
Abstract
Aythya marila is one of the few species of Anatidae, and the only Aythya to live in the circumpolar. However, there is a relative lack of research on genetics of this species. In this study, we reported and assembled the first high-quality chromosome-level genome assembly of A. marila. This genome was assembled using Nanopore long reads, and errors corrected using Illumina short reads, with a final genome size of 1.14 Gb, scaffold N50 of 85.44 Mb, and contig N50 of 32.46 Mb. 106 contigs were clustered and ordered onto 35 chromosomes based on Hi-C data, covering approximately 98.28% of the genome. BUSCO assessment showed that 97.0% of the highly conserved genes in aves_odb10 were present intact in the genome assembly. In addition, a total of 154.94 Mb of repetitive sequences were identified. 15,953 protein-coding genes were predicted in the genome, and 98.96% of genes were functionally annotated. This genome will be a valuable resource for future genetic diversity and genomics studies of A. marila.
Similar content being viewed by others


Background & Summary
Greater scaup (Aythya marila), the diving duck of the family Anatidae, which are distributed across Eurasia and North America. A. marila is one of the few Anatidae that breed in the Arctic, and the only duck belonging to the Aythya. A. marila which is a typical winter migrant. In winter, they migrate to the south to overwinter in shallow waters near the coast, estuaries, inland lakes, reservoirs, etc1,2,3. Little attention has been paid to this species because of extremely large range and population size. In fact, the population of A. marila has been declining in the past decades due to lack of effective conservation and reduced food quality4,5.
In recent years, with the development of high-throughput sequencing technology, genomes of more birds have been sequenced, providing a rich genetic resource for the study of species evolutionary mechanisms6,7,8,9,10. However, the published data does not include this species. Studies at the genetic level in this species were extremely rare due to lack of attention, and other few relevant studies were also related to diet, populations, and avian viruses4,5,11.
In this study, we present the first genome assembly of A. marila based on Nanopore sequencing and Hi-C sequencing technology. We used Nanopore sequencing data, Illumina sequencing data, and Hi-C sequencing data to assemble a genome (1,135.19 Mb) with scaffold N50 of 85.44 Mb, and 98.28% of the assembled bases were associated with the 35 chromosomes. This genome will be a valuable resource for many studies, including convergent evolution, population genomics, adaptive evolution, and comparative genomics, among others.
Materials and methods
Ethics statement
All animal experimental procedures were approved by the Biomedical Ethics Committee of Qufu Normal University (approval number: 2022001).
Sampling and sequening
The experimental sample is a wounded male duck found during the wild bird survey in Jiangsu, China, which died unexpectedly during rescue. We dissected the sample and used the muscle for genome sequencing. Additionally, six transcriptomic samples (heart, kidney, liver, lung, pancreas, and spleen) from the same individual were collected and stored in an ultra-low temperature refrigerator at −80 °C, until samples were used for extracted genomic DNA and RNA.
Using Illumina® TruSeq® Nano DNA Library Prep kits to generate sequencing libraries of genomic DNA, we constructed a paired-end library with an insertion size of 350 bp, which was sequenced using Illumina HiSeq 4000 platform. Finally, a total of 60.77 Gb data (coverage of 45.30×) of 150 bp paired-end reads were generated. Simultaneously, using the PromethION platform with single-molecule real-time sequencing to sequence long reads. 122.55 Gb data were obtained, which covered 91.36 fold of the greater scaup’s genome. To assemble a chromosome-level genome of A. marila, a high-through chrome configuration capture (Hi-C) library was built and sequenced using Illumina PE150 platform. In total, we obtained 63.43 Gb raw reads. The six transcriptomic samples were used for RNA and sequenced using Illumina NovaSeq 6000 Platform, 43.9 Gb raw reads of all samples were obtained and used for subsequent gene prediction.
Genome size estimation and assembly
To estimate the genome size, heterozygosity and the proportion of repetitive sequences in A. marila, we performed K-mer analysis using Illumina sequencing data by jellyfish (v2.2.7). While K = 17, the estimated genome size was 1,341.4 Mb, the heterozygosity was 0.47%, and the proportion of repetitive sequences was 42.28% (Table 1). Then, we used NextDenovo (v2.4.0) (https://github.com/Nextomics/NextDenovo) software to assemble the genome with Oxford nanopore technologies (ONT) long reads, and NextPolish12 (v1.3.1) was used to increase the precision of single base with Illumina short reads. We obtained a preliminarily assembly with size of 1,135.19 Mb after de novo assembly and base correction, which included 194 contigs, contig N50 was 34.42 Mb, and the proportion of GC was 41.94%.
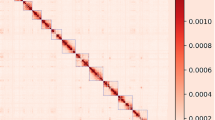
ALLHiC13 (v0.9.8) and Juicebox (v1.11.08) software were used to mount the contigs in preliminarily assembly onto chromosomes. Contigs were sorted, pruned, and optimized based on the signal strength after Hi-C data were utilized to identify the interaction signals between Contigs. The interaction signals were then shown as a heat map using ALLHiC (Fig. 1). Using Juicebox to correct the position of contigs according to the intensity of chromosome interaction, and finally get a chromosome-level genome assembly with scaffold N50 of 85.44 Mb (Table 2). A total of 106 contigs were mapped to 35 chromosomes with lengths ranging from 2.11 Mb to 207.93 Mb, and 98.28% of the sequences were mapped to the chromosomes (Table 3).

Visual heat map of the Greater Scaup’s Hi-C assembly. (a) The global heat map of all the chromosomes. (b) Heat maps of each chromosome.
Assessment of the genome assemblies
To assess the accuracy of genome assembly, we used Burrows-Wheeler aligner14 (BWA) (v0.7.8) to map Illumina reads to the genome of A. marila. The reads matching rate was approximately 98.80%, indicating a good agreement between the reads and the assembled genome. Merqury15 (v1.3) was ran to evaluate assembly quality value (QV), and a high QV (42.14) indicates that this assembly is of good quality. Benchmarking Universal Single-Copy Orthologs16 (BUSCO) (v5.4.4) (use option “--augustus”) and Core Eukaryotic Genes Mapping Approach17 (CEGMA) (v2.5) were also used to assess the integrity of the assembled genome. The BUSCO results showed that 97.0% of the complete BUSCOs and 0.8% of the fragmented BUSCOs were found in 8338 single-copy orthologs of avers_odb10, and 2.2% of BUSCOs was missing (Fig. 2). Moreover, 238 of 248 core eukaryotic genes were detected using CEGMA. The above results indicate that the genome assembly of A. marila was complete and of high quality.

The BUSCO evaluation result of the Greater Scaup’s genome assembly.
Comparison with tufted duck genome assembly
Mummer18 (v4.0.0) was used to identify the synteny between A. marila and tufted duck19 (Aythya fuligula) genomes to determine orthologous chromosome pairs, and we used TBtools20 (v1.112) to draw the synteny between their chromosomes. The synteny analysis shows that the chromosome sequences of the two species have good synteny, and each chromosome corresponds to multiple chromosomes of another species. There is one sex chromosome Z (Chr4) in A. marila, and it’s multiple autosome have some synteny with the sex chromosome W of A. fuligula (Fig. 3). Most of the chromosomes can correspond to the chromosome of the tufted duck, however both Chr31 and Chr32 shared synteny with chromosome 34 of the tufted duck, and the synteny was poor (Fig. 3d). Simultaneously, they also have synteny with some contigs. This may be due to the poor assembly quality caused by too small chromosomes size (Chr31, 2.05 Mb; Chr32, 1.03 Mb). In gap analysis, the chromosomes of A. fuligula genome assembly had a higher proportion of N bases (Fig. 3c). In general, the genome we assembled was of high quality.

Chromosome-level synteny analysis of A. marila and A. fuligula, Amar means A. marila, Aful means A. fuligula. (a) Chromosomes length of A. marila and A. fuligula (Mb). (b) GC ratio (%) by 500 kb window. (c) Gap ratio (%) by 500 kb window. (d) The synteny plot of remove the non-orthologous links.
Annotation of repetitive sequences
We used a combined approach of de novo prediction and homology-based prediction to annotate repetitive sequences in the genome of A. marila. For de novo prediction, we used Tandem Repeats Finder21 (TRF) (v4.09) to detect tandem repeat sequences in the genome. RepeatModeler (v2.0.3), RepeatScout22 (v1.0.6) and RECON (v1.08) were used to build a database of transposable element (TE) family for de nove prediction. RepeatProteinMask and RepeatMasker (v4.1.2-p1) were used for homology prediction with Repbase database23 and Dfam database24, the species parameter used was chicken. Ultimately, we identified 154.94 Mb of repetitive sequences by all methods, accounting for 13.65% of the assembled genome (Table 4).
Gene structure prediction
We used three methods to predict the protein-coding sequences in the genome of A. marila, ab initio prediction, homology-based prediction and RNA-Seq prediction. Three ab initio prediction software were used in the study, Augustus25 (v3.3.2), GlimmerHMM26 (v3.0.4), and Geneid27 (v1.4.5). For the six transcriptomic samples, the raw data were quality controlled and filtered out bad reads using fastp28 (v0.23.1) to obtain clean data. Then, the paired-end reads are assembled using SPAdes29 (v3.15.3). Next, the candidate coding regions in the transcript sequences are identified using TransDecoder30 (v5.5.0), and the sequences were clustered using CD-hit31 (v4.8.1) to remove redundant sequences. Eventually, the obtained protein sequences were used for subsequent prediction along with other downloaded sequences of related species. Genomes and annotation files of six related species19,32,33,34,35,36 (Anser cygnoides, Anas platyrhynchos, Aythya fuligula, Cygnus olor, Cygnus atratus, Gallus gallus) were downloaded from NCBI database to extract the longest transcript and translated them into protein sequences. Their protein sequences were matched to A. marila genome using Spaln37 (v2.4.6), and then the matched sequences were accurately spliced against the homologous protein-coding sequences using GeneWise38 (v2.4.1) software. Homology-based prediction results, RNA-Seq prediction results and ab initio prediction results are integrated using EvidenceModeler39 (EVM) (v1.1.1) to generate gene sets, and including masked repeats as input to the gene predictions. There were 19,257 genes after EVM integration. The BUSCO results showed that complete BUSCOs was 88.2%, fragmented BUSCOs was 1.5%, and missing BUSCOs was 10.3%, which was lower than the BUSCO results of genome (Fig. 4a). Thus, for some single-copy orthologs of aves_odb10, that ware complete in genome but missing or fragmented in predictive gene set, their annotation file were integrated into the gene set. Overall, a total of 19,533 protein-coding genes were predicted in A. marila genome, including 159 prematurely terminated genes and 273 partial genes. The average transcript length of this gene set was 23,126.79 bp, and the average coding sequence (CDS) length was 1,558.71 bp (Table 5). The BUSCO results showed 95.5% of the complete BUSCOs, 0.3% of the fragmented BUSCOs and 4.2% of the missing BUSCOs (Fig. 4b).

The evaluation result of BUSCO. (a) BUSCO result for 19,257 genes. (b) BUSCO result for 19,533 genes.
Gene function annotation
Next, function annotation of predicted genes for A. marila using diamond40 against SwissProt41, TrEMBL42, Non-Redundant Protein Sequence Database (NR), Gene Ontology42 (GO), and Kyoto Encyclopedia of Genes and Genomes Orthology43 (KO) databases with e-value cutoff of 1e-5. The InterPro database44 is also used for function annotation by classifying proteins into families and predicting domains and important sites with Interproscan45 (v5.53). Finally, 15,789 genes were annotated, and accounted for 81.50% of all predicted genes of A. marila (Table 6).
Filtering and verification of gene set
We filtered the predicted gene set, because the number of predicted genes was more than in the six related species, and more than 3,000 genes could not be function annotated. We used OrthoFinder to find the orthologs of A. marila and six related species. There were 4,086 unassigned genes, and 3,421 of them were not annotated in any database (Table 7). Most of these genes (3,417/3,421) were predicted using at least one of the de novo prediction software, and only four genes are supported by other evidence. The BUSCO test was performed after removed unassigned genes without function annotations, the results showed no effect compared with before removed (Table 8; Fig. 4b). These suggests that these genes may only have specific sequences and patterns, rather than real genes46. After filtering the unassigned genes without annotation and 159 prematurely terminated genes, 15,953 genes remain, including 182 partial genes, and 98.96% of them were annotated. The filtered gene set of A. marila was compared with the longest transcript set of related species, it has similar structure (Table 9 and Fig. 5). Except G. gallu, the average transcript length, average CDS length, average exons number, and average intron length of A. marila were smaller than those of others. Simultaneously, the average exon length of A. Marila was the longest among all species. However, these differences were not significant. This may be due to A. Marila has a higher proportion of short genes (Table 5a).

Comparison graph of the prediction gene in A. marila and related species. (a) Gene length distribution and comparison with other species. (b) Exon length distribution and comparison with other species. (c) Exon number distribution and comparison with other species. (d) CDS length distribution and comparison with other species. (e) Intron length distribution and comparison with other species.
Data Records
All sequencing data had been uploaded to NCBI database
The genomic Illumina sequencing data, Nanopore sequencing data and Hi-C sequencing data were deposited at the Sequence Read Archive database of NCBI (accession numbers: SRR2167222547, SRR2167222348, and SRR2167222449).
The transcriptome data of 6 samples were deposited at the Sequence Read Archive database of NCBI (accession numbers: SRR21700073 - SRR2170007850,51,52,53,54,55).
The genome assembly of A. marila were deposited at GenBank database under the accession JAOQIG00000000056.
The annotation results of repeats sequences, gene structure and functional prediction were deposited at the Figshare database57.
Technical Validation
Quality control was performed on Illumina, Nanopore, Hi-C, and RNA sequencing data. The Q20 of Illumina sequencing data were greater than 96% and Q30 were greater than 90%. The Q7 of Nanopore sequencing data were greater than 92%. The Q20 of Hi-C sequencing data were greater than 94% and Q30 were greater than 96%. And the Q20 of RNA sequencing data were greater than 97%, and Q30 were greater than 93%. These evidences suggest that the data are reliable and can be used in subsequent analyses (Tables 10, 11).
Code availability
No special codes or scripts were used in this work, all data processing commands and parameters were executed according to the manuals and protocols of the corresponding bioinformatics software.
References
Liu Y. & Chen S. H. The CNG Field Guide to the Birds of China [China]. Hunan Science & Technology Press, HuNan, (2021).
Youngman, J. A., Flaspohler, D. J., Knowlton, J. L. & Dombroski, L. J. Autumn waterbird migration over Lake Superior: Numbers, species, and timing. Journal of Great Lakes Research 43, 1186–1190 (2017).
Cervencl, A. & Alvarez-Fernandez, S. Winter distribution of Greater Scaup Aythya marila in relation to available food resources. Journal of Sea Research 73, 41–48 (2012).
Marchowski, D. et al. Effectiveness of the European Natura 2000 network to sustain a specialist wintering waterbird population in the face of climate change. Scientific Reports 10, 20286 (2020).
Marchowski, D. et al. The Importance of Non-Native Prey, the Zebra Mussel Dreissena polymorpha, for the Declining Greater Scaup Aythya marila: A Case Study at a Key European Staging and Wintering Site. PLoS One 10, e0145496 (2015).
Cole, T. L. et al. Genomic insights into the secondary aquatic transition of penguins. Nature Communications 13, 3912 (2022).
Luo, H. et al. Genomic insight into the nocturnal adaptation of the black-crowned night heron (Nycticorax nycticorax). BMC Genomics 23, 683 (2022).
Sætre, C. L. C. et al. A Chromosome-Level Genome Assembly of the Reed Warbler (Acrocephalus scirpaceus). Genome Biology and Evolution 13, evab212 (2021).
Recuerda, M. et al. Chromosome-Level Genome Assembly of the Common Chaffinch (Aves: Fringilla coelebs): A Valuable Resource for Evolutionary Biology. Genome Biology and Evolution 13, evab034 (2021).
Zhang, G. et al. Genomics: Bird sequencing project takes off. Nature 522, 34 (2015).
Wille, M., Muradrasoli, S., Nilsson, A. & Järhult, J. D. High Prevalence and Putative Lineage Maintenance of Avian Coronaviruses in Scandinavian Waterfowl. PLoS One 11, e0150198 (2016).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nature Plants 5, 833–845 (2019).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–60 (2009).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21, 245 (2020).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Marçais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLOS Computational Biology 14, e1005944 (2018).
Formenti, G., Uliano Da Silva, M. & Kraus, R. H. S. Aythya fuligula isolate bAytFul2, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:WNMM00000000 (2019).
Chen, C. et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Molecular Plant 13, 1194–1202 (2020).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573–580 (1999).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015).
Storer, J., Hubley, R., Rosen, J., Wheeler, T. J. & Smit, A. F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mobile DNA 12, 2 (2021).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res 33, W465–7 (2005).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–9 (2004).
Parra, G., Blanco, E. & Guigó, R. GeneID in Drosophila. Genome Res 10, 511–515 (2000).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A. & Korobeynikov, A. Using SPAdes De Novo Assembler. Current Protocols in Bioinformatics 70, e102 (2020).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols 8, 1494–1512 (2013).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Li, Q., Zhao, X. & Gao, G. Anser cygnoides breed Sichuan white goose isolate SCWG-2014, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:LABU00000000 (2017).
Li, J. Anas platyrhynchos breed Pekin duck isolate Z2, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JACGAL000000000 (2020).
Formenti, G., Fedrigo, O. & Kraus, R. Cygnus olor isolate bCygOlo1, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:WNMI00000000 (2021).
Cheng, Y., Bruxner, T. J. C. & Karawita, A. C. Cygnus atratus isolate AKBS03, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JABXOC000000000 (2020).
Fedrigo, O., Formenti, G. & Warren, W. Gallus gallus breed Cross of Broiler mother + white leghorn layer father isolate bGalGal1, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAENSK010000000 (2021).
Gotoh, O. A space-efficient and accurate method for mapping and aligning cDNA sequences onto genomic sequence. Nucleic Acids Research 36, 2630–2638 (2008).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res 14, 988–995 (2004).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology 9, R7 (2008).
Buchfink, B., Reuter, K. & Drost, H. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nature Methods 18, 366–368 (2021).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research 28, 45–48 (2000).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nature Genetics 25, 25–29 (2000).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Research 45, D353–D361 (2017).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Research 49, D344–D354 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Scalzitti, N., Jeannin-Girardon, A., Collet, P., Poch, O. & Thompson, J. D. A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms. BMC Genomics 21, 293 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21672225 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21672223 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21672224 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21700073 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21700074 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21700075 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21700076 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21700077 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21700078 (2023).
Zhou, S. Aythya marila isolate BB-2018, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAOQIG000000000 (2023).
Zhou, S. A high-quality chromosomal-level genome assembly of Greater Scaup (Aythya marila). figshare. https://doi.org/10.6084/m9.figshare.22500445.v1 (2023).
Acknowledgements
This research was supported by the National Natural Science Fundation of China (32200407, 31872242, 32070405, 32270444, and 32170530).
Author information
Authors and Affiliations
Contributions
Shengyang Zhou, Tian Xia, and Honghai Zhang designed this study; Xiaodong Gao, Xibao Wang, Lupeng Shi and Yuehuan Dong dissected and collected the samples. Tianshu Lyu and Lidong Wang extracted DNA. Shengyang Zhou, Tian Xia and Xiaodong Gao performed data analysis. Shengyang Zhou wrote the paper. All authors had read, revised, and finally agreed to submit this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, S., Xia, T., Gao, X. et al. A high-quality chromosomal-level genome assembly of Greater Scaup (Aythya marila). Sci Data 10, 254 (2023). https://doi.org/10.1038/s41597-023-02142-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02142-x
