
Abstract
Macleaya cordata produces a variety of benzylisoquinoline alkaloids (BIAs), such as sanguinarine, protopine, and berberine, which are potential anticancer drugs and natural growth promoters. The genes encoding the berberine bridge enzyme (BBE) were isolated from M. cordata and Papaver somniferum, and then the two genes were overexpressed in M. cordata. Through liquid chromatography with triple-quadrupole mass spectrometry analysis, it was determined that McBBE-OX caused higher levels of (S)-norcoclaurine, (S)-coclaurine, (S)-N-cis-methylcoclaurine, (S)-reticuline, (S)-tetrahydrocolumbamine, (S)-tetrahydroberberine, (S)-cheilanthifoline, and (S)-scoulerine than PsBBE-OX, empty vector or control treatments. qRT-PCR analysis demonstrated that the introduced genes in the transgenic lines were all highly expressed. However, the levels of sanguinarine (SAN) and chelerythrine (CHE) in all the transgenic lines were slightly lower than those in the wild-type lines, possibly because the overexpression of McBBE causes feedback-inhibition. This is the first report on the overexpression of potential key genes in M. cordata, and the findings are important for the design of metabolic engineering strategies that target BIAs biosynthesis.
Similar content being viewed by others

Introduction
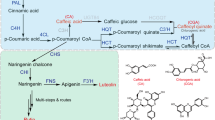
Benzylisoquinoline alkaloids (BIAs) are a large and structurally diverse group of natural products, most of which occur in the families Papaveraceae, Ranunculaceae, Lauraceae, Rutaceae and Menispermaceae1. Some BIAs have important clinical medicinal benefits; for example, morphine is a narcotic drug, berberine (BBR) has been used to treat bacterial diarrhoea2, and protopine (PRO) and sanguinarine (SAN) were potential anticancer drugs and have real potential as effective antischistosomal drugs3,4,5. More importantly, SAN has been widely used in livestock as an alternative to antibiotic growth promoters6,7. Recently, the market demand for BIAs has increased steadily every year. However, the use of BIAs is significantly restricted because of its low levels in plants. Fortunately, the synthetic pathways and enzymes of BIAs in the Papaveraceae family of plants have been elucidated in previous studies8,9. BIA biosynthesis begins with tyrosine; then, two tyrosine derivatives, dopamine and 4-hydroxyphenylacetaldehyde, are condensed to form (S)-norcoclaurine. Then, (S)-norcoclaurine is converted into (S)-reticuline through several steps. If we consider the BIA synthetic pathway to be a network, (S)-reticuline is the key branch-point intermediate of BIAs. This compound is located at a crucial crossroads site, and the N-methyl group of (S)-reticuline is converted into the methylene bridge moiety of (S)-scoulerine by the berberine bridge enzyme (BBE) (Fig. 1). In fact, (S)-scoulerine is the key intermediate in many BIA-producing plants, and BBE is the most critical rate-limiting enzyme in the entire BIA synthesis pathway10.

The metabolic pathway of sanguinarine and chelerythrine. Berberine bridge enzyme (BBE) was studied in this work. 6OMT, norcoclaurine 6-O-methyltransferase; CNMT, coclaurine-N-methyltransferase; NMCH, N-methylcoclaurine hydroxylase; 4OMT, 4′-O-methyltransferase; BBE, berberine bridge enzyme; CFS, cheilanthifoline synthase; SPS, stylopine synthase; TNMT, tetrahydroprotoberberine cis-N-methyltransferase; MSH, (S)-cis-N-methylstylopine 14-hydroxylase, P6H, protopine 6-hydroxylase; DBOX, dihydrobenzophenanthridine oxidase; TDC, (S)-canadine synthase; SMT, (S)-scoulerine 9-O- methyltransferase.
In recent years, many studies have engineered microbes and plants to produce BIAs11,12,13,14,15,16,17. Microbial-based production of BIAs requires the introduction of some heterologous plant genes and numerous genetic modifications to increase productivity. However, the advances in microbial-based methods have significantly shortened production time and although microbial methods are not affected by the environment, the production of BIAs still needs an external supply of precursors15. In a recent study, researchers combined different enzymes from various species and, through pathway and strain optimization, accomplished full opiate biosynthesis in yeast16. This work solved the problem of BIA production needing precursors, but the synthesis step was too long, and the final product concentration was hardly sufficient for industrial production. For this reason, many studies have used transgenic methods to increase production, especially overexpression of potential key enzymes. The most famous examples are the strategies used to increase artemisinin yield in Artemisia annua. Although semi-synthesis of artemisinin was successfully achieved in yeast18, many groups still enhance the yield of artemisinin by transgenic methods19. In addition, overexpression of synthesis genes has also been used to increase levels of ginsenoside, phytosterols, scopolamine and terpenoid indole alkaloids20,21,22. In addition, in a previous study, the content of BIAs in California poppy was increased through overexpression of the P. somniferum BBE gene (PsBBE), and this experiment indicated that BBE is a potential key enzyme to enhance the content of BIAs12.
Macleaya cordata is a perennial herb that belongs to the Papaveraceae family and is used for the commercial production of BIAs, including SAN, PRO, BBR; the herb contains many trace BIAs that have beneficial medicinal properties, such as (S)-reticuline and (S)-scoulerine. Recently, the whole genome of M. cordata has been de novo sequenced and validated for SAN biosynthesis9. In addition, a transformation and regeneration system to produce transgenic M. cordata has been constructed23. These genomics data and transgenic methods provide the opportunity for enhanced production of BIAs through plant metabolic engineering. In this study, we overexpressed BBE genes isolated from M. cordata and P. somniferum (McBBE and PsBBE) in M. cordata. Then, we investigated whether the overexpression of McBBE or PsBBE in M. cordata could affect the biosynthesis of BIAs. Finally, we investigated the metabolite profiles through ESI/QQQ MS analysis and detected the expression levels of 11 genes involved in the synthesis of SAN and CHE. To our knowledge, this is the first study using overexpression of BBE in transgenic M. cordata with increased alkaloid production.
Results
Establishment of Transgenic Plants with the McBBE/PsBBE Gene
We subjected leaf and stem explants to vacuum infiltration with Agrobacterium tumefaciens harbouring McBBE- or PsBBE- and GUS (β-glucuronidase)-overexpression vectors, respectively for 10 min, and then the explants were placed into co-cultivation medium (100 μM acetosyringone) for 3 days. Then, the explants were transferred to selection medium (75 mg/l kanamycin and 400 mg/l Timentin)23. To determine whether the transgenic lines were established successfully, GUS (β-glucuronidase) histochemical analysis was performed. The GUS histochemical results showed the presence of blue colour in the McBBE-OX and PsBBE-OX plants. In contrast, the wild-type lines did not show GUS activity (Fig. 2A–D). Polymerase chain reaction was performed using GUS, nptII (kanamycin resistance gene) and PsBBE primers for the transgenic lines, one wild type plant (negative control) and pCMBIA2301, pCMBIA2301 + McBBE and pCMBIA2301 + PsBBE plasmid vectors (positive control). The results showed that the reporter gene GUS and the nptII gene were detected in all three vectors and in all the transgenic lines (Fig. 2E–G). Only the 2301 + PsBBE vector and the PsBBE-OX line displayed PsBBE-specific fragments. No PCR amplification product was detected for the wild-type plants.

(A–D) GUS assay of leaf and stem section of transgenic and non-transgenic M. cordata plants. (A,B) Staining of GUS activity in overexpression McBBE-OX plants. (C) Staining of GUS activity in transgenic PsBBE-OX plant. (D) Staining of GUS activity in non-transgenic plant. (E–G) Amplify of GUS, nptII, PsBBE genes in transgenic and non-transgenic plants, marker 2 kb plus ladder and vector1 (pCMBIA-2301), vector2 (pCMBIA-2301 + PsBBE), vector3 (pCMBIA-2301 + McBBE) were used to positive control, WT negative control. (E) Amplify GUS gene (750 bp). (F) Amplify nptII gene (364 bp). (G) Amplify PsBBE gene (750 bp). Vec1-Vec3 means vector1 to vector3. WT means wild type.
Modulation of BIA biosynthesis by overexpression of McBBE and PsBBE in Macleaya cordata and transcriptional activation of several genes encoding BIA biosynthetic enzymes
We analysed the BIA content of 3-month-old plants using liquid chromatography with triple-quadrupole mass spectrometry (LC-QQQ MS) to detect whether BIA content was increased in the transgenic plants. Compared with those in the WT plants, in the McBBE-OX plants, the levels of (S)-norcoclaurine, (S)-N-cis-methylcoclaurine, (S)-reticuline, (S)-scoulerine, (S)-cheilanthifoline and (S)-tetrahydrocolumbamine were increased by 1.75-, 5.8-, 3-, 16-, 6- and 74-fold, respectively (P < 0.05). However, stylopine, PRO, dihydrosanguinarine (DHSAN), cis-N-methyltetrahydroberberine, allocryptopine (ALL), and chelerythrine (CHE) showed decreased concentrations upon the overexpression of McBBE (not significant, P > 0.05) (Fig. 3). However, there were no significant differences (P > 0.05) in the metabolic profiles among PsBBE-OX, EV and WT plants.

The analysis of alkaloid levels (from (S)-norcoclaurine to SAN and CHE) in transgenic (pcambia2301 + McBBE, pcambia2301 + PsBBE, pcmbia2301) and WT M. cordata plants were negative control. Data represent means ± SD (n = 3). Asterisks denote the significant changes (* means P < 0.05, ** means P < 0.01, *** means P < 0.005, **** means P < 0.001). McMSH and McSPS genes in this pathway still uncharacterized, we mark it in blue. EV means empty vector, WT means wild type.
Compared with that in the WT lines, the expression of the Mc4OMT, Mc6OMT, McCNMT, McNMCH, McBBE and McSMT genes in the McBBE-OX lines was significantly increased (Fig. 4). However, some other genes, McTDC, McP6H, and McDBOX, were significantly decreased. Since the McBBE gene was overexpressed in McBBE-OX lines, it is understandable that PsBBE-OX lines showed different results from McBBE-OX lines in the expression level of the McBBE gene. However, the results for PsBBE-OX plants were quite different from those for McBBE-OX lines and were more similar to those for the EV and WT lines, except regarding the expression of McTDC, McP6H and McDBOX.

The analysis of gene expression levels in transgenic lines (pcambia2301 + McBBE, pcambia2301 + PsBBE, pcmbia2301) and WT lines. Data represent means ± SD (n = 3). Asterisks denote the significant changes (* means P < 0.05, ** means P < 0.01, *** means P < 0.005, **** means P < 0.001). EV means empty vector, WT means wild type.
Discussion
One method to enhance target compounds in a secondary metabolic pathway is to overexpress the potential key enzyme at the genetic level. The BBE gene is responsible for a rate-limiting step in the biosynthetic pathway of BIA and affects the final production of BIA12. In the present study, the two BBE genes from M. cordata and P. somniferum were integrated into separate plant lines to analyse their impact on the BIA metabolic network. However, the levels of SAN in all transgenic plants (McBBE-OX, PsBBE-OX, and EV) were slightly lower than those in wild-type plants. This difference can be attributed to major changes in the levels of some intermediate metabolites; in particular, in the McBBE-OX line, some of the downstream metabolites, such as (S)-scoulerine, (S)-cheilanthifoline and (S)-tetrahydrocolumbamine, were significantly risen. Additionally, McBBE and McSMT gene expression was increased in the McBBE-OX line (P < 0.05), consistent with the metabolic profile. Then, we analysed the upstream metabolite profiles ((S)-norcoclaurine, (S)-coclaurine, (S)-N-cis-methylcoclaurine, and (S)-reticuline) and the expression levels of upstream genes (Mc6OMT, McCNMT, McNMCH and Mc4OMT) in the different transgenic lines (Figs 3 and 4). Compared with those in the PsBBE-OX, EV and WT lines, the expression levels of upstream genes (Mc6OMT, McCNMT, McNMCH and Mc4OMT) were significantly increased (P > 0.05) in the McBBE-OX lines. Correspondingly, the metabolite levels ((S)-norcoclaurine, (S)-coclaurine, (S)-N-cis-methylcoclaurine, and (S)-reticuline) in McBBE-OX lines were also higher than those in the other transgenic lines. A possible reason for the reduction in SAN content and the increase in reticuline content is that positive feedback derived from higher expression of the McBBE gene activates the expression of upstream genes. Furthermore, the reduction in the expression of downstream genes (McTDC, McP6H, and McDBOX) blocked the carbon flow and caused the upstream intermediate substances to accumulate slowly. A similar result was observed in 1-deoxy-D-xylulose 5-phosphate synthase (DXS)-overexpressing tissues in Catharanthus roseus24. These unintended consequences often appear when engineering single enzymes to increase flux. For example, overexpression of phytoene synthase in tomato increases lycopene content but downregulates the gibberellin pathway, ultimately resulting in dwarf plants. However, overexpression of BBE in E. californica hairy root culture increases total benzophenanthridines and slightly increases SAN content12, indicating that there are difference between species even when the same metabolic pathways are disturbed. On the other hand, the gene expression level of McTDC was significantly decreased in McBBE-OX lines, while the content of (S)-tetrahydroberberine in the McBBE group was the highest among all the groups (Fig. 3). Similarly, there were no differences between transgenic plants and wild-type plants in McCFS and McTNMT mRNA expression, but (S)-cheilanthifoline and (S)-tetrahydroberberine levels were significantly higher in McBBE-OX plants than in WT plants. The possible reason is that the accumulation of a large number of synthetic precursors in the McBBE-OX line resulted in higher levels of downstream products and caused a corresponding impact on metabolites. Through analysis of gene expression, we found that 9 genes were significantly influenced in transgenic plants, which affected the metabolite profiles (Figs 3 and 4). Another unexpected observation in this study was that transformation with PsBBE did not increase any BIAs. However, the overexpression of PsBBE in Eschscholzia californica significantly increases the levels of the end products12. Because the focus of this study was to use overexpression of McBBE to enhance BIA content, we will study this phenomenon in the PsBBE group in a future study.
Previous researchers have succeeded in achieving high production of BIAs by overexpressing the key enzyme in Papaveraceae plants12,13,14. However, it is difficult to cultivate many Papaveraceae plants; for example, the cultivation of poppy is regulated by the government. In contrast, M. cordata is free of addictive compounds and has a good prospect of cultivation9. The alkaloid profile in transgenic M. cordata was clearly made more diverse through overexpression of the key step gene and introduction of the exogenous gene. This article is the first to report the successful overexpression of key genes in M. cordata.
Materials and Methods
cDNA Synthesis and PsBBE synthetic
RNA from M. cordata root was isolated using the RNA prep pure Plant Kit (TIANGEN, CHINA) and reverse transcribed into cDNA using the PrimeScriptTM II 1st Strand cDNA Synthesis Kit (TAKARA, CHINA) according to the manufacturer’s instructions. The cDNA was used as a template for subsequent vector construction. The PsBBE (GenBankAF025430) synthetic from (Genscript, China).
Plant Materials
The line originated from Fujian province (FJ1, available as seeds upon request; geographic coordinates: 27°46′23.408″N and 117°25′12.560″E) and cultivated at Hunan Agricultural University. Then further propagated via tissue culture to transgenic, metabolic analysis, and qRT–PCR, which would ensure the samples used in the following experiments have the same genetic background. All the tissue culture and the genetic transformation protocol for M. cordata was according to the previous paper23.
Plant Expression Vector Construction
The plant expression vector pCAMBIA2301 (Fig. 5) was purchased from (Miaolingbio, China). McBBE and PsBBE was amplified using the primer listed in Table 1 by polymerase (NEB, Q5® High-Fidelity DNA Polymerase, England). Then purified PCR products were subsequently cloned into pCAMBIA2301 using the In-Fusion Cloning Kit (Clontech, USA), and the pCAMBIA2301 was used as the empty vector control. These three vectors were introduced into A. tumefaciens strain GV3101, respectively for transformation in M. cordata.

The plant expression vector pCAMBIA2301, pCAMBIA2301 + PsBBE, pCAMBIA2301 + McBBE used for M. cordata transformation. The T-DNA region contains the antibiotic selection marker, NPTII, driven by CaMV35 promoter and polyA signal. The uidA (GUS) reporter is under CaMV35S promoter and nopalinesynthase (Nos) terminator.
Genomic DNA Isolation and PCR Amplification
Genomic DNA was isolated from putative transgenic lines and wild type lines using TIANGEN DNeasy Plant Mini Kit (China, TIANGEN). The GUS (750 bp), npt II (364 bp) and PsBBE (750 bp) primers were used in PCR analysis to confirm the integration of T-DNA in transgenic lines formation (Table 2). Plasmid of pcambia2301 (positive control), pcambia2301-McBBE and pcambia2301-PsBBE and wild type (negative control). The PCR amplification program (China, TAKARA): denaturation at 94 °C for 5 min, followed by 30 cycles, 94 °C for 1 min, 58 °C for 30 second, and 72 °C for 30 second, final extension 72 °C for 5 min. The products were amplified products were analyzed by 1% (w/v) agarose gels prepared in 0.5XTBE (Tris/Borate/EDTA) buffer.
GUS Staining Analysis
We performed GUS histochemical staining in wild-type lines (WT) and putative transgenic lines. All tissues were infiltrated in phosphate buffer (pH 7.0, 0.1 M), EDTA (pH 8.0, 0.5 M), mannitol (0.3 M), X-Gluc (pH 7.0, 0.1 M) and 0.5% Triton X-100 for overnight at 37 °C. Then, Stained tissues were cleared in 70% ethanol.
Metabolite Extraction and LC-QQQ MS Analysis
The WT and putative transgenic lines were collected after 3-month-old days of culture and ground into a fine powder using liquid nitrogen and then freeze-dried. Then, use ultrasonic extraction for 30 min at room temperature 1 mL of methanol, followed by ultrasonic extraction for 60 min at room temperature to isolate metabolites from 50 mg tissues. After filtration through a 0.22-mm membrane filter (Pall, USA), the solution was quantitative analyzed by LC/triple-quadrupole (QQQ) MS. An ultra-HPLC Agilent 1290 instrument coupled to a QQQ mass spectrometer (6460 A, Agilent) with a BEH C18 column (2.1 3 100 mm, 1.8 mm; Waters, Ireland) was used for the determination of 18 target alkaloids. The quantitative analyzed for metabolite according to our previous research9. The LC-QQQ MS data were processed using the Agilent Mass hunter Quantitative Analysis software (B.07.00). For absolute quantification analysis, the method was validated using the mixed standard solution, which was diluted with methanol to produce a at least 5 points and was used to evaluate absolute quantification of the target compound.
Gene Expression Analysis by qPCR
Total RNA was isolated from putative transgenic line and wild-type of M. cordata using MiniBEST Plant RNA Extraction Kit (TaKaRa). The quality of RNA was checked by agarose gel electrophoresis and quantity was confirmed by Qubit 2.0. One micrograms of total RNA from each sample was reverse transcribed into cDNA using the PrimeScript RT reagent kit with gDNA eraser (TaKaRa, Dalian, China). The resulting cDNA products were diluted to 100 μL used as templates for subsequent experiments. PCR was performed on an ABI 7300 using FastStart Universal SYBR Green Master (ROX) according to the manufacturer’s instructions. The total volume of Quantitative real-time PCR assay was performed in a 20 μL (10 μL of PCR Mix, 0.5 μL of specific primers, 4 μL of cDNA and 5 μL of water). The qPCR cycling conditions were as follows: 95 °C for 15 min; 95 °C, 15 s; 55 °C 15 s; 72 °C 20 s, with 40 cycles. In this method, three replicates were performed in all cases. Relative gene expression was performed using the comparative 2−ΔΔCTmethod. We have been proved that only one gene involved in two methylation steps (4OMT and 6OMT) in previous study. Therefore, the primers used in 4OMT and 6OMT are the same. All the primer sequence (Table 3) was checked via blast analysis and the 18S gene as the internal reference.
Statistical Analysis
All the transgenic experiments were carried out in triplicate and each treatment contained 100 explants. All metabolic content and qPCR experiments were analyzed by one-way analysis of variance (ANOVA). The data were statistically analyzed using the GraphPad prism statistical software (version 7.0, USA). Differences between means were determined by analysis of variance with Tukey’s test on the level of significance declared at P < 0.05.
References
Ziegler, J. et al. Evolution of morphine biosynthesis in opium poppy. Phytochemistry 70, 1696–1707 (2009).
Tang, J. et al. Berberine and Coptidis rhizoma as novel antineoplastic agents: a review of traditional use and biomedical investigations. Journal of ethnopharmacology 126, 5–17 (2009).
Lee, K. H. et al. Regulation of glutamate level in rat brain through activation of glutamate dehydrogenase by Corydalis ternata. Experimental & molecular medicine 37, 371 (2005).
Zhang, S.-M. & Coultas, K. A. Identification of plumbagin and sanguinarine as effective chemotherapeutic agents for treatment of schistosomiasis. International Journal for Parasitology: Drugs and Drug Resistance 3, 28–34 (2013).
Slaninová, I., Pěnčíková, K., Urbanová, J., Slanina, J. & Táborská, E. Antitumour activities of sanguinarine and related alkaloids. Phytochemistry reviews 13, 51–68 (2014).
Vieira, S. L. et al. Studies with sanguinarine like alkaloids as feed additive in broiler diets. Revista Brasileira de Ciência Avícola 10, 67–71 (2008).
Lee, K.-W., Kim, J.-S., Oh, S.-T., Kang, C.-W. & An, B.-K. Effects of dietary sanguinarine on growth performance, relative organ weight, cecal microflora, serum cholesterol level and meat quality in broiler chickens. The Journal of Poultry Science 52, 15–22 (2015).
Beaudoin, G. A. & Facchini, P. J. Benzylisoquinoline alkaloid biosynthesis in opium poppy. Planta 240, 19–32 (2014).
Liu, X. et al. The Genome of Medicinal Plant Macleaya cordata Provides New Insights into Benzylisoquinoline Alkaloids Metabolism. Molecular plant 10, 975–989 (2017).
Bird, D. A. & Facchini, P. J. Berberine bridge enzyme, a key branch-point enzyme in benzylisoquinoline alkaloid biosynthesis, contains a vacuolar sorting determinant. Planta 213, 888–897 (2001).
Schläger, S. & Dräger, B. Exploiting plant alkaloids. Current opinion in biotechnology 37, 155–164 (2016).
Park, S.-U., Yu, M. & Facchini, P. J. Modulation of berberine bridge enzyme levels in transgenic root cultures of California poppy alters the accumulation of benzophenanthridine alkaloids. Plant molecular biology 51, 153–164 (2003).
Inui, T., Tamura, K.-i., Fujii, N., Morishige, T. & Sato, F. Overexpression of Coptis japonica norcoclaurine 6-O-methyltransferase overcomes the rate-limiting step in benzylisoquinoline alkaloid biosynthesis in cultured Eschscholzia californica. Plant and cell physiology 48, 252–262 (2007).
Inui, T. et al. Improvement of benzylisoquinoline alkaloid productivity by overexpression of 3′-hydroxy-N-methylcoclaurine 4′-O-methyltransferase in transgenic Coptis japonica plants. Biological and Pharmaceutical Bulletin 35, 650–659 (2012).
Fossati, E. et al. Reconstitution of a 10-gene pathway for synthesis of the plant alkaloid dihydrosanguinarine in Saccharomyces cerevisiae. Nature communications 5, 3283 (2014).
Galanie, S., Thodey, K., Trenchard, I. J., Interrante, M. F. & Smolke, C. D. Complete biosynthesis of opioids in yeast. Science 349, 1095–1100 (2015).
Nakagawa, A. et al. Total biosynthesis of opiates by stepwise fermentation using engineered Escherichia coli. Nature communications 7, 10390 (2016).
Paddon, C. J. et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 496, 528 (2013).
Shi, P. et al. Promotion of artemisinin content in Artemisia annua by overexpression of multiple artemisinin biosynthetic pathway genes. Plant Cell, Tissue and Organ Culture (PCTOC) 129, 251–259 (2017).
Lee, M.-H. et al. Enhanced triterpene and phytosterol biosynthesis in Panax ginseng overexpressing squalene synthase gene. Plant and Cell Physiology 45, 976–984 (2004).
Hashimoto, T., Yun, D.-J. & Yamada, Y. Production of tropane alkaloids in genetically engineered root cultures. Phytochemistry 32, 713–718 (1993).
Canel, C. et al. Effects of over-expression of strictosidine synthase and tryptophan decarboxylase on alkaloid production by cell cultures of Catharanthus roseus. Planta 205, 414–419 (1998).
Huang, P. et al. Establishment of an efficient Agrobacterium-mediated genetic transformation method in Macleaya cordata. Scientia Horticulturae 226, 302–306 (2017).
Peebles, C. A. et al. The expression of 1-deoxy-D-xylulose synthase and geraniol-10-hydroxylase or anthranilate synthase increases terpenoid indole alkaloid accumulation in Catharanthus roseus hairy roots. Metabolic engineering 13, 234–240 (2011).
Acknowledgements
This work was supported by the National Key R&D Program of China (2017YFD0501500 and 2016YFD0501308), the Natural Science Foundation of Hunan Province (2016JJ4040), China Hunan Provincial Science& Technology Department (2016SK3002).
Author information
Authors and Affiliations
Contributions
P.H. and J.Z. conceived and designed research. P.H., W.L., M.X., L.X., R.J., P.W., H.L., Z.T. and Q.Z. conducted experiments. W.L., M.X. analyzed data. P.H. wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, P., Liu, W., Xu, M. et al. Modulation of benzylisoquinoline alkaloid biosynthesis by overexpression berberine bridge enzyme in Macleaya cordata. Sci Rep 8, 17988 (2018). https://doi.org/10.1038/s41598-018-36211-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-36211-8
This article is cited by
-
Evaluation of reference genes for qRT-PCR studies in the colchicine producing Gloriosa superba L.
Plant Biotechnology Reports (2023)
-
Effects of codon optimization, N-terminal truncation and gene dose on the heterologous expression of berberine bridge enzyme
World Journal of Microbiology and Biotechnology (2022)
-
The effects of protopine 6-hydroxylase (P6H) overexpression on benzylisoquinoline alkaloids in Macleaya cordata
Plant Cell, Tissue and Organ Culture (PCTOC) (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
