
Abstract
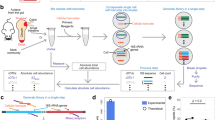
Bacteria often function as a community, called the microbiota, consisting of many different bacterial species. The accurate identification of bacterial types and the simultaneous quantification of the cells of each bacterial type will advance our understanding of microbiota; however, this cannot be performed by conventional 16S rRNA sequencing methods as they only identify and quantify genes, which do not always represent cells. Here, we present a protocol for our developed method, barcoding bacteria for identification and quantification (BarBIQ). In BarBIQ, the 16S rRNA genes of single bacterial cells are amplified and attached to a unique cellular barcode in a droplet. Sequencing the tandemly linked cellular barcodes and 16S rRNA genes from many droplets (representing many cells with unique cellular barcodes) and clustering the sequences using the barcodes determines both the bacterial type for each cell based on 16S rRNA gene and the number of cells for each bacterial type based on the quantity of barcode types sequenced. Single-base accuracy for 16S rRNA sequencing is achieved via the barcodes and by avoiding chimera formation from 16S rRNA genes of different bacteria using droplets. For data processing, an easy-to-use bioinformatic pipeline is available (https://github.com/Shiroguchi-Lab/BarBIQ_Pipeline_V1_2_0). This protocol allows researchers with experience in molecular biology but without bioinformatics experience to perform the process in ~2 weeks. We show the application of BarBIQ in mouse gut microbiota analysis as an example; however, this method is also applicable to other microbiota samples, including those from the mouth and skin, marine environments, soil and plants, as well as those from other terrestrial environments.
Key points
-
This protocol describes barcoding bacteria for identification and quantification, a method for identifying and quantifying bacteria in microbiota samples.
-
Barcoding bacteria for identification and quantification uses a barcoding and single-step droplet-based amplification strategy that enables it to more precisely determine bacterial species than conventional 16S rRNA sequencing methods do.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
The sequencing data generated using the protocol in the relevant study are available from the National Center for Biotechnology Information Sequence Read Archive under accessions PRJNA639639, PRJNA639647, PRJNA780331, and PRJNA780361. A demonstration dataset is available at github (https://github.com/Shiroguchi-Lab/BarBIQ_Pipeline_V1_2_0).
Code availability
The pipeline (v1.2.0) for BarBIQ is available at GitHub (https://github.com/Shiroguchi-Lab/BarBIQ_Pipeline_V1_2_0).
References
Zmora, N., Suez, J. & Elinav, E. You are what you eat: diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 16, 35–56 (2019).
Lamont, R. J., Koo, H. & Hajishengallis, G. The oral microbiota: dynamic communities and host interactions. Nat. Rev. Microbiol. 16, 745–759 (2018).
Chen, Y. E., Fischbach, M. A. & Belkaid, Y. Skin microbiota–host interactions. Nature 553, 427–436 (2018).
Trevathan-Tackett, S. M. et al. A horizon scan of priorities for coastal marine microbiome research. Nat. Ecol. Evol. 3, 1509–1520 (2019).
Jansson, J. K. & Hofmockel, K. S. The soil microbiome—from metagenomics to metaphenomics. Curr. Opin. Microbiol. 43, 162–168 (2018).
Trivedi, P., Leach, J. E., Tringe, S. G., Sa, T. & Singh, B. K. Plant–microbiome interactions: from community assembly to plant health. Nat. Rev. Microbiol. 18, 607–621 (2020).
Thompson, L. R. et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463 (2017).
Jin, J. et al. High-throughput identification and quantification of single bacterial cells in the microbiota. Nat. Commun. 13, 863 (2022).
Olsen, G. J., Lane, D. J., Giovannoni, S. J., Pace, N. R. & Stahl, D. A. Microbial ecology and evolution: a ribosomal RNA approach. Annu. Rev. Microbiol. 40, 337–365 (1986).
Pace, N. R., Stahl, D. A., Lane, D. J. & Olsen, G. J. in Advances in Microbial Ecology 1–55 (Springer, 1986).
Mccaig, A. E., Glover, L. A. & Prosser, J. I. Molecular analysis of bacterial community structure and diversity in unimproved and improved upland grass pastures. Appl. Environ. Microbiol. 65, 1721–1730 (1999).
Kroes, I., Lepp, P. W. & Relman, D. A. Bacterial diversity within the human subgingival crevice. Proc. Natl Acad. Sci. Usa. 96, 14547–14552 (1999).
Navas-Molina, J. A. et al. Advancing our understanding of the human microbiome using QIIME. Methods Enzymol. 531, 371–444 (2013).
Callahan, B. J., McMurdie, P. J. & Holmes, S. P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11, 2639–2643 (2017).
Kembel, S. W., Wu, M., Eisen, J. A. & Green, J. L. Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PLoS Comput. Biol. 8, e1002743 (2012).
Louca, S., Doebeli, M. & Parfrey, L. W. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 6, 41 (2018).
Vandeputte, D. et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 551, 507–511 (2017).
Props, R. et al. Absolute quantification of microbial taxon abundances. ISME J. 11, 584–587 (2017).
Rao, C. et al. Multi-kingdom ecological drivers of microbiota assembly in preterm infants. Nature 591, 633–638 (2021).
Nguyen, N., Warnow, T., Pop, M. & White, B. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. npj Biofilms Microbiomes 2, 16004 (2016).
McDonald, D. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618 (2012).
Cole, J. R. et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642 (2014).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Lan, F., Demaree, B., Ahmed, N. & Abate, A. R. Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding. Nat. Biotechnol. 35, 640–646 (2017).
Chijiiwa, R. et al. Single-cell genomics of uncultured bacteria reveals dietary fiber responders in the mouse gut microbiota. Microbiome 8, 1–14 (2020).
Zheng, W. et al. High-throughput, single-microbe genomics with strain resolution, applied to a human gut microbiome. Science 376, eabm1483 (2022).
Lloréns-Rico, V., Simcock, J. A., Huys, G. R. B. & Raes, J. Single-cell approaches in human microbiome research. Cell 185, 2725–2738 (2022).
Callahan, B. J. et al. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 47, e103 (2019).
Kinoshita, Y., Niwa, H., Uchida-Fujii, E. & Nukada, T. Establishment and assessment of an amplicon sequencing method targeting the 16S-ITS-23S rRNA operon for analysis of the equine gut microbiome. Sci. Rep. 11, 1–12 (2021).
Ogawa, T., Kryukov, K., Imanishi, T. & Shiroguchi, K. The efficacy and further functional advantages of random-base molecular barcodes for absolute and digital quantification of nucleic acid molecules. Sci. Rep. 7, 13576 (2017).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Acknowledgements
We thank K. Fukuhara for helping technical development in library preparation and sequencing. This work was supported by JST/PRESTO JPMJPR15F3 (K.S.), the Nakatani Foundation (K.S.), the Japan Society for the Promotion of Science KAKENHI grant numbers 17K19364 (K.S.) and 26115719 (K.S.), and Incentive Research Project from RIKEN (J.J.). J.J. was supported by the Japan Society for the Promotion of Science Postdoctoral Fellowships for Foreign Researchers in Japan (P17389).
Author information
Authors and Affiliations
Contributions
K.S. developed the initial version of the library preparation and sequencing system for BarBIQ. J.J. developed the pipeline for BarBIQ and the final version of whole BarBIQ. R.Y. developed some steps of the ecDNA separation and gel purification for BarBIQ. J.J., R.Y. and K.S. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
K.S., J.J. and R.Y. are co-inventors on a patent application based on this work filed by RIKEN.
Peer review
Peer review information
Nature Protocols thanks Wataru Suda and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Jin, J. et al. Nat. Commun. 13, 863 (2022): https://doi.org/10.1038/s41467-022-28426-1
Ogawa, T. et al. Sci. Rep. 7, 13576 (2017): https://doi.org/10.1038/s41598-017-13529-3
Supplementary information
Supplementary Information
Supplementary Methods 1 and 2.
Supplementary Table 1
Information of oligomers and barcodes.
Supplementary Table 2
16S rRNA sequences of bacteria in mock community.
Supplementary Table 3
Quantification of bacteria in mock community.
Supplementary Table 4
16S rRNA sequences of bacteria in cecal samples.
Supplementary Table 5
Quantification of bacteria in cecal samples.
Supplementary Data 1
Example of ‘sample sheet’ for a MiSeq run.
Supplementary Data 2
16S rRNA sequences of cecal samples in fasta format with predicted taxonomies.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jin, J., Yamamoto, R. & Shiroguchi, K. High-throughput identification and quantification of bacterial cells in the microbiota based on 16S rRNA sequencing with single-base accuracy using BarBIQ. Nat Protoc 19, 207–239 (2024). https://doi.org/10.1038/s41596-023-00906-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-023-00906-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
