(D)J recombinations in solid tumor specimen exome files: Evidence for high level B-cell infiltrates in breast cancer. Hum Vaccin Immunother 13, 501–506, https://doi.org/10.1080/21645515.2016.1246095 (2017).
Li, Y. et al. Immunoglobulin superfamily genes are novel prognostic biomarkers for breast cancer. Oncotarget 8, 2444–2456, https://doi.org/10.18632/oncotarget.13683 (2017).
Pandey, J. P., Namboodiri, A. M., Mohan, S., Nietert, P. J. & Peterson, L. Genetic markers of immunoglobulin G and immunity to cytomegalovirus in patients with breast cancer. Cell Immunol 312, 67–70, https://doi.org/10.1016/j.cellimm.2016.11.003 (2017).
Whiteside, T. L. & Ferrone, S. For breast cancer prognosis, immunoglobulin kappa chain surfaces to the top. Clin Cancer Res 18, 2417–2419, https://doi.org/10.1158/1078-0432.CCR-12-0566 (2012).
Marchan, R. Lung and breast cancer research: immunoglobulin Kappa C hits the headlines. EXCLI J 11, 237–239 (2012).
Pandey, J. P., Namboodiri, A. M., Kurtenkov, O. & Nietert, P. J. Genetic regulation of antibody responses to human epidermal growth factor receptor 2 in breast cancer. Hum Immunol 71, 1124–1127, https://doi.org/10.1016/j.humimm.2010.08.009 (2010).
Millis, S. Z. et al. Predictive Biomarker Profiling of >6000 Breast Cancer Patients Shows Heterogeneity in TNBC, With Treatment Implications. Clin Breast Cancer 15, 473–481 e473, https://doi.org/10.1016/j.clbc.2015.04.008 (2015).
De Laurentiis, M. et al. Treatment of triple negative breast cancer (TNBC): current options and future perspectives. Cancer treatment reviews 36(Suppl 3), S80–86, https://doi.org/10.1016/S0305-7372(10)70025-6 (2010).
R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2016).
Acknowledgements
We would like to express our sincere appreciation for the reviewers who provided us with helpful suggestions for revising the manuscript. Grant support from Songshan Branch of Tri-Service General Hospital, National Defense Medical Center (201704).
Author information
Authors and Affiliations
Contributions
Y.T. Chang analyzed the data and designed the study. J.C., H.M., Y.J., Y.T. Chiang and C.W. gave information and clinical suggestions. C.M. and H.M. supervised the study. C.T. and C.E. helped analyzed the data in different approaches. C.Y. provided the integrated microarray data.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hsu, HM., Chu, CM., Chang, YJ. et al. Six novel immunoglobulin genes as biomarkers for better prognosis in triple-negative breast cancer by gene co-expression network analysis. Sci Rep 9, 4484 (2019). https://doi.org/10.1038/s41598-019-40826-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-40826-w
This article is cited by
-
Establishment and validation of an aging-related risk signature associated with prognosis and tumor immune microenvironment in breast cancer
European Journal of Medical Research (2022)
-
Identification of early biomarkers in saliva in genetically engineered mouse model C(3)1-TAg of breast cancer
Scientific Reports (2022)
-
A prospective study revealing the role of an immune-related eRNA, WAKMAR2, in breast cancer
Scientific Reports (2021)
-
Genetic co-expression networks contribute to creating predictive model and exploring novel biomarkers for the prognosis of breast cancer
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
