
Abstract
Smoking is a major cause of respiratory conditions. To date, the genetic pleiotropy between smoking behavior and lung function/chronic obstructive pulmonary disease (COPD) have not been systematically explored. We leverage large data sets of smoking behavior, lung function and COPD, and addressed two questions, (1) whether the genetic predisposition of nicotine dependence influence COPD risk and lung function; and (2) the genetic pleiotropy follow causal or independent model. We found the genetic predisposition of nicotine dependence was associated with COPD risk, even after adjusting for smoking behavior, indicating genetic pleiotropy and independent model. Two known nicotine dependent loci (15q25.1 and 19q13.2) were associated with smoking adjusted lung function, and 15q25.1 reached genome-wide significance. At various suggestive p-value thresholds, the smoking adjusted lung function traits share association signals with cigarettes per day and former smoking, substantially greater than random chance. Empirical data showed the genetic pleiotropy between nicotine dependence and COPD or lung function. The basis of pleiotropic effect is rather complex, attributable to a large number of genetic variants, and many variants functions through independent model, where the pleiotropic variants directly affect lung function, not mediated by influencing subjects’ smoking behavior.
Similar content being viewed by others

Introduction
Chronic obstructive pulmonary disease (COPD) is the third cause of death in the US after cancers and cardiovascular diseases1 and is among the leading causes of hospitalization in industrialized countries2,3. It was recently estimated that the absolute number of COPD cases in developed countries will increase by more than 150% from 2010 to 20304, yet there is no curative therapy for COPD1,5. A lack of understanding of the molecular mechanisms in the pathogenesis of COPD has hampered efforts to develop new biomarkers and effective therapies.
COPD has multiple risk factors and complex etiology, where cigarette smoking and genetic susceptibility are among the main risk factors1,6. Tobacco smoking accounted for about 5.1 million deaths globally in 2004, and it is observed recent increases in smoking prevalence in developing countries7. But only 20–25% of smokers develop clinically significant airflow obstruction8. Smoking behavior is partially genetically determined, and at least genome-wide significant associated loci were identified in European ancestry subjects9,10, where the strongest and most consistent association reported is at the 15q25.1 locus (CHRNA3-CHRNA5-CHRNB4 gene cluster). Genes associated with smoking quantity were enriched for cholinergic receptors, sensory perception of smell, and Retinoid Binding11.
In parallel, there is strong evidence that genetic variants (both common and rare) contribution to COPD and pulmonary function. Candidate gene and genome-wide association studies (GWAS) have identified genetic variants associated with COPD12,13,14,15,16. The latest GWAS was performed by the International COPD Genetics Consortium (ICGC)17 identified 22 COPD susceptibility loci at genome-wide significance. Exome sequencing study found rare variants on CHRNA3, CHRNA5, and CHRNB4 genes were associated with COPD18. Lung function (e.g. FEV1, FVC and FEV1/FVC ratio) are traits closely related to COPD, in fact, lung function parameters are often used in defining COPD cases6. Three key parameters are commonly used in characterizing lung function: expired volume in 1st second (FEV1), forced vital capacity (FVC) and FEV1/FVC. FVC is the volume of air that can forcibly be blown out after full inspiration19, and is the most basic maneuver in spirometry tests. FEV1 is the volume of air exhaled in the first second during forced exhalation after maximal inspiration19. FEV1/FVC is the ratio of these two values, where the ratio ≥ 80% is considered normal19. To date, 97 independent genetic loci were identified influencing lung function traits (FEV1, FVC and FEV1/FVC)6.
Genetic pleiotropy is the phenomenon where a DNA variant influences multiple traits20. Proposed by our group and others, available GWAS summary data can be used to detect genetic pleiotropy and pinpoint the variants, gene and pathways underlying the shared etiology21,22,23. Given smoking is a strong cause of COPD and lung function decline, genetic variants associated with smoking behavior or nicotine dependence (ND) are likely also in association with lung function (LF), which can be viewed as genetic pleiotropy. The genetic pleiotropy could have two possible mechanisms: (1) a genetic locus causes ND and then in turn causes LF decline (Fig. 1A); and (2) a genetic locus causes ND and LF decline independently (Fig. 1B). These two mechanisms can be distinguished by testing the association between genetic locus and smoking adjusted LF (ie, LFadj). If mechanism (1) is true, the ND locus will show no association with LFadj (Fig. 1C).

Models of genetic pleiotropy.

QQplot showed SNPs associated with nicotine dependence are enriched for small p values in lung function GWAS. The y axis of the plot of the top row (plot A, B, C and D) denotes the observed p value of smoking adjusted FEV1 in UKBB study; the y axis of the plot of the middle row (plot E, F, G and H) denotes the observed p value of smoking adjusted FVC in UKBB study; the y axis of the plot of the bottom row (plot I, J, K and L) denotes the observed p value of smoking adjusted FEV1/FVC Ratio in UKBB study. In the 1st column (plot A, E and I), we investigated on SNPs associated with cigarettes per day (CPD) at ≤1e-2 (red dots), ≤1e-3 (blue dots), ≤1e-4 (green dots), and ≤1e-5 (black dots); in the 2nd column (plot B, F and J), we investigated on SNPs associated with ever smoking (EVERSMK) at ≤1e-2 (red dots), ≤1e-3 (blue dots), ≤1e-4 (green dots), and ≤1e-5 (black dots); in the 3rd column (plot C, G and K), we investigated on SNPs associated with former smoking (FORMER) at ≤1e-2 (red dots), ≤1e-3 (blue dots), ≤1e-4 (green dots), and ≤1e-5 (black dots); in the 4th column (plot D, H and L), we investigated on SNPs associated with log of smoking onset (ONSET) at ≤1e-2 (red dots), ≤1e-3 (blue dots), ≤1e-4 (green dots), and ≤1e-5 (black dots).
A recent GWAS meta-analysis employed 48,943 UK Biobank individuals in discovery phase and 95,375 individuals in follow-up phase, and increased the independent signals for lung function from 54 to 976. Importantly, the study carefully adjusted for smoking behavior, and among smokers, the pack-year was also adjustment6. The GWAS results of smoking-adjusted lung function offer a unique opportunity to decipher the genetic pleiotropy between nicotine dependence and function as well as the etiological mechanisms.
Results
Genetic predisposition of nicotine dependence is associated with COPD risk
We leverage the individual level phenotype (COPD affected status and smoking behavior) and genotype data of COPDgene cohort (Materials and Methods) to test association between genetic predisposition of nicotine dependence (quantitatively summarized as polygenic score, or PGS) and COPD risk. On each COPDgene study subject, we computed the PGS for ND CPD trait (i.e., PGSND-CDP). The PGSND-CDP compiled at 1e-3 and 1e-4 p-value thresholds were associated with that COPD affected status at p-value 0.025 and 1.03e-6, respectively (Table 1), indicating significant pleiotropy of ND and COPD. The positive association coefficient indicate genetic predisposition of higher cigarettes per day leads to higher COPD risk (Table 1). PGS of former smoking (ie, PGSND-FORMER) were also associated with COPD, and the negative coefficient suggested the genetic predisposition of smoking cessation reduced COPD risk. Next, we adjusted the smoking covariates and found the PGSND-CDP and PGSND-FORMER were still associated with COPD (Tables 2 and 3). Importantly, the association p value and coefficient were little changed before and after adjustment, indicating an independent pleiotropy model (Fig. 1B), where the effect of PGSND-CDP and PGSND-FORMER on COPD risk was not mediated by smoking behavior of the study participants.
Nicotine dependence genome-wide significant loci and their association with lung function (LF)
Firstly, we retrieved six independent ND GWAS loci of genome-wide significance (p < 5e-8) in European ancestry24 from the NHGRI-EBI catalog25, and examined the association between these loci and smoking adjusted lung function (LFadj) in UKBB meta-analysis cohort (Table 4). In UKBB study, the smoking status was adjusted with the regression model in the form of indicator variables denoting current smoker, and denoting former smokers; as well as quantitative variable denoting pack-year (in smokers). At nominal p value (≤0.05), two ND loci showed association with at least one LFadj traits (FEV1, FVC, and FEV1/FVC Ratio). The lead SNP, rs1051730, of the 15q25.1 locus was strongly associated with all three LFadj trait, and its association with smoking adjusted FEV1 reached genome-wide significant (p = 5e-8), further, the 19q13.2 locus, known in association with COPD26, is also associated with smoking adjusted FEV1 (Table 4), suggesting the 15q25.1 and 19q13.2 loci could influence ND and LF independently (Fig. 1B). In contrast, four genome-wide significant ND loci (10q23.32, 8p11.21, 11p14.1, 9q34.2) showed no association with LFadj traits (Table 4 and Fig S1), suggesting causal pleiotropy model (Fig. 1 A), where the genetic variants influence LF, mediated by smoking behavior.
Identification of SNPs associated with lung function and nicotine dependence at suggestive threshold
There is a general consensus among GWAS studies that a p-value less than 5e-8 corresponds to genome-wide significance24. NHGRI-EBI GWAS catalog focuses on highly significant loci, but true association loci may only show suggestive p values given the limited power in GWAS. We leverage the summary data of with UKBB smoking adjusted LF (LFadj) and TAG ND (Materials and Methods). Applied various p thresholds (ranging from 1e-8 to 1e-2), we identified SNPs in associated with LFadj or ND (Table S1). The UKBB LFadj GWAS yield many SNPs highly significantly associated with smoking adjusted FEV1, FVC and FEV1/FVC Ratio. On the other hand, the TAG ND study captured strong genetic signals for CPD (cigarettes per day) and moderate signal for former smoking (Table S1). But no SNP associated ever smoking or log-onset at p < 1e-7 level, indicating these two traits either are not controlled by genetic factors or the TAG study did not have sufficient power (Table S1).
Among the ~2 million SNPs tested in both LFadj and ND GWAS studies, the proportion of associated SNPs at any given p-value threshold was greater than alpha level (Table 5). For example, at p-value ≤ 1e-3 threshold, 11,716 SNPs and 2,791 SNPs were associated with FEV1 and CPD, respectively, and 71 SNPs were associated with both trait, which is substantially greater than random chance (enrichment fold = 4.61). In fact, we observed excess overlapping of FEV1 and CPD GWAS SNPs were across multiple p-value thresholds (Table 5 and Fig. 2). Further, at p value ≤ 1e-3 threshold, 139 SNPs were associated with both FEV1 and former smoking (Table 6), 11.8 folds of enrichment than random chance. While, GWAS signals of ever smoking and smoking onset showed little or very modest overlap with LFadj GWAS (Tables S2 and S3). The SNPs associated with at least one of the nicotine dependence and lung function traits (cigarettes per day, former smoking, FEV1, FVC, FEV1/FVC) at p value ≤ 1e-3 level were listed in Table S4; while SNPs associated with all of the five nicotine dependence and lung function traits were detailed in Table S4.
Association direction of shared LFadj and ND SNPs
The shared LFadj/ND SNPs can be stratified into categories according whether the allele associated with higher LFadj trait is also associated with higher ND trait or ND event odds (ie, consistent direction SNP) or vice versa (ie, divergent direction SNP). We stratified the shared LFadj/ND SNPs by directions, and found all three LFadj traits primarily have divergent direction with CDP (Table 5). At 1e-3 threshold, 71 SNPs are shared by FEV1 and CPD, where 65 SNPs (91.55%) with divergent directions (binomial p value = 4.34e-13), indicating alleles lead to higher smoking quantity (cigarettes per day) tends to associated with lower LFadj traits. In contrast, the LFadj traits primarily have consistent direction with former smoking (Table 6). At 1e-3 threshold, 193 SNPs were shared by FEV1 and former smoking, and only 21 SNPs (10.88%) with divergent directions but 172 SNPs are of a consistent direction (binomial p-value = 4.34e-13), indicating alleles lead to smoking cessation tend to associated with higher LFadj traits.
Further, we stratified the SNPs into the consistent direction SNP and divergent SNP subcategories, regardless of GWAS test p-values criteria (Tables S5–S8). We found the overlap of CPD and LFadj GWAS signals primarily occur among the SNPs of divergent allele direction, and observed little overlap among the SNPs of consistent allele direction. For example, at a GWAS p-value < 1e-3, SNPs of divergent allele direction for FEV1 and CPD showed substantial overlap (6.94 fold enrichment), in contrast, the overlap for SNPs of consistent allele direction were not different from random chance.
Gene ontology influenced by the shared SNPs of LFadj and ND
Shared LFadj/ND SNPs may undergo complicated pathways and lead to disease risk. Identifying the genes influenced by such shared SNPs would be the key step elucidating the SNPs’ function and pathogenic pathways. To investigate potential functional impacts of LFadj/ND shared SNPs, we used a “relax” p-value threshold criterion of 1e-2 for GWAS. Among the 826 shared FEV1 - CPD SNPs, 375 are characterized by having consistent direction and 451 are characterized by divergent direction. Based on SNP position and dbSNP annotation27, we identified genes located with on or near the share GWAS SNPs of LFadj and ND, and then carried out the Gene Ontology (GO) cellular processes enrichment analysis using METACORE suite (Fig. 3). The top 10 enriched processes (p-value ≤ 1e-6) were regulation of synapse vesicles as well as regulation of respiratory systems, highly relevant to lung function and nicotine dependence.

Genes harbor or close to share SNPs between cigarettes per day and smoking adjusted FEV1 are enriched for certain Gene Ontology cellular processes.
Co-localization of 15q25.1 locus underlying LFadj and ND
SNPs on 15q25.1 locus are associated with both LFadj and ND at genome-wide significant level (Table 4). Also, the CHRNA3-CHRNA5-CHRNB4 gene cluster in this locus is functional relevant to both traits. However, given that neighboring SNPs were often in tight linkage disequilibrium (LD), the overlap of GWAS signals do not guarantee that the disease risks of the two traits are caused by the same variant. Recently developed methods allow more advanced integration of GWAS summary data to co-localize GWAS signals28. The co-localization methods28 evaluated 5 hypotheses (Materials and Methods), where we were particularly interested in hypothesis 4 (H4: the two phenotypic traits were caused by the same SNP in the locus), and H4 posterior probability over 0.75 was considered as supporting evidence to the corresponding hypothesis29,30. All the three LFadj trait (FEV1, FVC and FEV1/FVC Ratio) were co-localized at 15q25.1 (Table 7), indicating they are controlled by the same genetic variant. The cigarettes per day and former smoking traits were also co-localized with LFadj (Table 8). For example, between CPD and FEV1, the coloc H4 posterior probability was 0.908, and between former smoking and FEV1, the coloc H4 posterior probability was 0.942. The clear co-localization of LFadj and ND at 15q25.1 suggested same genetic variant influence both nicotine dependence and lung function, through an independent pleiotropy model.
Discussion
In this report, we leverage the latest large GWAS data sets and investigated the genetic pleotropic effect between nicotine dependence and respiratory outcomes (ie, lung function and COPD). It is known smoking is a major cause of reduced lung function and COPD8. Smoking behavior is at least partially controlled by genetic factors7,9. To date, the pleiotropic effects of genetic risk of smoking behavior and lung function/COPD have not been systematically explored.
Employing several analytical approaches, this paper addressed two questions, (1) whether the genetic predisposition of nicotine dependence influence COPD risk and lung function; and (2) the genetic pleiotropy follow causal or independent model (Fig. 1). On COPDgene cohort, we found the polygenic score of nicotine dependence (ie, PGSND) was associated with COPD case/control status, demonstrating the genetic pleiotropy of the two conditions. We also investigated the association between PGSND and COPD while adjusting smoking behavior. Interestingly, the crude and adjusted results were very similar, indicating a mainly independent pleiotropy model. That is the shared genetic factors directly modify COPD risk, not mediated by influencing the individual’s smoking behavior.
We zoomed in the known ND loci of genome-wide significance (Table 4), and found both causal and independent pleiotropy models may exist in certain loci. Two genome-wide significant ND loci (15q25.1 and 19q13.2) were associated with LFadj (smoking adjusted lung function), supporting independent pleiotropy model. While, four ND loci (10q23.32, 8p11.21, 11p14.1 and 9q34.2) were not associated with smoking adjusted lung function. At various p value threshold (1e-6 to 1e-1), we found the LFadj traits share association SNP with cigarettes per day and former smoking substantially more than random chance, indicating a large number of genetic variants contribute to the genetic pleiotropy. Importantly, the lung function and cigarettes per day mainly share SNPs of divergent direction, meaning genetic predisposition of higher smoking dosage leads to lower lung function. In contrast, the lung function and former mainly share SNPs of consistent direction, meaning genetic predisposition of smoking cessation leads to higher lung function.
In summary, we used empirical data of largest cohorts to date and showed the genetic pleiotropy between nicotine dependence and COPD or lung function. The pleiotropic effect exist even COPD status or lung function is adjusted for smoking behavior. Further, we found the pleiotropic effect is attributable not only to the genome-wide significant loci, but also loci associated to ND and COPD/LF at suggestive p value (e.g. 1e-3), suggesting a large number of variants influence both ND and respiratory outcome, and among which many variants functions through independent genetic pleiotropy model.
Materials and Methods
Genome-wide meta-analyses on nicotine dependence (ND)
The Tobacco and Genetics (TAG) Consortium conducted GWAS meta-analyses across 16 studies10. We examined four carefully harmonized smoking phenotypes: smoking initiation (ever versus never been a regular smoker), age of smoking initiation, smoking quantity (number of cigarettes smoked per day, CPD) and smoking cessation (former versus current smokers) among people of European ancestry. Smoking cessation contrasted former versus current smokers, where current smokers reported at interview that they presently smoked and former smokers had quit smoking at least 1 year before interview. Smokers who had quit smoking for less than 1 year at interview were excluded from the analysis to minimize misclassification. Genotype imputation resulted in a common set of ~2.5 million SNPs that entered the GWAS and meta-analysis, where summary statistics were used in this report.
COPDGene dataset
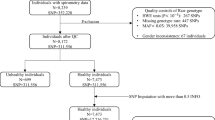
Individual level genotype and phenotype data of COPDgene study were retrieved from dbGap (accession: phs000179.v1.p1). COPD case/control status, indicator variable of current smoking, indicator variable of former smoking, quantitative variable of smoking duration, and SNP genotype data were available on 4,903 individuals. We conducted genotype imputation using HRC reference31, and in total, 19,932,879 SNPs of high quality score entered the current study. In polygenic score analysis, the smoking status variable (indicator and quantitative variables) were adjusted within the regression model.
UK Biobank Lung Function (LF) GWAS
Recently we reported a large GWAS study on lung function using the UK biobank samples6. Genome-wide association analyses of forced expired volume in 1 s (FEV1), forced vital capacity (FVC) and FEV1/FVC were undertaken in 48,943 individuals from the UK BiLEVE study7 who were selected from the extremes of the lung function distribution in UK Biobank (total n = 502,682). Association tests were conducted on 27,624,732 variants, where linear regression of age, age2, sex, height, the first ten principal components of genetic ancestry and pack-years of smoking (in smokers), and summary statistics were used in this report.
Polygenic Score
We analyzed GWAS summary statistics data from the TAG nicotine dependence study, COPDgene dataset case control status, and COPDgene dataset smoking related covariate: smoking status (binary variable) and smoking duration (continuous variable). We computed the nicotine dependence polygenic score (PGS) on each COPDgene study subject in following steps: (1) identify shared SNPs in TAG GWAS summary data and COPDgene imputed genotype; (2) align alleles strands to the 1000 G panel (hg19), and adjust β coefficients TAG nicotine dependence GWAS accordingly; (3) filter TAG GWAS data by p value threshold (1e-3 and 1e-4 as shown in Table 5) and prune the SNPs by linkage disequilibrium (LD) based on 1000 G EUR reference; lastly (4) for each ND traits, and for each p value threshold, we computed a PGS for every subject as a linear combination of the imputed doses of the selected coefficients. Then we tested the association between nicotine dependent PGS and COPD case/control status using a logistic regression model with or without adjusting for smoking covariates.
Identification of SNPs associated with both LF and ND
The effect size attributable to individual genetic variants for a given complex disorder is typically modest, suggesting that individual genetic variants may only explain a very small amount of the genetic risk and heritability of complex disorders21,31. Therefore, genetic contributions to complex conditions such as LF and ND are likely derived from a large number of genetic causal variants, each contributing a small genetic risk. We surveyed a number of p value thresholds, and identified SNPs that are associated with LFadj and ND in order to more comprehensively capture SNPs with modest effect sizes. For a shared SNP, we term it “consistent allele direction” if a specific allele that is associated with increased LFadj traits and that specific SNP allele is also associated with higher ND trait value or event odds. We term “divergent allele direction” for SNP where a specific allele associated with increased lung function, and lower ND trait value or ND event odds.
Pathway Enrichment Analyses
To further characterize the regulatory nature, enrichment analysis of the genes influenced by shared SNPs were performed using the METACORE integrated software suite (http://thomsonreuters.com/metacore/).
Co-localization of lung function and nicotine dependence GWAS top SNPs
LFadj and ND GWAS results were used in co-localization analysis, which is performed using COLOC version 2.3–6 in R28. Our analysis focuses on the 15q25.1 locus. Default priors of the software were used. In total, 5 hypotheses were evaluated. H0: No association with either trait 1 or trait 2; H1: Association with trait 1, not with trait 2; H2: Association with trait 2, not with trait 1; H3: Association with trait 1 and trait 2, two independent SNPs; H4: Association with trait 1 and trait 2, one shared SNP. Genes that demonstrated a high posterior probability of hypothesis 4 (PP.H4 > 75%) indicate the disease risk and placenta gene expression were controlled by the same genetic variant; and genes that demonstrated a high posterior probability of hypothesis 3 (PP.H3 > 75%) indicate the disease risk and placenta gene expression were controlled by distinct genetic variant at the locus.
References
Martinez, F. D. Early-Life Origins of Chronic Obstructive Pulmonary Disease. The New England journal of medicine 375, 871–878, https://doi.org/10.1056/NEJMra1603287 (2016).
Sanchez, M., Vellanky, S., Herring, J., Liang, J. & Jia, H. Variations in Canadian rates of hospitalization for ambulatory care sensitive conditions. Healthc Q 11, 20–22 (2008).
The Human and Economic Burden of COPD: A Leading Cause of Hospital Admission in Canada. Canadian Thoracic society (2010).
Khakban, A. et al. The Projected Epidemic of COPD Hospitalizations Over the Next 15 Years: A Population Based Perspective. Am J Respir Crit Care Med, https://doi.org/10.1164/rccm.201606-1162PP (2016).
Sly, P. D. & Bush, A. From the Cradle to the Grave: The Early-Life Origins of Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 193, 1–2, https://doi.org/10.1164/rccm.201509-1801ED (2016).
Wain, L. V. et al. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet 49, 416–425, https://doi.org/10.1038/ng.3787 (2017).
Wain, L. V. et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. Lancet Respir Med 3, 769–781, https://doi.org/10.1016/S2213-2600(15)00283-0 (2015).
Lokke, A., Lange, P., Scharling, H., Fabricius, P. & Vestbo, J. Developing COPD: a 25 year follow up study of the general population. Thorax 61, 935–939, https://doi.org/10.1136/thx.2006.062802 (2006).
Liu, J. Z. et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nat Genet 42, 436–440, https://doi.org/10.1038/ng.572 (2010).
Thorgeirsson, T. E. et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet 42, 448–453, https://doi.org/10.1038/ng.573 (2010).
Harari, O. et al. Pathway analysis of smoking quantity in multiple GWAS identifies cholinergic and sensory pathways. PloS one 7, e50913, https://doi.org/10.1371/journal.pone.0050913 (2012).
Bossé, Y. Updates on the COPD gene list. Int J Chron Obstruct Pulmon Dis 7, 607–631, https://doi.org/10.2147/COPD.S35294copd-7-607 (2012).
Cho, M. H. et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet 42, 200–202, https://doi.org/10.1038/ng.535 (2010).
Cho, M. H. et al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum Mol Genet 21, 947–957, https://doi.org/10.1093/hmg/ddr524 (2012).
Pillai, S. G. et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet 5, e1000421, https://doi.org/10.1371/journal.pgen.1000421 (2009).
Cho, M. H. et al. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med 2, 214–225, https://doi.org/10.1016/S2213-2600(14)70002-5 (2014).
Hobbs, B. D. et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat Genet 49, 426–432, https://doi.org/10.1038/ng.3752 (2017).
Zhao, Z. et al. Exon sequencing identifies a novel CHRNA3-CHRNA5-CHRNB4 variant that increases the risk for chronic obstructive pulmonary disease. Respirology 20, 790–798, https://doi.org/10.1111/resp.12539 (2015).
Ranu, H., Wilde, M. & Madden, B. Pulmonary function tests. The Ulster medical journal 80, 84–90 (2011).
Gratten, J. & Visscher, P. M. Genetic pleiotropy in complex traits and diseases: implications for genomic medicine. Genome medicine 8, 78, https://doi.org/10.1186/s13073-016-0332-x (2016).
Hao, K. et al. Shared genetic etiology underlying Alzheimer’s disease and type 2 diabetes. Molecular aspects of medicine 43–44, 66–76, https://doi.org/10.1016/j.mam.2015.06.006 (2015).
Han, B. et al. A method to decipher pleiotropy by detecting underlying heterogeneity driven by hidden subgroups applied to autoimmune and neuropsychiatric diseases. Nat Genet 48, 803–810, https://doi.org/10.1038/ng.3572 (2016).
Pickrell, J. K. et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet 48, 709–717, https://doi.org/10.1038/ng.3570 (2016).
Stranger, B. E., Stahl, E. A. & Raj, T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics 187, 367–383, https://doi.org/10.1534/genetics.110.120907 (2011).
Welter, D. et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 42, D1001–1006, https://doi.org/10.1093/nar/gkt1229 (2014).
Ryan, D. M. et al. Smoking dysregulates the human airway basal cell transcriptome at COPD risk locus 19q13.2. PloS one 9, e88051, https://doi.org/10.1371/journal.pone.0088051 (2014).
Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29, 308–311 (2001).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10, e1004383, https://doi.org/10.1371/journal.pgen.1004383 (2014).
Peng, S. et al. Expression quantitative trait loci (eQTLs) in human placentas suggest developmental origins of complex diseases. Hum Mol Genet 26, 3432–3441, https://doi.org/10.1093/hmg/ddx265 (2017).
Huang, K. L. et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nature neuroscience 20, 1052–1061, https://doi.org/10.1038/nn.4587 (2017).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 48, 1279–1283, https://doi.org/10.1038/ng.3643 (2016).
Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet 42, 441–447, https://doi.org/10.1038/ng.571 (2010).
Acknowledgements
We thank Drs. Alison Goate and Manav Kapoor for the valuable discussions and suggestions. This work is partially supported by NIH 1R41DA042464–01, NIH 1U01HD079068-01, National Natural Science Foundation of China (Grant No. 21477087, 91643201) and by the Ministry of Science and Technology of China (Grant No. 2016YFC0206507).
Author information
Authors and Affiliations
Contributions
Z.J., P.S., C.H., D.N.A. and H.K. are responsible for designing and conducting the study. Z.J., P.S., Y.N., D.N.A., and H.K. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, J., Peng, S., Cheng, H. et al. Genetic Pleiotropy between Nicotine Dependence and Respiratory Outcomes. Sci Rep 7, 16907 (2017). https://doi.org/10.1038/s41598-017-16964-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16964-4
This article is cited by
-
Meta-analysis of sample-level dbGaP data reveals novel shared genetic link between body height and Crohn’s disease
Human Genetics (2021)
-
Genetic architecture of cardiometabolic risks in people living with HIV
BMC Medicine (2020)
-
GWAS of smoking behaviour in 165,436 Japanese people reveals seven new loci and shared genetic architecture
Nature Human Behaviour (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
