
Abstract
Nonalcoholic steatohepatitis (NASH) might soon become the leading cause of end-stage liver disease and indication for liver transplantation worldwide. Fibrosis severity is the only histological predictor of liver-related morbidity and mortality in NASH identified to date. Moreover, fibrosis regression is associated with improved clinical outcomes. However, despite numerous clinical trials of plausible drug candidates, an approved antifibrotic therapy remains elusive. Increased understanding of NASH susceptibility and pathogenesis, emerging human multiomics profiling, integration of electronic health record data and modern pharmacology techniques hold enormous promise in delivering a paradigm shift in antifibrotic drug development in NASH. There is a strong rationale for drug combinations to boost efficacy, and precision medicine strategies targeting key genetic modifiers of NASH are emerging. In this Perspective, we discuss why antifibrotic effects observed in NASH pharmacotherapy trials have been underwhelming and outline potential approaches to improve the likelihood of future clinical success.
Similar content being viewed by others

Introduction
Nonalcoholic fatty liver disease (NAFLD) is characterized by fat accumulation within the liver (hepatic steatosis) without secondary causes, including substantial alcohol intake, medications or inherited metabolic conditions1. In some individuals, steatosis is associated with cellular injury (ballooned hepatocytes) and lobular inflammation — termed nonalcoholic steatohepatitis (NASH). NASH can lead to progressive fibrosis and sometimes cirrhosis with a consequent risk of hepatic decompensation and hepatocellular carcinoma (HCC) (global incidence 0.5–2.6% per year)2. HCC can also occur in the presence of non-cirrhotic NAFLD, but this is rare (0.1–1.3 per 1,000 patient-years)2. NAFLD most commonly occurs in the context of the metabolic syndrome that is characterized by the presence of two or more of the following conditions: insulin resistance and type 2 diabetes mellitus (T2DM), obesity, hypertension and hypercholesterolaemia (although a ‘lean’ NAFLD variant phenotype is also recognized)3. Notably, there is currently a vigorous debate and an international Delphi consensus process about renaming NAFLD, using terminology perceived as less stigmatizing whilst ensuring positive diagnosis and patient engagement4,5. The global burden of NAFLD (regardless of the definition) is increasing at an alarming rate5. A systematic review and meta-analysis published in 2022 suggested an overall worldwide prevalence of 30%6 to 32.4%7, although there is marked geographical variation. Additionally, strong evidence is emerging linking NAFLD with social deprivation and food insecurity8. Between 1990 and 2017, the number of patients with decompensated NAFLD cirrhosis doubled (although the relabelling of previous ‘cryptogenic’ cirrhosis cases as NAFLD might have contributed), and it is the most rapidly increasing indication for liver transplantation in the USA9,10. These observations pinpoint the urgent need to develop effective therapeutic interventions to stem the rising tide of NAFLD-associated morbidity and mortality.
Although the past four decades have witnessed an explosion in the biological understanding of NAFLD and the mechanisms driving progression to cirrhosis, this has not translated to an approved therapy that can directly modulate fibrosis and improve clinical outcomes in patients (Box 1). By contrast, two antifibrotic drugs are approved for treating idiopathic pulmonary fibrosis (nintedanib and pirfenidone) that can reduce the decline in lung function11. Here, we reflect on the current therapeutic landscape in NAFLD, offer our perspective on the challenges that stand in the way of developing an effective antifibrotic and propose possible solutions that take advantage of new and emerging technologies.
Why is fibrosis important in NASH?
The stages of NASH-related fibrosis range from absent (stage F0) to cirrhosis (stage F4). In general, fibrosis progression to cirrhosis and adverse liver-related outcomes in NASH is slow and unpredictable, with the actual fibrosis progression rate (FPR) uncertain. Nevertheless, the FPR seems to be substantially higher in NASH than in isolated steatosis, corresponding to fibrosis progression by one stage over 7 years and 14 years, respectively12. An FPR of only 0.03 stages was recently calculated from 1,419 participants treated with placebo undergoing per protocol repeat biopsy in 35 randomized controlled trials in NASH13. However, the generalizability of using clinical trial datasets for natural history insights is limited by selection bias and rigid entry criteria. Analysis of paired biopsy series suggests that NASH is a dynamic, bidirectional disease with almost as many patients regressing by up to two stages of fibrosis as seen in biopsy samples taken a year apart as patients progressing14, although the inherent sampling error of liver biopsies might explain this.
Nevertheless, the importance of fibrosis (but no other histological features) in predicting outcomes in NAFLD has been highlighted by several studies15,16,17, with advancing fibrosis stage heightening the risk of future liver-related morbidity (for example, decompensation events and HCC) and liver-related and all-cause mortality. It is, therefore, alarming that in a prospective study published in 2022, conducted in Southern California, 14% of 501 patients with T2DM aged ≥50 years had advanced fibrosis and 6% had cirrhosis18. Furthermore, in a large meta-analysis including 12 cohort studies involving 25,252 patients with established cardiovascular disease, higher levels of non-invasive fibrosis biomarker tests were related to an increased risk of cardiovascular events, cardiovascular mortality and all-cause mortality19. The association of cardiovascular diseases with liver fibrosis is also observed in the general population, although direct mechanistic pathways are not defined20,21. Interestingly, a decision-analytic model simulating the natural history of NAFLD showed that among patients aged 65 years, an estimated 10-year non-liver-related mortality was higher than liver-related mortality in all fibrosis stages22. This raises the intriguing possibility that targeting liver fibrosis might lead to improvements in mortality independent of a reduction in liver-related events, which will require well-conducted, long-term (>5 years at least) clinical trials.
Contrastingly, data from two large (negative) drug trials including 1,135 patients with compensated NAFLD cirrhosis indicated that a reduction in fibrosis is associated with improved clinical outcomes. Specifically, after a median follow-up of 16.6 months, patients whose fibrosis regressed had a sixfold reduction in risk of liver-related events23. Moreover, the strong concordance between histological evidence of cirrhosis regression with decreases in non-invasive measures of fibrosis burden in this study, such as the Enhanced Liver Fibrosis (ELF) score and liver stiffness by transient elastography, underscores the potential of non-invasive tests for longitudinal disease monitoring.
Given that fibrosis progression and regression determine prognosis in NASH, there is an urgent unmet medical need (and multibillion dollar market) for effective antifibrotic therapies.
Weight loss is the cornerstone of NAFLD therapy but hard to achieve and sustain
Weight reduction achieved through lifestyle intervention leads to histological improvements in NASH. Fibrosis regression occurs in patients who manage to lose ≥10% of their body weight24,25, although most find this difficult to achieve and sustain. Numerous diets have been promoted for NAFLD with systematic reviews and randomized controlled trials favouring a Mediterranean diet26, which has been proven to benefit liver, metabolic and cardiovascular health27. Physical activity has multisystem health benefit28 even without weight loss, but recommendations regarding the type, intensity and frequency are less clear. Moreover, when counselling patients, it might be best to discuss simple strategies to increase ‘movement’ rather than ‘exercise’, given that age and comorbidities (obesity, cardiovascular disease, osteoarthritis) might be limiting, and investment in equipment or gym memberships might be unrealistic or off-putting. Even moderate physical activity could reprogramme key pathophysiological mechanisms29. Indeed, observational data have shown that insufficient physical activity is an independent predictor of fibrosis in NAFLD30,31. However, liver inflammation and fibrosis end points have largely been overlooked in lifestyle intervention studies. Notably, in a single-arm 16-week intervention study, diet and moderate-intensity exercise reduced body weight and decreased hepatic venous pressure gradient (HVPG) in 50 patients with overweight, cirrhosis and portal hypertension32. Notwithstanding, the limited success of volitional lifestyle measures in real-world practice has provided a strong rationale for developing disease-modifying therapies.
Currently, bariatric surgery is only an option for a minority of selected patients with severe obesity. However, it can lead to substantial weight loss (between 14% and 25%), durable improvements in histological NASH and fibrosis33, and reduced risk of major adverse liver and cardiovascular outcomes34 and many cancers35. However, delivering such a complex intervention requires substantial resources and is therefore inaccessible to most of the global NAFLD population. Additionally, emerging data suggest that bariatric surgery is associated with an increased prevalence of alcohol use disorder and alcohol-related liver disease; potential candidates should be rigorously assessed before undergoing such surgery36.
A less-invasive approach is insertion of an endoscopic intragastric balloon (IGB) (using an adjustable fluid-filled balloon), which reduces stomach capacity, and delays gastric emptying, thereby inducing weight loss37. An open-label study of IGB placement in 21 patients with fibrotic NASH, in combination with a prescribed diet and exercise programme, induced significant weight loss (mean difference −14.4 ± 7.9 kg; P = 0.01) and metabolic improvements (for example, mean difference in haemoglobin A1c −1.2 ± 0.5; P = 0.02). Although histological NASH improved in 80% of participants (median baseline NAFLD activity score (NAS) of 4 versus median follow-up NAS of 1; P < 0.001) 6 months after IGB placement, effects on hepatic fibrosis were variable38. This study was limited by its small size, lack of a control group and the short duration of follow-up; further studies are clearly needed.
What are the limitations of our current approach to drug development?
Are our preclinical models fit for purpose?
Traditional liver fibrosis drug development starts with basic in vitro assays using isolated hepatic stellate cells (HSCs) (or cell lines) to study phenotypic effects and then employs animal (usually mouse) models to determine efficacy and toxicity. However, this framework has inherent limitations in replicating the complexity of human pathophysiology in vivo.
In vitro and ex vivo models
Simple cell culture models are limited by their non-physiological conditions in lacking both cell–cell and cell–substrate interactions, which influence innate cellular responses in healthy and diseased milieux39. Additionally, primary cells adapt in vitro to favour their optimal phenotypic state for survival in culture conditions, drifting from their normal physiological behaviours in native tissue. Hence mono-cell, bi-cell or even tri-cell culture systems are limited in predicting drug-induced responses in complex, anatomically organized multicellular tissues.
Self-assembling stem cell-derived or organoid-derived tissues and bioprinted tissues simulate some of the complexity of a liver tissue microenvironment; however, these platforms are still in their infancy40,41,42. Advances in designing organoid-engineered human multicellular three-dimensional NASH models have shown promise, recapitulating features of steatosis, inflammation and fibrosis, including a biophysical readout of organoid stiffening that reflects the fibrosis severity43. Moreover, co-culturing pluripotent stem cell (PSC)-derived lineages might provide greater reproducibility in constructing liver-like microstructures. However, these models lack a physiologically relevant vasculature and the immune components of NASH.
Liver-on-a-chip (LoC) systems overcome some of the limitations of organoids. They can be engineered from parenchymal and non-parenchymal cells to recapitulate anatomical features of the liver, such as hepatic zonation and lobe-like structures44. Exposing LoCs containing multiple cell types to lipids has been reported to induce steatosis, hepatocyte ballooning, tumour necrosis factor (TNF) and α-smooth muscle actin expression, indicative of a NASH phenotype45. Moreover, the therapeutic effects of several NASH drug candidates have been demonstrated using these models46.
A caveat in the use of LoCs is the need to dissociate and purify individual cell types from human liver tissues before their reassembly into synthetic liver structures47. This processing inevitably introduces epigenetic alterations and activation of stress pathways that alter the biology of the different cellular constituents. There are also challenges with batch-to-batch variability of LoCs, which might be overcome by using induced PSC lines as a reproducible, standardized source of different cell lineages. Moreover, despite impressive advances, LoCs do not fully recapitulate the physiological, anatomical and cellular complexity of the liver tissue. Alternatively, human ex vivo precision-cut liver slice (PCLSs) systems successfully model fibrogenesis and have demonstrated antifibrotic therapy efficacy48. PCLS retain architectural and zonal spatial contexts of parenchymal and non-parenchymal liver cells, resident Kupffer cells and lymphocytes, and they might represent a valuable tool for studying human innate immunity49.
Animal models
Mouse or rat models offer the advantage that the biological actions, pharmacology, efficacy and toxicity of drugs can be determined in a living animal. However, there is a lack of agreement on which NAFLD model (if any)50 provides the closest approximation of human disease. Genetic models might complicate drug discovery as they will not recapitulate mechanisms in patients. A systematic review catalogued a total of 3,920 NAFLD models (including dietary, chemical, genetic and combination models) across 4,540 published studies51, and found inconsistencies in terminology and study design. In addition, the study demonstrated substantial heterogeneity in replicating human NAFLD phenotypic characteristics, which, when further confounded by interlaboratory variability52, severely compromises the reproducibility of in vivo experiments. NAFLD models incorporating a baseline (pretreatment) liver biopsy resemble clinical trial design, as they control for phenotypic variation through stratification of fibrosis severity and balance treatment allocation whilst enabling assessment of drug response in individual animals53.
To overcome this uncertainty, experts should agree on a standardized model that the industry can adopt, with continued iterative improvements advised by the academic community. An ideal model (besides providing metabolic, immune, fibrogenic, carcinogenic and ideally angiogenic characteristics of human NASH) would pose the relevant clinical challenges of late-stage disease (for example, addressing the association between hepatic fibrosis and increased cardiac-related mortality). Inherent differences between mouse and human immune systems, including variations in innate and adaptive immune compartments, mean that mice will not accurately mimic the immunological components of human NASH. In addition, the sanitized environment and divergent microbiota of laboratory mouse models limit the translational value and reproducibility of the models. This was circumvented by implanting laboratory-strain embryos into wild (non-laboratory) mice to restore natural microbiota and pathogens54. The resulting ‘wilding’ mice replicated human disease more faithfully than conventional models. Moreover, they accurately predicted two (non-NASH) clinical trial failures (an anti-CD28 monoclonal antibody for T cell expansion and an anti-TNF treatment during septic shock), in which conventional mouse studies had predicted success. A further factor is ageing, which increases the risk of fibrosis; most mouse studies use very young animals in which tissue repair and immunity are more robust, and a lifetime of exposure to environmental stressors is not modelled. Humanized mice offer a potential solution and can include human hepatic cells55 and partial reconstitution of a human immune system; however, they are technically challenging and expensive.
Additional limitations of mouse models relate to the lack of consideration of biological sex and genetic and ethnic variations in the human population that influence disease progression. When combined, these limitations considerably affect our faith in animal models to predict drug mechanisms and efficacy, and argue strongly for using human-based models early in the drug development process.
Are we targeting the right mechanisms for fibrosis regression at the right time?
In addition to the technical challenges discussed above, does the current reductionist approach, positioning HSCs as the central instigators of (NASH) fibrosis, remain valid? We still lack concrete evidence that HSCs are the pivotal fibrogenic cells in human NASH; indeed, the cross-tissue heterogeneity of fibroblasts in perturbed states, including fibrosis and cancer, is increasingly recognized56. Moreover, given that fibrosis is a highly conserved wound-healing response, manipulation of HSC activation could lead to unanticipated effects such as impaired hepatic regeneration.
Discoveries concerning the role of cell–cell crosstalk between epithelial, myeloid and mesenchymal cells in fibrosis underscore the limitations of focusing on individual cell types, and suggest that key intercellular networks that trigger or promote disease progression might be tractable treatment targets to inhibit in NASH-related fibrosis57. The functional heterogeneity and dynamic plasticity of cell lineages in the liver, including HSCs58, highlight yet further levels of complexity that are not yet accounted for in our preclinical drug discovery platforms.
Table 1 outlines the current prospective single-agent pharmacotherapy approaches under evaluation. However, one fundamental question remains: are we targeting the correct mechanisms underpinning fibrogenic NASH?
Hepatocellular senescence seems to be a key stimulator of steatosis and fibrosis; however, senolytic drugs have not yet advanced to clinical development. An early-phase trial (NCT05506488) evaluating the combination of the tyrosine kinase inhibitor dasatinib and the antioxidant quercetin, which decreases senescent cells in diabetic kidney disease59, will provide important proof-of-principle for their use in fibrotic NASH60,61,62. Future work will clarify the importance of other fibrogenic mechanisms in NASH63, including extrahepatic drivers (for example, the microbiome and gut–liver axis) and direct hepatic triggers (for example, pro-inflammatory modes of hepatocyte cell death such as ferroptosis64) to establish the safest and most potent approaches.
Another key question is when in the disease process is it best to intervene with antifibrotics (and when is it simply too late)? Certain architectural changes, that are likely to be irreversible (for example, vascularized septae)65,66 plus the reduced regenerative capacity in advanced cirrhosis, argue for pre-cirrhosis as the optimal initial stage at which antifibrotics should be clinically utilized. Otherwise, as evidenced by the multiple trial failures in NAFLD-related cirrhosis, the likelihood of success is low67. As a histological surrogate end point is not established for drug trials in NAFLD-related cirrhosis, a long-term composite clinical outcome end point (including all-cause mortality) is required68. Although decreased portal hypertension (using HVPG) is associated with improved clinical outcomes in patients with both compensated and decompensated cirrhosis69,70, variability is a potential issue, although possibly over-stated69. However, it is not yet accepted by regulatory agencies despite being evaluated as a primary end point in several large studies in NAFLD-related cirrhosis71,72,73.
How can we improve the fibrosis drug discovery process?
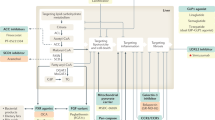
To mitigate against failure, we must look critically at: (1) how we are selecting drug targets and/or combinations; (2) whether we are utilizing preclinical models (and readouts used to determine drug efficacy) with sufficient proximity to patients; and (3) optimizing the discovery biology that can be extracted from human data, including existing clinical trial materials and electronic health record data (Fig. 1). The precision and depth of quantitative biological information generated from single-cell omics technologies, which includes spatial analyses in intact tissues for transcripts, proteins, post-translational modifications and metabolic factors, could pinpoint the key fibrogenic cell interactions (and best potential therapeutic targets) in NASH74. The evolution of artificial intelligence and high-performance computing can now combine carefully archived biopsy and serum or plasma samples with clinical data and focus the drug discovery process on the patient rather than on laboratory-based experiments. Indeed, integrated multimodal multiscale human resources such as the national-level SteatoSITE NAFLD Data Commons could transform drug target discovery and validation. Moreover, the enormous repertoire of human tissue within clinical trial biobanks offers previously unimaginable opportunities, such as the interrogation of those samples using state-of-the-art single-cell omics and artificial intelligence. This process will enable investigators to understand how drugs that have failed in trials might have influenced liver cell biology (for example, HSC activation and hepatocyte function) far beyond the typical end points mandated by regulators and trialists. Such approaches can re-evaluate our pharmacological theory, identify more or less susceptible patient subgroups and uncover new avenues towards a more stratified approach to drug development. The application of advanced techniques to serial biopsy samples (selected to represent the transitions of NAFLD from steatosis to F1–F4 fibrosis stage) might generate surprises regarding the cellular triggers and mediators of fibrosis and the signalling events that influence disease progression and regression. From these serial biopsy studies, novel molecular targets can emerge for which we can have a high degree of confidence in designing drugs with precision for suppressing fibrosis at a specific disease stage.

At all stages of the drug development process (discovery, preclinical testing and clinical evaluation), a plethora of patient-centric approaches are now available to pinpoint the most effective drug candidates for nonalcoholic steatohepatitis (NASH)-related fibrosis. Based on a priori biological knowledge, hypothesis-driven approaches are now further enhanced by multiomics analysis, artificial intelligence (AI) and machine learning methods to improve drug target discovery. Innovative clinical trial designs using hierarchical stratifications (for example, recruit-by-genotype, augmented digital pathology read-outs, and real-world outcomes and end points) offer great potential to enhance success. Together, these refined approaches are likely to yield a paradigm shift in the development and delivery of effective diagnostic tests and treatments to improve liver-related and extra-hepatic outcomes in patients with NASH. NAFLD, nonalcoholic fatty liver disease.
Preclinical drug testing should then ideally employ human tissue models that, as accurately as possible, reflect the pathobiology of fibrosis. Innovations in modelling whole-body physiology (using advanced tissue chip systems to recapitulate interdependent organ systems linked by vascular flow) offer a potential glimpse of the future75. However, in the opinion of the authors, human three-dimensional PCLS models are currently the closest replicants of a human liver, with the caveat that they lack a peripheral blood supply and whole-body context48. An essential advantage of PCLS is that variables influencing drug efficacy and toxicity, such as ageing, biological sex, lifestyle, ethnicity and genetics, can be accounted for at the preclinical stage of drug development. Notwithstanding, establishing a PCLS platform is challenging, requiring a laboratory with proximity to hospital operating theatres to avoid hypoxic damage during the collection, transport and processing of resected tissues. For scalability, the PCLS platform requires miniaturization to at least 96-well format, and the design of fibrosis assays that lend themselves to automation76.
How can we improve clinical trials and enhance their outputs?
A positive outcome on a prespecified end point (either NASH resolution or fibrosis improvement) in a well-designed placebo-controlled phase II trial is likely to increase the probability of a successful phase III study. This was illustrated in phase II studies of obeticholic acid77 and resmetirom78 that were predictive of statistically significant effects on NASH resolution and fibrosis at phase III (REGENERATE trial (n = 1,968, NCT02548351), P = 0.0002 for improvement in fibrosis of one or more stages with 25 mg obeticholic acid versus placebo; and MAESTRO-NASH trial (n = 966, NCT03900429), P < 0.0001 for improvement of one or more stages in fibrosis with 100 mg resmetirom versus placebo)77,78. Nevertheless, multiple clinical factors have been identified as potential obstacles to successful development of drugs for the treatment of NASH79,80. Key issues include a high placebo response rate (estimated at 22%)81, in part attributable to biopsy sampling variability and inexact assessment of fibrosis using standard histopathological metrics; regression to the mean (a statistical phenomenon that can affect data interpretation when the outcome measure has high variability); the Hawthorne effect (the tendency for study participants to change their behaviour simply as a result of being observed); and the lack of a standardized approach to diet and exercise across trials82. Additionally, uncertainty around the optimal duration of drug exposure required for fibrosis improvement (especially if the mechanism of action is indirect) and the existence of multiple (undefined) disease subtypes that might vary in responsiveness to a specific drug further complicate the evaluation of new candidates. There also remains an unknown hierarchy of targets, such that certain mechanisms of action might be too far ‘downstream’. Finally, undisclosed alcohol use, which has been highlighted in patients suspected of having NAFLD, is a potential confounder but could be routinely tested for using highly sensitive and specific direct markers of alcohol intake, such as phosphatidyl ethanol in the blood or ethyl glucuronide in the hair and urine83,84.
Refinement of clinical trial design
Given the track record of drug failures in NASH (owing to lengthy development time, difficulty in recruiting participants, complex disease biology with multiple potential therapeutic targets and a bulging pipeline of early drug candidates worthy of evaluation) there is a strong argument for pursuing new approaches to streamline clinical trials. One approach involves the ongoing quest by large research consortia such as LITMUS and NIMBLE for suitable non-invasive fibrosis biomarkers that could supplant the need for liver biopsies in NASH trials. Biopsies are a barrier to participant recruitment, largely account for the high and costly screening failure rate of trials (~60–70%) and represent an imperfect surrogate end point85.
Another aspect involves optimization of the selection of participants, for example enriching studies for patients harbouring known genetic polymorphisms that are associated with an accelerated disease course and adverse outcomes. Adopting new trial designs (for example, platform trials) to efficiently evaluate multiple drugs through adaptive randomization has been proposed in NASH86. However, unlike in oncology, platform trials are inherently more challenging to implement in a highly heterogeneous and slowly progressive condition such as NASH, in which molecular subtypes (suitable for specific targeted therapies) have not yet been defined and robust end points (for example, survival) generally do not apply.
Finally, NASH drugs are currently assessed in near-ideal test conditions in highly selected non-diverse populations, often with low levels of deprivation. This approach does not represent routine clinical practice. By contrast, pragmatic effectiveness trials measure the benefit that a treatment produces in patients in everyday real-world settings. Current data showing high rates of advanced fibrosis in patients aged ≥50 years with T2DM create a potential opportunity for real-world studies to determine whether metabolic interventions prevent progression to cirrhosis18.
Histological assessment of fibrosis in NAFLD
Medicine regulatory authorities currently require a pathologist to stage biopsies using ordinal scales87. Despite widespread adoption and notionally clearly defined stages, interpretation remains necessary. The development of fibrosis is a dynamic and non-linear continuum. One limitation of ordinal fibrosis scales is that they impose a small number of scores on this continuum88. Observer-defined scoring also introduces an unavoidable intra-observer and interobserver variation of histological features — this negative effect on NAFLD clinical trials has been specifically demonstrated89. Digital quantification ought to offer an objective alternative and solution to observer-related error. However, although an observer can easily compensate for inherent variation in stain quality and intensity, such variation represents a marked challenge for current computational methods, and routinely encountered artefact is also troublesome90. Stain-free methods to quantify liver scarring and NASH features have also been developed and show promise in detecting drug-induced tissue changes that conventional scoring methods miss91.
Irrespective of the method of evaluating a needle core biopsy, the validity of judging the condition of the whole organ from a minute sample is questionable, especially considering that all histological features of the disease (such as NASH) are known to be heterogeneously distributed92. A study in which two biopsies of the right liver lobe were obtained at the same time from 51 patients with a suspected diagnosis of NAFLD demonstrated discordance between the scores for histological features in the biopsy samples from the same patient assigned by a single pathologist. For example, hepatocyte ballooning was not present in one of the paired biopsy samples but present in the other in 9 of the 51 patients (18%). The effect of this histological intrahepatic heterogeneity on the overall identification of features required for a diagnosis of NASH was also examined; ballooning was absent from both biopsy samples from the same patient in 14 patients, present in both biopsies in 28 patients, and present in one biopsy alone in the remaining 9, such that ballooning would have been missed in 9 of the 37 patients (24%) if only a single biopsy had been obtained and assessed. Further, only 30 of the 51 patients had the same assigned NASH-CRN fibrosis score on both of the paired biopsy samples, with a difference of at least one scale point in 21 patients (41%) and two or more scale point differences in biopsy samples from 6 patients. Ultimately, alternative methods that non-invasively and dynamically assess the architecture of the whole organ (for example, MRI) are imperative as a gold standard expert subjective assessment of scarring using ordinal scales results in artefactual information loss and is inherently inexact, and ‘objective’ computational methods struggle with real-world conditions93. Indeed, a 30% reduction in MRI proton density fat fraction is associated with a five times higher odds of NASH resolution94. Given the challenges associated with liver biopsy, the European Medicines Agency and US Food and Drug Administration (FDA) support the development of non-invasive biomarkers to potentially replace histology as less burdensome and more reliable end points in future trials87.
Future therapeutic considerations
Combination therapies
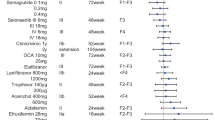
Many drug monotherapies continue to be evaluated in phase II trials, with specific compounds targeting a range of disease mechanisms, including insulin resistance, lipid metabolism, lipotoxicity and oxidative stress, inflammation, cell death and fibrosis95. The following agents are being tested for their effectiveness in improving NASH-related fibrosis in phase III trials: aramchol (stearoyl-CoA desaturase 1 inhibitor), resmetirom (thyroid hormone receptor-β agonist), obeticholic acid (farnesoid X receptor (FXR) agonist), lanifibranor (pan-peroxisome proliferator-activated receptor agonist) in non-cirrhotic NASH, and belapectin (galectin 3 inhibitor) in NASH-related cirrhosis (although the primary end point is the prevention of oesophageal varices rather than improvement in fibrosis). These compounds, including their mechanism of action, are listed in Table 1.
To date, the effect of single agents on histological fibrosis has been modest. Although a press release from the pivotal phase III MAESTRO-NASH trial of resmetirom (NCT04197479) reported positive topline data for NASH and fibrosis primary efficacy end points, the focus in the field is increasingly shifting towards combination therapies (Table 2).
An ideal drug combination should be safe, well tolerated and possess orthogonal activities to amplify treatment efficacy. So far, the choice of combination regimens has been serendipitous rather than underpinned by hard science; initial trials have only shown limited effects on non-invasive fibrosis biomarkers96,97 but will inform future development. In addition, novel computational and high-throughput preclinical combinatorial screening methods could be employed to improve the likelihood of clinical success75. Key challenges will be identifying which patient subphenotypes are likely to respond best to certain combinations, defining the chronology of regimens (that is, overlapping, outlasting or additive), and navigating a more complex route to regulatory approval98. Combinations might also be utilized strategically to mitigate unwanted effects, such as dyslipidaemia associated with FXR agonists and acetyl-CoA carboxylase inhibitors. These iatrogenic effects might potentially increase cardiovascular risk in an already atherogenic patient population99,100.
A precision medicine paradigm for NASH therapy
As our ability to assimilate a holistic picture of NAFLD evolves (based on demographics, comorbidities, disease staging, and detailed genetic and metabolic risk assessment — incorporating multiomics), treatment approaches could increasingly be tailored to individual patients. In parallel, genetic and molecular advances have paved the way for novel interventions, including precision medicines that can modulate the activity of specific genes associated with NASH. Genetically validated targets include polymorphisms associated with high-risk NASH phenotypes, such as variants associated with all-cause cirrhosis (for example, PNPLA3, TM6SF2, HSD17B13 and MARC1)101,102. For example, loss-of-function variants in HSD17B13 are associated with reduced risk of progression to NASH and cirrhosis103 and hepatocyte-targeted small interfering RNA (siRNA)-mediated knockdown of HSD17B13, mimicking genetic loss of function, is currently being tested in early clinical trials in NASH-related fibrosis. ASO-mediated silencing of Pnpla3 improved all features of NAFLD, including fibrosis, in mice fed a NASH-inducing diet104 and early studies of an investigational ASO medicine (AZD2693) in patients with pre-cirrhotic NASH (NCT04483947), homozygous for the PNPLA3*148M risk allele, are ongoing.
Cell-specific therapies for fibrosis
The next generation of therapeutics for fibrosis in NASH might also include cell-specific approaches such as vitamin A-coupled lipid nanoparticles delivering siRNA against HSC heat shock protein 47 (a collagen chaperone), which reversed advanced fibrosis in mouse models105 and showed initial antifibrotic effects in patients with successfully eradicated hepatitis C and advanced fibrosis106. The investigational agent BMS-986263 is now being evaluated in NAFLD-related cirrhosis (NCT04267393).
Additionally, advances in immunotherapy offer increasingly targeted approaches; for example, chimeric antigen receptor (CAR) T cells might act as ‘guided missiles’ to treat fibrotic diseases. These tools have been developed to engage specific receptor moieties such as urokinase-type plasminogen activator receptor-specific CAR T cells that ablated senescent HSCs and reduced fibrosis in a mouse NASH model107. Moreover, transient antifibrotic CAR T cells were generated in vivo by delivering modified mRNA in T cell-targeted lipid nanoparticles108. When administered to mice with cardiac fibrosis (these in vivo-reprogrammed CAR T cells were designed to bind to fibroblast activation protein on activated cardiac fibroblasts), these so-called FAPCAR T cells reduced cardiac fibrosis and improved cardiac function. This transient in vivo approach potentially circumvents the risk of persistent antifibrotic CAR T cells in the setting of future injuries and the ability to titrate dosing and re-dose as needed.
Further fine-tuning will be required to determine the best context-specific cell-surface targets, to minimize off-target effects and the disruption of effective tissue regeneration and repair107,108.
Conclusions
In 2008 the renowned biologist Sydney Brenner said: “We don’t have to look for a model organism anymore. Because we are the model organisms”. These words have proved prescient, and now that NASH is giving up its pivotal secrets through ever more sophisticated human-based technologies and datasets, the future of drug development in NASH looks brighter109. However, in contrast to oncology, where several examples of FDA-approved drugs and companion diagnostics are embedded in clinical practice, navigating the path towards precision medicine for a complex disease like NASH is more challenging. Moreover, the cause of death in most patients with NASH is cardiovascular disease or non-hepatic malignancy rather than liver disease, so individualized outcome and treatment prediction models are needed110,111.
Although advanced methods of liver tissue analysis will be useful for target identification, the need for liver biopsies to stage patients for initiation of treatment or assessment of efficacy is likely to be superseded by reliable non-invasive tests. Moving forward, we anticipate more efficient clinical trial design, including genotype-driven approaches, with the approval of new drug monotherapies or combination regimens for subgroups of patients with specific genetic or metabolic risk profiles. Meanwhile, the cost-effectiveness and affordability of future NASH therapies (especially when lined up against diet and exercise) remain the elephant in the room112. The initial wave of new NASH drugs will face unchartered reimbursement territory and could encounter strict prior authorization from payers tied to the (histological) enrolment criteria of pivotal trials.
Finally, drug repurposing was at the forefront of efforts to identify effective therapies during the COVID-19 pandemic and highlighted the need for a standardized translational drug development platform113. Several studies114,115 indicate that similar approaches could deliver unexpected success in the face of a NAFLD pandemic.
References
Loomba, R., Friedman, S. L. & Shulman, G. I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184, 2537–2564 (2021).
Huang, D. Q., El-Serag, H. B. & Loomba, R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 18, 223–238 (2021).
Albhaisi, S., Chowdhury, A. & Sanyal, A. J. Non-alcoholic fatty liver disease in lean individuals. JHEP Rep. 1, 329–341 (2019).
Eslam, M. et al. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 158, 1999–2014.e1 (2020).
Lazarus, J. V. et al. Advancing the global public health agenda for NAFLD: a consensus statement. Nat. Rev. Gastroenterol. Hepatol. 19, 60–78 (2022).
Younossi, Z. M. et al. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review. Hepatology 77, 1335–1347 (2023).
Riazi, K. et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 7, 851–861 (2022).
Tamargo, J. A. et al. Food insecurity is associated with magnetic resonance-determined nonalcoholic fatty liver and liver fibrosis in low-income, middle-aged adults with and without HIV. Am. J. Clin. Nutr. 113, 593–601 (2021).
Heimbach, J. Debate: A bridge too far – liver transplantation for nonalcoholic steatohepatitis will overwhelm the organ supply. Liver Transplant. 20, S32–S37 (2014).
Shaker, M., Tabbaa, A., Albeldawi, M. & Alkhouri, N. Liver transplantation for nonalcoholic fatty liver disease: new challenges and new opportunities. World J. Gastroenterol. 20, 5320–5330 (2014).
Maher, T. M. & Strek, M. E. Antifibrotic therapy for idiopathic pulmonary fibrosis: time to treat. Respir. Res. 20, 205 (2019).
Singh, S. et al. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 13, 643–654.e9 (2015).
Roskilly, A. et al. Fibrosis progression rate in a systematic review of placebo-treated nonalcoholic steatohepatitis. Liver Int. 41, 982–995 (2021).
Kleiner, D. E. et al. Association of histologic disease activity with progression of nonalcoholic fatty liver disease. JAMA Netw. Open 2, e1912565 (2019).
Angulo, P. et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 149, 389–397.e10 (2015).
Sanyal, A. J. et al. Prospective study of outcomes in adults with nonalcoholic fatty liver disease. N. Engl. J. Med. 385, 1559–1569 (2021).
Vilar-Gomez, E. et al. Fibrosis severity as a determinant of cause-specific mortality in patients with advanced nonalcoholic fatty liver disease: a multi-national cohort study. Gastroenterology 155, 443–457.e17 (2018).
Ajmera, V. et al. A prospective study on the prevalence of NAFLD, advanced fibrosis, cirrhosis and hepatocellular carcinoma in people with type 2 diabetes. J. Hepatol. https://doi.org/10.1016/J.JHEP.2022.11.010 (2022).
Yan, Z. et al. Liver fibrosis scores and prognosis in patients with cardiovascular diseases: a systematic review and meta-analysis. Eur. J. Clin. Invest. 52, e13855 (2022).
Ostovaneh, M. R. et al. Association of liver fibrosis with cardiovascular diseases in the general population: the Multi-Ethnic Study of Atherosclerosis (MESA). Circ. Cardiovasc. Imaging 11, e007241 (2018).
Huang, D. Q. et al. Shared mechanisms between cardiovascular disease and NAFLD. Semin. Liver Dis. 42, 455–464 (2022).
Chhatwal, J. et al. Analysis of a simulation model to estimate long-term outcomes in patients with nonalcoholic fatty liver disease. JAMA Netw. Open 5, E2230426 (2022).
Sanyal, A. J. et al. Cirrhosis regression is associated with improved clinical outcomes in patients with nonalcoholic steatohepatitis. Hepatology 75, 1235–1246 (2022).
Promrat, K. et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 51, 121–129 (2010).
Vilar-Gomez, E. et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 149, 367–378.e5 (2015).
Zelber-Sagi, S., Salomone, F. & Mlynarsky, L. The Mediterranean dietary pattern as the diet of choice for non-alcoholic fatty liver disease: evidence and plausible mechanisms. Liver Int. 37, 936–949 (2017).
Dinu, M., Pagliai, G., Casini, A. & Sofi, F. Mediterranean diet and multiple health outcomes: an umbrella review of meta-analyses of observational studies and randomised trials. Eur. J. Clin. Nutr. 72, 30–43 (2018).
Booth, F. W., Roberts, C. K. & Laye, M. J. Lack of exercise is a major cause of chronic diseases. Compr. Physiol. 2, 1143–1211 (2012).
Bianchi, A. et al. Moderate exercise inhibits age-related inflammation, liver steatosis, senescence, and tumorigenesis. J. Immunol. 206, 904–916 (2021).
Kim, D., Konyn, P., Cholankeril, G. & Ahmed, A. Physical activity is associated with nonalcoholic fatty liver disease and significant fibrosis measured by FibroScan. Clin. Gastroenterol. Hepatol. 20, e1438–e1455 (2022).
Sherry, A. et al. Physical activity is inversely associated with hepatic fibro-inflammation: a population-based cohort study using UK Biobank data. JHEP Rep. 5, 100622 (2022).
Berzigotti, A. et al. Effects of an intensive lifestyle intervention program on portal hypertension in patients with cirrhosis and obesity: the SportDiet study. Hepatology 65, 1293–1305 (2017).
Lassailly, G. et al. Bariatric surgery provides long-term resolution of nonalcoholic steatohepatitis and regression of fibrosis. Gastroenterology 159, 1290–1301.e5 (2020).
Aminian, A. et al. Association of bariatric surgery with major adverse liver and cardiovascular outcomes in patients with biopsy-proven nonalcoholic steatohepatitis. J. Am. Med. Assoc. 326, 2031–2042 (2021).
Rustgi, V. K. et al. Bariatric surgery reduces cancer risk in adults with nonalcoholic fatty liver disease and severe obesity. Gastroenterology 161, 171–184.e10 (2021).
Alvarado-Tapias, E. et al. Bariatric surgery is associated with alcohol-related liver disease and psychiatric disorders associated with AUD. Obes. Surg. 33, 1494–1505 (2023).
Barrichello, S. et al. The effect of the intra-gastric balloon on gastric emptying and the DeMeester score. Obes. Surg. 30, 38–45 (2020).
Bazerbachi, F. et al. Intragastric balloon placement induces significant metabolic and histologic improvement in patients with nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 19, 146–154.e4 (2021).
Kaur, S. et al. In vitro models for the study of liver biology and diseases: advances and limitations. Cell Mol. Gastroenterol. Hepatol. 15, 559–571 (2023).
Mun, S. J. et al. Generation of expandable human pluripotent stem cell-derived hepatocyte-like liver organoids. J. Hepatol. 71, 970–985 (2019).
Guan, Y. et al. A human multi-lineage hepatic organoid model for liver fibrosis. Nat. Commun. 12, 6138 (2021).
Kizawa, H., Nagao, E., Shimamura, M., Zhang, G. & Torii, H. Scaffold-free 3D bio-printed human liver tissue stably maintains metabolic functions useful for drug discovery. Biochem. Biophys. Rep. 10, 186–191 (2017).
Wang, S. X., Yan, J. S. & Chan, Y. S. Advancements in MAFLD modeling with human cell and organoid models. Int. J. Mol. Sci. 23, 11850 (2022).
Du, K. et al. Modeling nonalcoholic fatty liver disease on a liver lobule chip with dual blood supply. Acta Biomater. 134, 228–239 (2021).
Freag, M. S. et al. Human nonalcoholic steatohepatitis on a chip. Hepatol. Commun. 5, 217–233 (2020).
Deguchi, S. & Takayama, K. State-of-the-art liver disease research using liver-on-a-chip. Inflamm. Regen. 42, 62 (2022).
Feaver, R. E. et al. Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. JCI Insight 1, e90954 (2016).
Paish, H. L. et al. A bioreactor technology for modeling fibrosis in human and rodent precision-cut liver slices. Hepatology 70, 1377–1391 (2019).
Wu, X. et al. Precision-cut human liver slice cultures as an immunological platform. J. Immunol. Methods 455, 71–79 (2018).
Teufel, A. et al. Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology 151, 513–525.e0 (2016).
Im, Y. R. et al. A systematic review of animal models of NAFLD finds high-fat, high-fructose diets most closely resemble human NAFLD. Hepatology 74, 1884–1901 (2021).
von Herrath, M. et al. Case reports of pre-clinical replication studies in metabolism and diabetes. Cell Metab. 29, 795–802 (2019).
Oldham, S., Rivera, C., Boland, M. L. & Trevaskis, J. L. Incorporation of a survivable liver biopsy procedure in mice to assess non-alcoholic steatohepatitis (NASH) resolution. J. Vis. Exp. https://doi.org/10.3791/59130 (2019).
Rosshart, S. P. et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365, eaaw4361 (2019).
Bissig-Choisat, B. et al. A human liver chimeric mouse model for non-alcoholic fatty liver disease. JHEP Rep. 3, 100281 (2021).
Buechler, M. B. et al. Cross-tissue organization of the fibroblast lineage. Nature 593, 575–579 (2021).
Wallace, S. J., Tacke, F., Schwabe, R. F. & Henderson, N. C. Understanding the cellular interactome of non-alcoholic fatty liver disease. JHEP Rep. 4, 100524 (2022).
Dobie, R. et al. Single-cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis. Cell Rep. 29, e8 (2019).
Hickson, L. T. J. et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine 47, 446–456 (2019).
Wang, T. W. et al. Blocking PD-L1–PD-1 improves senescence surveillance and ageing phenotypes. Nature 611, 358–364 (2022).
Ogrodnik, M. et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 8, 15691 (2017).
Meijnikman, A. S. et al. Evaluating causality of cellular senescence in non-alcoholic fatty liver disease. JHEP Rep. 3, 100301 (2021).
Schwabe, R. F., Tabas, I. & Pajvani, U. B. Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology 158, 1913–1928 (2020).
Tsurusaki, S. et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 10, 449 (2019).
Chedid, A. Regression of human cirrhosis. Arch. Pathol. Lab. Med. 124, 1591–1593 (2000).
Desmet, V. J. & Roskams, T. Cirrhosis reversal: a duel between dogma and myth. J. Hepatol. 40, 860–867 (2004).
Fallowfield, J. A., Jimenez-Ramos, M. & Robertson, A. Emerging synthetic drugs for the treatment of liver cirrhosis. Expert. Opin. Emerg. Drugs 26, 149–163 (2021).
European Medicines Agency. Reflection paper on regulatory requirements for the development of medicinal products for chronic non-infectious liver diseases (PBC, PSC, NASH) (EMA, 2018).
Bai, W. et al. Test–retest reliability and consistency of HVPG and impact on trial design: a study in 289 patients from 20 randomized controlled trials. Hepatology 74, 3301–3315 (2021).
Turco, L. et al. Lowering portal pressure improves outcomes of patients with cirrhosis, with or without ascites: a meta-analysis. Clin. Gastroenterol. Hepatol. 18, 313–327.e6 (2020).
Chalasani, N. et al. Effects of belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology 158, 1334–1345.e5 (2020).
Garcia-Tsao, G. et al. Randomized placebo-controlled trial of emricasan for non-alcoholic steatohepatitis-related cirrhosis with severe portal hypertension. J. Hepatol. 72, 885–895 (2020).
Harrison, S. A. et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology 155, 1140–1153 (2018).
Adler, M. et al. Principles of cell circuits for tissue repair and fibrosis. iScience 23, 100841 (2020).
Ronaldson-Bouchard, K. et al. A multi-organ chip with matured tissue niches linked by vascular flow. Nat. Biomed. Eng. 6, 351–371 (2022).
Govaere, O. et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 12, eaba4448 (2020).
Younossi, Z. M. et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 394, 2184–2196 (2019).
Harrison, S. A. et al. Effects of resmetirom on noninvasive endpoints in a 36-week phase 2 active treatment extension study in patients with NASH. Hepatol. Commun. 5, 573–588 (2021).
Ratziu, V. & Friedman, S. L. Why do so many NASH trials fail? Gastroenterology https://doi.org/10.1053/J.GASTRO.2020.05.046 (2020).
Drenth, J. P. H. & Schattenberg, J. M. The nonalcoholic steatohepatitis (NASH) drug development graveyard: established hurdles and planning for future success. Expert. Opin. Investig. Drugs 29, 1365–1375 (2020).
Rowe, I. A. & Parker, R. The placebo response in randomized trials in nonalcoholic steatohepatitis simply explained. Clin. Gastroenterol. Hepatol. 20, e564–e572 (2022).
Glass, O. et al. Standardisation of diet and exercise in clinical trials of NAFLD-NASH: recommendations from the Liver Forum. J. Hepatol. 73, 680–693 (2020).
Staufer, K. et al. Ethyl glucuronide in hair detects a high rate of harmful alcohol consumption in presumed non-alcoholic fatty liver disease. J. Hepatol. 77, 918–930 (2022).
Viel, G. et al. Phosphatidylethanol in blood as a marker of chronic alcohol use: a systematic review and meta-analysis. Int. J. Mol. Sci. 13, 14788–14812 (2012).
Harrison, S. A., Allen, A. M., Dubourg, J., Noureddin, M. & Alkhouri, N. Challenges and opportunities in NASH drug development. Nat. Med. 29, 562–573 (2023).
Pericàs, J. M. et al. Platform trials to overcome major shortcomings of traditional clinical trials in non-alcoholic steatohepatitis? Pros and cons. J. Hepatol. https://doi.org/10.1016/J.JHEP.2022.09.021 (2022).
Loomba, R. et al. Expert panel review to compare FDA and EMA guidance on drug development and endpoints in nonalcoholic steatohepatitis. Gastroenterology 162, 680–688 (2022).
Kleiner, D. E. et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321 (2005).
Davison, B. A. et al. Suboptimal reliability of liver biopsy evaluation has implications for randomized clinical trials. J. Hepatol. 73, 1322–1332 (2020).
Astbury, S. et al. Reliable computational quantification of liver fibrosis is compromised by inherent staining variation. J. Pathol. Clin. Res. 7, 471–481 (2021).
Naoumov, N. V. et al. Digital pathology with artificial intelligence analyses provides greater insights into treatment-induced fibrosis regression in NASH. J. Hepatol. 77, 1399–1409 (2022).
Ratziu, V. et al. Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology 128, 1898–1906 (2005).
Loomba, R. et al. Multicenter validation of association between decline in MRI-PDFF and histologic response in NASH. Hepatology 72, 1219–1229 (2020).
Huang, D. Q. et al. Clinical utility of combined MRI-PDFF and ALT response in predicting histologic response in nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. https://doi.org/10.1016/J.CGH.2022.08.036 (2022).
Ratziu, V., Francque, S. & Sanyal, A. Breakthroughs in therapies for NASH and remaining challenges. J. Hepatol. 76, 1263–1278 (2022).
Loomba, R. et al. Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology 73, 625–643 (2021).
Alkhouri, N. et al. Safety and efficacy of combination therapy with semaglutide, cilofexor and firsocostat in patients with non-alcoholic steatohepatitis: a randomised, open-label phase II trial. J. Hepatol. 77, 607–618 (2022).
Ianevski, A., Timonen, S., Kononov, A., Aittokallio, T. & Giri, A. K. SynToxProfiler: an interactive analysis of drug combination synergy, toxicity and efficacy. PLoS Comput. Biol. 16, e1007604 (2020).
Lawitz, E. J. et al. Fenofibrate mitigates hypertriglyceridemia in nonalcoholic steatohepatitis patients treated with cilofexor/firsocostat. Clin. Gastroenterol. Hepatol. https://doi.org/10.1016/J.CGH.2021.12.044 (2022).
Pockros, P. J. et al. CONTROL: a randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. 39, 2082–2093 (2019).
Parisinos, C. A. et al. Genome-wide and Mendelian randomisation studies of liver MRI yield insights into the pathogenesis of steatohepatitis. J. Hepatol. 73, 241–251 (2020).
Emdin, C. A. et al. A missense variant in mitochondrial amidoxime reducing component 1 gene and protection against liver disease. PLoS Genet. 16, e1008629 (2020).
Abul-Husn, N. S. et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N. Engl. J. Med. 378, 1096–1106 (2018).
Lindén, D. et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol. Metab. 22, 49–61 (2019).
Sato, Y. et al. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat. Biotechnol. 26, 431–442 (2008).
Lawitz, E. J. et al. BMS-986263 in patients with advanced hepatic fibrosis: 36-week results from a randomized, placebo-controlled phase 2 trial. Hepatology 75, 912–923 (2022).
Amor, C. et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020).
Rurik, J. G. et al. CAR T cells produced in vivo to treat cardiac injury. Science 375, 91–96 (2022).
Wang, S. et al. An autocrine signaling circuit in hepatic stellate cells underlies advanced fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 15, eadd3949 (2023).
Baratta, F. et al. Heterogeneity of non-alcoholic fatty liver disease (NAFLD): implication for cardiovascular risk stratification. Atherosclerosis 357, 51–59 (2022).
Herring, W. L. et al. Evaluation of emerging NASH therapies: the impact of treatment efficacy profiles on long-term health outcomes. J. Comp. Eff. Res. https://doi.org/10.2217/CER-2021-0194 (2022).
Bell, J. On the path to patients, NASH drugs may hit a payer roadblock. BioPharma Dive https://www.biopharmadive.com/news/nash-drugs-payer-pushback-price-diet-exercise/554245/ (2019).
Assmus, F. et al. Need for a standardized translational drug development platform: lessons learned from the repurposing of drugs for COVID-19. Microorganisms 10, 1639 (2022).
Bhattacharya, D. et al. Repositioning of a novel GABA-B receptor agonist, AZD3355 (lesogaberan), for the treatment of non-alcoholic steatohepatitis. Sci. Rep. 11, 20827 (2021).
Lei, Y. et al. Disulfiram ameliorates nonalcoholic steatohepatitis by modulating the gut microbiota and bile acid metabolism. Nat. Commun. 13, 6862 (2022).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04767529?term=efruxifermin&draw=2&rank=2 (2023).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT05039450?term=efruxifermin&draw=2&rank=1 (2023).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04929483?term=Enliven&draw=2&rank=1 (2022).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04210245?term=alpine+4&draw=2&rank=1 (2022).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04321343?term=NCT04321343&draw=2&rank=1 (2023).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04906421?term=FASCINATE-2&draw=2&rank=1 (2023).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/results/NCT04480710?term=AMBITION&draw=2&rank=1 (2022).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04849728?term=NATiV3&draw=2&rank=1 (2023).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02548351?term=REGENERATE+NASH&draw=2&rank=1 (2023).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03439254?term=REVERSE+NASH&draw=2&rank=1 (2022).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03900429?term=MAESTRO+NAsh&draw=2&rank=1 (2022).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT05500222?term=MAESTRO+NAsh&draw=1&rank=2 (2023).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04822181?term=ESSENCE+NASH&draw=2&rank=1 (2023).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04104321?term=armor+nash&draw=2&rank=1 (2022).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04365868 (2023).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT05506488?cond=NASH+with+Fibrosis&draw=2&rank=2 (2023).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04321031?term=PF-06865571+nash&draw=2&rank=1 (2023).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04065841?term=elivate+nash&draw=2&rank=1 (2022).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT05327127?cond=NASH+with+Fibrosis&draw=2&rank=24 (2022).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04971785?term=NCT04971785&draw=2&rank=1 (2023).
Friedman, S. L., Roll, F. J., Boyles, J. & Bissell, D. M. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc. Natl Acad. Sci. USA 82, 8681–8685 (1985).
Arenson, D. M., Friedman, S. L. & Bissell, D. M. Formation of extracellular matrix in normal rat liver: lipocytes as a major source of proteoglycan. Gastroenterology 95, 441–447 (1988).
Maher, J. J., Friedman, S. L., Roll, F. J. & Bissell, D. M. Immunolocalization of laminin in normal rat liver and biosynthesis of laminin by hepatic lipocytes in primary culture. Gastroenterology 94, 1053–1062 (1988).
Iredale, J. P. et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J. Clin. Invest. 102, 538–549 (1998).
Issa, R. et al. Apoptosis of hepatic stellate cells: involvement in resolution of biliary fibrosis and regulation by soluble growth factors. Gut 48, 548–557 (2001).
Wright, M. C. et al. Gliotoxin stimulates the apoptosis of human and rat hepatic stellate cells and enhances the resolution of liver fibrosis in rats. Gastroenterology 121, 685–698 (2001).
Author information
Authors and Affiliations
Contributions
D.A.M., P.N.B. and J.A.F. researched data for the article. All authors contributed substantially to discussion of the content. D.A.M., P.N.B., A.M.E., T.J.K. and J.A.F. wrote the article. All authors reviewed and/or edited the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
P.N.B. has received educational honoraria from Takeda. P.N.B. also served as a consultant and is employed by Resolution Therapeutics. J.A.F. serves as a consultant or advisory board member for Ipsen, Redx Pharma, River 2 Renal Corp., Stimuliver, Galecto Biotech, Resolution Therapeutics and Global Clinical Trial Partners. J.A.F. has received research funding from Genentech and Intercept Pharmaceuticals. T.J.K. serves as a consultant or received speaker fees from Resolution Therapeutics, Clinovate Health, Perspectum Diagnostics and Incyte Corporation. A.M.E. serves as a consultant or received speaker fees from GSK, Gilead and SOBI. D.A.M. is a director, major shareholder and salaried CSO of FibroFind Ltd, has received research funding from GSK and collaborates with AstraZeneca on novel treatments for advanced hepatocellular carcinoma. R.L. serves as a consultant to Aardvark Therapeutics, Altimmune, Anylam/Regeneron, Amgen, Arrowhead Pharmaceuticals, AstraZeneca, Bristol Myers Squibb, CohBar, Eli Lilly, Galmed, Gilead, Glympse bio, Hightide, Inipharma, Intercept, Inventiva, Ionis, Janssen Inc., Madrigal, Metacrine Inc., NGM Biopharmaceuticals, Novartis, Novo Nordisk, Merck, Pfizer, Sagimet, Theratechnologies, 89 bio, Terns Pharmaceuticals and Viking Therapeutics. In addition, R.L.’s institutions received research grants from Arrowhead Pharmaceuticals, AstraZeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Eli Lilly, Galectin Therapeutics, Galmed Pharmaceuticals, Gilead, Intercept, Hanmi, Intercept, Inventiva, Ionis, Janssen, Madrigal Pharmaceuticals, Merck, NGM Biopharmaceuticals, Novo Nordisk, Merck, Pfizer, Sonic Incytes and Terns Pharmaceuticals. R.L. is also co-founder of LipoNexus Inc.
Peer review
Peer review information
Nature Reviews Gastroenterology & Hepatology thanks Yury Popov, Salvatore Petta and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
LITMUS: https://litmus-project.eu/
NCT02548351: https://clinicaltrials.gov/ct2/show/NCT02548351
NCT03900429: https://clinicaltrials.gov/ct2/show/NCT03900429
NCT04197479: https://clinicaltrials.gov/ct2/show/NCT04197479
NCT04267393: https://clinicaltrials.gov/ct2/show/NCT04267393
NCT04483947: https://clinicaltrials.gov/ct2/show/NCT04483947
NCT05506488: https://clinicaltrials.gov/ct2/show/NCT05506488
NIMBLE: https://www.fnih.org/our-programs/biomarkers-consortium/programs/nimble
SteatoSITE: https://steatosite.com/
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Brennan, P.N., Elsharkawy, A.M., Kendall, T.J. et al. Antifibrotic therapy in nonalcoholic steatohepatitis: time for a human-centric approach. Nat Rev Gastroenterol Hepatol 20, 679–688 (2023). https://doi.org/10.1038/s41575-023-00796-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41575-023-00796-x
This article is cited by
-
Emerging advanced approaches for diagnosis and inhibition of liver fibrogenesis
The Egyptian Journal of Internal Medicine (2024)
-
Integrating social nutrition principles into the treatment of steatotic liver disease
Communications Medicine (2023)
-
Comprehensive analysis of epigenetic and epitranscriptomic genes’ expression in human NAFLD
Journal of Physiology and Biochemistry (2023)
