
Abstract
Long-wavelength near-infrared light-emitting diodes (NIR LEDs) with peak emission wavelengths beyond 900 nm are of critical importance for various applications including night vision, biomedical imaging, sensing and optical communications. However, the low radiance and poor operational stability of state-of-the-art long-wavelength NIR LEDs based on soft materials remain the most critical factors limiting their practical applications. Here we develop NIR LEDs emitting beyond 900 nm with improved performance through the rational manipulation of p doping in all-inorganic tin perovskites (CsSnI3) by retarding and controlling the crystallization process of perovskite precursors in tin-rich conditions. The resulting NIR LEDs exhibit a peak emission wavelength at 948 nm, high radiance of 226 W sr−1 m−2 and long operational half-lifetime of 39.5 h at a high constant current density of 100 mA cm−2. Our demonstration of efficient and stable NIR LEDs operating at high current densities may also open up new opportunities towards electrically pumped lasers.
Similar content being viewed by others


Main
Near-infrared (NIR) light-emitting diodes (LEDs) are of great interest for diverse applications including night vision, biomedical imaging, sensing and optical communications1,2,3,4. Present-day NIR emitters primarily rely on III–V epitaxial hard inorganic semiconductors (such as GaAs and AlInGaAs)5,6. However, the harsh and complicated high-temperature processing increases their cost and limits their integration in multifunctional photonic platforms. Therefore, great efforts have been made to develop alternative soft NIR emitters that can overcome these drawbacks. Although substantial strides have been made for short-wavelength NIR LEDs with emission peak below 900 nm (refs. 7,8,9), long-wavelength NIR LEDs over 900 nm based on organic semiconductors and colloidal quantum dots (QDs) still face primary challenges of low radiance and poor operational stability (Table 1), limiting their practical applications10,11,12,13,14.
Metal halide perovskites are promising candidates for LEDs due to their excellent optical and electrical properties15,16,17,18,19. Tin-based halide perovskites (THPs), especially all-inorganic black orthorhombic (γ)-CsSnI3, are promising for use in NIR LEDs due to their low bandgap (∼1.3 eV), high hole mobilities and excellent intrinsic inorganic structural stability20,21,22,23,24,25,26,27,28,29,30,31,32. However, the easy oxidation of Sn2+ to Sn4+ usually results in p doping with high background hole densities, leading to strong non-radiative recombinations33,34.
In spite of the strong p doping in THPs, we notice that doping is not necessarily detrimental towards device performance, as long as the doping can be controlled. For example, electronic doping in conventional III–V-group inorganic semiconductors is one of the most powerful tools in established technologies, allowing for the fine-tuning of their functionalities and creating the base for modern microelectronics. It has been demonstrated that intentional electronic doping in semiconductors could boost emission properties as a result of the radiative recombinations of doped holes with free electrons35,36,37,38. Inspired by the development of electronic doping in inorganic semiconductors for improved LED performance, we, thus, believe that the intrinsic p-doping characteristic of THPs could be beneficial for achieving high-performance NIR LEDs through elaborately controlling the hole-doping and trap densities.
Here we report long-wavelength NIR LEDs peaking at 948 nm with high radiance and long operational stability based on CsSnI3, achieved by simultaneously exploiting the beneficial effect of intrinsic p doping in THPs and reducing the carrier trap density. A facile yet effective strategy is developed to control the intrinsic p-doping density and reduce the trap density by controlling the crystallization process of CsSnI3 precursors in tin-rich conditions, leading to improved radiative efficiency and stability. Leveraging these benefits, we demonstrate NIR LEDs with a high radiance of 226 W sr−1 m−2 and an operational half-lifetime of 39.5 h at a high constant current density of 100 mA cm−2, both significantly exceeding the previously reported soft-materials-based NIR LEDs with emission peaks over 900 nm.
Device structure and performance
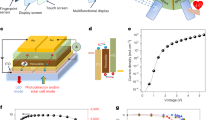
We fabricate CsSnI3-based NIR LEDs with the following device architecture: indium tin oxide (ITO) glass substrate anode, a poly(3,4-ethylenedioxythiophene):poly(styrene-sulfonate) (PEDOT:PSS) hole injection layer, an active CsSnI3 emission layer, a 1,3,5-tris(N-phenylbenzimidazol-2-yl) benzene (TPBi) electron transport layer and a LiF/Al cathode (Fig. 1a). CsSnI3 films are prepared by spin coating from SnI2 and CsI precursors with additives of N-phenylthiourea (NPTU) and SnF2 (denoted as N-S-CsSnI3). SnF2 has been widely used in THP-based solar cells, providing a tin-rich condition to reduce the intrinsic hole-doping density mainly due to Sn vacancies. NPTU helps to retard the crystallization process, which—as we show later—further manipulates the intrinsic p-doping density as well as reduces the carrier trap density. Control CsSnI3 films treated without additives (C-CsSnI3) and with SnF2 (S-CsSnI3) are also prepared for comparison. A cross-sectional scanning electron microscopy (SEM) image (Supplementary Fig. 1) of the device shows the formation of discrete submicrometre-structured perovskite crystals in the emission layer. The thickness of the perovskite film was determined to be around 40–50 nm. The emission peak of the electroluminescence (EL) spectra of N-S-CsSnI3 LEDs is centred at ∼948 nm, which is redshifted compared with that of ∼920 and ∼942 nm for C- and S-CsSnI3 LEDs (Supplementary Fig. 2). In addition, a reduced full-width at half-maximum (FWHM) of 71 nm is obtained for N-S-CsSnI3 LEDs, much smaller than 135 and 81 nm shown by C- and S-CsSnI3 LEDs, respectively. We also observe a negligible shift in the EL spectra under different operating voltages (Fig. 1b and Supplementary Fig. 3), demonstrating good EL spectral stability.

a, Energy-level diagram. b, The EL spectra at different operating voltages. c,d, Current density–voltage–radiance (c) and histograms of maximum radiance (d) of N-S-CsSnI3 LEDs. e, Operational stability of LEDs prepared under different conditions at a constant current density of 100 mA cm−2. The stability of N-S-CsSnI3 LEDs are measured with (dark red line) and without (orange line) a nitrogen flow for comparison.
The N-S-CsSnI3 LEDs (Fig. 1c and Supplementary Fig. 4) can operate at a high current density of over 2,800 mA cm−2, and show a corresponding maximum radiance of 226 W sr−1 m−2, which is much larger than that of 14 and 103 W sr−1 m−2 for C- and S-CsSnI3 LEDs (Supplementary Fig. 5, 6). The radiance is also much higher than that of other long-wavelength NIR LEDs with emission peaks over 900 nm, including noble-metal Pt(ii)-based organic materials, PbS QDs, Ag2S QDs and mixed Pb–Sn halide perovskites10,11,12,13,14,20. In addition, the N-S-CsSnI3 LEDs show a low efficiency roll-off, with the maximum external quantum efficiency of ∼2.63% at a high radiance of 142 W sr−1 m−2 (Supplementary Fig. 4). The maximum radiance and peak external quantum efficiency histograms of N-S-CsSnI3 LEDs (Fig. 1d and Supplementary Fig. 7) show good reproducibility, demonstrating the effectiveness of the NPTU/SnF2 incorporation strategy.
Another challenge for THPs is their poor stability, which hinders their practical applications. Notably, N-S-CsSnI3 LEDs exhibit a particularly long operational stability with a half-lifetime (T50) of 39.5 h at a constant current density of 100 mA cm−2, greatly improved compared with C-CsSnI3 LEDs (0.6 h) and S-CsSnI3 LEDs (9.8 h) (Fig. 1e). The T50 value for N-S-CsSnI3 LEDs represents the record stability for NIR LEDs with an emission peak of over 900 nm, significantly longer than that of 3 min to 1 h for other NIR LEDs operating at a much smaller constant current density of around 10 mA cm−2 (Table 1)13,20. This value for N-S-CsSnI3 LEDs can be extended to 125 h when measured under a constant nitrogen flow, indicating that strategies on thermal management would be helpful to further improve the device stability.
Controlled growth of CsSnI3 films
To understand the underlying reasons for improved device performance after the incorporation of NPTU/SnF2, we perform a range of experiments to investigate the film formation processes. The controlled and retarded crystallization process of CsSnI3 is clearly revealed (Fig. 2a and Supplementary Fig. 8). The colour of C-CsSnI3 quickly changes from light yellow to inhomogeneous dark brown within 10 min after spin coating (without annealing), indicating the fast and uncontrollable crystallization process. This crystallization process is slowed down in S-CsSnI3 and further retarded in N-S-CsSnI3, which changes from colourless to yellow with high transparency within 10 min after spin coating. These results demonstrate that the combination of SnF2 and NPTU could effectively retard the nucleation and control the crystallization process, leading to the formation of high-quality crystals. After annealing, the N-S-CsSnI3 films show a homogeneous dark brown colour across the entire film. The effect of additives on crystallization is also evidenced from the SEM images (Fig. 2b and Supplementary Fig. 9), where increasing the amount of NPTU leads to a decrease in the areal density of the crystals. Compared with the continuous film, the discontinuous morphology with isolated perovskite grains in the N-S-CsSnI3 films could boost the outcoupling efficiency of the devices, as demonstrated in FAPbI3-based LEDs8,9.

a, Photographs of CsSnI3 films prepared under different conditions before and after annealing, where 0, 3 and 10 min indicate the films before annealing. b, SEM images of S-CsSnI3 (i), N(5%)-S-CsSnI3 (ii), N(40%)-S-CsSnI3 (iii) and N(100%)-S-CsSnI3 (iv) films. Scale bars, 1 μm. c, XRD spectra of CsSnI3 films. The diffraction peaks from the ITO substrate are marked as green cycles. d, 13C NMR spectra of NPTU, NPTU + CsI, NPTU + SnI2 and NPTU + CsI + SnI2 in a DMSO-d6 solution. e, Proportion of Sn4+ of CsSnI3 films prepared under different conditions.
The X-ray diffraction (XRD) measurements reveal the black γ-CsSnI3 structure of all the films (Fig. 2c). The intensities of the characteristic peaks at 14.4° and 29.1° in N-S-CsSnI3 show significant improvement compared with those of C- and S-CsSnI3, indicating improved crystallinity (Supplementary Fig. 10)39. We identify the interaction mechanism between NPTU and CsSnI3 using 1H and 13C nuclear magnetic resonance (NMR) spectroscopy. The resonance signal of δ = 181.42 ppm arising from the C=S group in NPTU undergoes a chemical shift of Δδ ≈ 0.24 to δ = 181.18 ppm and Δδ ≈ 0.29 to δ = 181.13 ppm after interacting with the SnI2 and CsI + SnI2 mixture, respectively (Fig. 2d). Such variations indicate the occurrence of chemical interactions between the C=S group and SnI2. In contrast, a smaller chemical shift of Δδ ≈ 0.12 ppm of the C=S group is observed after the addition of CsI. This comparison indicates that the interaction between NPTU and SnI2 is much stronger than that between NPTU and CsI, ascribed to the stronger Lewis acid–base interaction between Sn2+ and NPTU compared with the relatively weak hydrogen bonding between the N–H group and I−. The stronger interaction between NPTU and SnI2 has also been confirmed by the 1H NMR and attenuated total reflectance Fourier transform infrared spectroscopy data (Supplementary Figs. 11–14)40,41.
The X-ray photoelectron spectra (XPS) further reveal the surface composition of CsSnI3 and confirm their chemical interactions with NPTU. The S2p XPS peak of N-S-CsSnI3 slightly shifts towards a higher binding energy compared with that of pure NPTU, indicating the lower electronegativity of S in N-S-CsSnI3 (Supplementary Figs. 15–17). The proportion of Sn4+ was fairly high with the peak intensity comparable to that of Sn2+ in C-CsSnI3 (Fig. 2e and Supplementary Fig. 17), suggesting the serious oxidation at the perovskite surface and grain boundaries. The easy oxidation of Sn2+ into Sn4+ is usually considered as the major source of trap-state formation in THPs. The Sn4+ content of N-S-CsSnI3 is significantly reduced compared with C- and S-CsSnI3. These results indicate that NPTU could prevent undesired tin oxidation due to the strong Lewis acid–base interaction of N–C=S⋯Sn2+, which can increase the electron density around Sn2+, offering excellent protection from being oxidized.
Controlled p doping and reduced trap density
Detailed optical and photophysical characterizations have revealed that NPTU/SnF2 could control the p-doping density and reduce the trap density of CsSnI3. C-CsSnI3 shows a broad absorption band with an onset at ∼1,000 nm and a broadband photoluminescence (PL) emission peak at ∼915 nm with a large FWHM of more than 150 nm (Fig. 3a). In contrast, S- and N-S-CsSnI3 show an ∼30 nm redshifted emission peak centred at ∼945 nm with reduced FWHM values of 82 and 74 nm, respectively. The time-resolved PL spectra of N-S-CsSnI3 show distinct monoexponential decay and increased carrier lifetime of 2.28 ns compared with that of 0.49 and 1.37 ns for C- and S-CsSnI3, respectively (Fig. 3b and Supplementary Table 1)42,43,44,45. The blueshift and broadening of the PL spectra as well as the short carrier lifetime of C-CsSnI3 are indicative of a large concentration of hole charge carrier accumulation near the valence band maximum due to the strong self p doping, which can be explained based on a dynamic Burstein–Moss shift (Supplementary Fig. 18). The Hall effect measurements indicate significantly reduced p-doping density of CsSnI3 after the incorporation of SnF2 and NPTU (Fig. 3c and Supplementary Fig. 19), qualitatively agreeing with the results from ultraviolet photoelectron spectroscopy measurements (Supplementary Fig. 20).

a,b, Absorption and PL spectra (a) and time-resolved PL spectra and corresponding fitting spectra (b) of CsSnI3 films. c, Charge carrier densities of CsSnI3 films determined by Hall effect measurements. The error bars correspond to standard errors in the results calculated from the Hall effect measurement. d, Excitation-density-dependent PLQY of CsSnI3 films. It should be noted that for the PLQY measurement, the CsSnI3 films are covered with a thick PMMA layer, and then encapsulated with glass assisted by an ultraviolet curing adhesive. As such, the measured PLQY is external PLQY, rather than internal PLQY.
The carrier recombination dynamics in THPs can be described by using a kinetic model that simulates the generation and recombination of charge carriers in the presence of both p doping and deep carrier traps (Supplementary Fig. 21). Through a qualitative analysis of the influence of p doping and trap carrier densities on the carrier recombination dynamics, we can learn how they affect the measurable figures of merit such as the internal photoluminescence quantum yield (PLQY) and the carrier lifetime (τ) (ref. 38). By simulating the excitation-density-dependent internal PLQY with hole-doping densities from 1014 to 1020 cm−3 and trap densities from 1015 to 1018 cm−3 (Supplementary Figs. 22–24), we learn that (1) the trap and hole-doping densities play a competing role in determining the carrier recombination dynamics and optical properties of CsSnI3; (2) the presence of free background holes due to p doping may improve the PLQY and boost the radiative recombination with photogenerated electrons—this is true for increasing doping densities until Auger recombination becomes dominant; and (3) when the trap density is comparable to or higher than the doping density, trap-mediated recombination processes dominate and mostly influence the PLQY.
The excitation-density-dependent PLQY values of CsSnI3 films are recorded and interpreted with the support of the abovementioned kinetic model. C-CsSnI3 shows a constant, low PLQY in a wide range of excitation densities (Fig. 3d), which could be ascribed to the severe Auger recombinations due to the high hole-doping density of more than 1020 cm−3. On the contrary, S- and N-S-CsSnI3 show improved PLQY in the whole excitation-density range with gradually reduced hole-doping density, which is visible in the emergence of trap-filling processes at low excitation densities that are completely hidden by Auger recombinations in C-CsSnI3. By comparing S- and N-S-CsSnI3, the trap density is reduced after the incorporation of NPTU, as revealed by the shift in the trap-filling-induced PLQY increase towards a lower-excitation-density region, as well as temperature-dependent PL (Supplementary Figs. 25–27), femtosecond transient absorption (fs-TA) spectra (Supplementary Figs. 28–34).
Electronic structure calculations
Considering the possible influence of the hole-doping density on the Auger recombination process, we perform electronic structure calculations to understand the mechanism behind the Auger recombination in CsSnI3. With the aim of highlighting the differences in THP and its well-known lead counterpart, we lay out the relation between the Auger recombination rate and phase-space volume for Auger processes (Ω). The electronic structure calculations of black γ-CsSnI3 and CsPbI3 reveal that the value of Ω for CsSnI3 should be much lower compared with CsPbI3, and consequently, owing to similarities between the two compounds, the recombination rates should follow the same trend. Figure 4a,b maps the peaks in Ω to energy levels in the band structures of CsPbI3 and CsSnI3. The arrows illustrate the typical eeh and hhe mechanisms, with the empty and filled circles representing holes and electrons, respectively. These eigenstates marked by numbers 1, 2, 3 and 4 correspond to the convention used in equation (1). We note that within a reasonable proximity to the band extrema, the number of available eigenstates, interpreted as points on the energy bands, are significantly larger in the case of a lead-based perovskite compared with its tin-based counterpart, leading to a large phase-space volume for CsPbI3. At the same time, the peaks in the spectrum of Ω (Fig. 4c,d) match fairly well with the density of available eigenstates for the three-body process in the band structure (Fig. 4a,b). These results imply that CsSnI3 probably has a much lower Auger recombination rate than CsPbI3. The calculated much smaller phase-space volume indicates that the high hole-doping density in CsSnI3 could be beneficial for its light emission properties. In particular, it has been previously suggested that the rate of Auger recombination in cubic CsSnI3 should be lower than that in cubic CsPbI3, owing to features in the band structure that are also quantified here via a calculation of Ω (ref. 46).

a,b, Dispersions near the band extrema of CsPbI3 (a) and CsSnI3 (b) following the path X–Γ–Y. The representative transitions involved in the eeh or hhe mechanisms are depicted within both a and b using unidirectional arrows. c,d, Calculated available phase space for the direct Auger recombination process for CsSnI3 and CsPbI3 via the eeh (c) and hhe (d) mechanisms as a function of Δ-scissors-adjusted gap. The dashed vertical lines in c and d represent the experimental bandgaps of CsSnI3 (green) and CsPbI3 (red) and are marked by Eg,Sn and Eg,Pb, respectively. The horizontal dashed red (CsPbI3) and green (CsSnI3) lines in a and b indicate the value of Δ-scissors-adjusted gap (eV) with respect to the conduction band minima (eeh) or valence band maxima (hhe) for which the peaks in the respective spectra of Ω are observed. For the range of Δ-scissors-adjusted bandgap in c and d, Ω {hhe} is nearly four orders of magnitude larger than Ω {eeh}.
Discussions
Based on the discussions above, we can conclude that CsSnI3 probably shows an intrinsically much lower Auger recombination rate than CsPbI3, indicating that the intrinsic p-doping characteristic of CsSnI3 could lead to improved PLQY due to the radiative recombination of background holes with the photogenerated electrons. The combination of these features provides rich opportunities for fabricating high-performance NIR LEDs that can operate at high current densities. As such, the record radiance and low efficiency roll-off of CsSnI3 NIR LEDs could be ascribed to the capability of withstanding high current densities above 2,800 mA cm–2 due to the low Auger recombination rate, the elaborately controlled p doping and reduced trap densities realized via the NPTU/SnF2 incorporation strategy.
The improved device stability can be ascribed to different concomitant phenomena. First, we have demonstrated an intrinsic lower Auger recombination rate for CsSnI3 with respect to CsPbI3. In addition, the possibility of controlling the p-doping concentration allows us to be in such a condition where the carrier density generated at 100 mA cm−2 is relatively small, with respect to the free hole density, and the latter is small enough to boost the radiative band-to-band recombination instead of fully entering the Auger recombination regime. Second, the energy barrier for ion migration is much larger in THP than that of its lead-based counterparts47. It is important to notice that in lead-based perovskites, photoinstability has been attributed to the trapping of carriers at halide interstitial deep defects, which eventually leads to the formation of I2 molecules and promotes halide migration. However, this does not apply to THPs, as its defect chemistry is primarily determined by Sn, resulting in remarkable photostability even under high excitation densities47. The degradation of the device may be attributed to the damage occurring at the interface between the transport layer and emission layer.
Conclusion
In summary, we have demonstrated highly bright and stable long-wavelength NIR LEDs based on all-inorganic lead-free CsSnI3. A facile yet effective strategy is developed to elaborately control the intrinsic p-doping density and reduce the trap density by retarding the crystallization process of CsSnI3. The long-wavelength NIR LEDs based on CsSnI3 not only demonstrate high radiance but also exhibit excellent stability, which is more than one order of magnitude longer than that of the best-known long-wavelength NIR LEDs fabricated with other kinds of soft material. Our work addresses the critical challenges of the low radiance and poor operational stability of long-wavelength NIR LEDs, representing a key step towards their practical applications. Moreover, the strategy to control the p-doping and trap densities can be extended to the design of other highly emissive and stable THP for light-emitting applications, including electrically pumped lasers.
Methods
Materials
Chemicals listed below are commercially available and used without further purification. Tin(ii) iodide (SnI2, 99.999%), tin(ii) fluoride (SnF2, 99.000%), caesium iodide (CsI, 99.999%) and NPTU (>98.000%) were purchased from Sigma-Aldrich. PEDOT:PSS (AI 4083) was purchased from Ossila. All the other materials used for the device fabrication were purchased from Sigma-Aldrich.
Electronic structure calculations
The electronic structure calculations of black γ-CsSnI3 and CsPbI3 were performed using the Vienna ab initio simulation package using the projector augmented wave method. The PBEsol functional in the framework of density functional theory was used for geometry optimization, whereas the hybrid HSE functional with 50% exact Hartree–Fock exchange for short-range interactions and a screening parameter of ω = 0.20 Bohr−1 was implemented to evaluate the band structure. We use 50% exact Hartree–Fock exchange as that provides the best conformity between the HSE and GW band structures, the latter requiring a very high computational cost for a dense sampling of the Brillouin zone, which is essential to calculate meaningful values of Ω. To fully account for relativistic effects, the spin–orbit interaction was included in all the calculations.
Preparation of perovskite solution and films
The control precursor solution without additives (denoted as C-CsSnI3) was prepared by dissolving 112 mg SnI2 and 73 mg CsI in 1 ml DMSO (0.3 M: the concentration was determined by the Sn content). The precursor solution with SnF2 (denoted as S-CsSnI3) was prepared by dissolving 112.0 mg SnI2, 73.0 mg CsI and 4.7 mg SnF2 in 1 ml DMSO (0.3 M, 10% SnF2). The NPTU solution was prepared by dissolving 152 mg of NPTU in 1 ml DMSO (1 M). As for the NPTU-added precursor solution, a certain amount of NPTU (1 M) was added in the S-CsSnI3 precursor solution to achieve the desired composition (denoted as N-S-CsSnI3). The solution was stirred and filtered before spin coating. Emissive layers of CsSnI3 with different compositions were spin coated in a nitrogen-filled glovebox (concentrations of H2O and O2, <0.1 ppm) via a two-step process, 1,000 r.p.m. for 10 s (acceleration speed, 200 r.p.m. s–1) and then 5,000 r.p.m. for 60 s (acceleration speed, 800 r.p.m. s–1), followed by annealing at 100 °C for 10 min to form the active emission layer. The optimized doping content for NPTU is 40% (mole fraction compared with Sn). For the PL, low-temperature-dependent PL, time-resolved PL, ultraviolet–visible absorption spectra, fs-TA spectra and XRD measurements, 80 mg ml–1 polymethyl methacrylate (PMMA)/chlorobenzene solution was spin coated at 6,000 r.p.m. for 50 s on top of the perovskite layer.
Optical characterization
The ultraviolet–visible absorption spectra were measured using a PerkinElmer model LAMBDA 900a. Steady-state PL spectral measurements were performed on an Andor Shamrock 303i spectrograph equipped with an Andor Newton electron-multiplying charge-coupled device instrument (DU970N-UVB). During the measurement, the charge-coupled device detector was cooled to –45 °C. The sample was excited by a Thorlabs collimated laser-diode-pumped DPSS laser module (CPS532), and a 550 nm long-pass filter was used to protect the detector. The wavelength of the system was calibrated by a mercury lamp. The intensity of the spectra was calibrated by a standard halogen lamp (AvaLight-HAL-S-Mini) from Avantes. A laser (405 nm, 75 ps pulse width) was used as the excitation source at 10 MHz repetition rate (Edinburgh Instruments) for the time-resolved PL spectra measurement. The laser was focused onto the sample by an air objective (numerical aperture, 0.7; spot size, ∼1 μm; Mitutoyo). The PL data of the sample were collected by the same objective and sent to an avalanche photodiode (timing resolution, ∼40 ps; ID Quantique) in a time-correlated single-photon counting system (timing jitter, ∼3 ps; qutools) with an instrument response function of ∼56 ps. Fluence-dependent PLQY values were acquired by focusing the second harmonic of an Yb:KGW laser (Light Conversion Pharos, 515 nm, 30 fs pulse duration, 1 kHz repetition rate) on thin films of CsSnI3 within a fluence range between 4.0 × 1013 and 2.5 × 1020 cm−3, and collecting the emitted PL on a fibre spectrometer (Ocean Optics Maya Pro 2000). Relative PLQY measurements were calibrated by scaling the resulting PLQY curve to an absolute PLQY measurement conducted in an integrating sphere (Labsphere). For the PLQY measurement, the CsSnI3 films were covered with a thick PMMA layer, and then encapsulated with glass assisted by an ultraviolet curing adhesive.
Low-temperature-dependent PL spectra
The samples were mounted in a closed-cycle cryostat (DE-202AE) from Advanced Research Systems. The cooling was performed by a helium compressor (ARS-4HW) from the same company. The temperature control was performed by a Lake Shore 330 autotuning temperature controller. During the measurement, the cryostat was kept in a vacuum. The PL excitation and detection were performed by an objective, a 45°-angled 552 nm long-pass dichroism glass and optical fibres. The laser light was coupled into a fibre, out of a collimator, then reflected by the glass and finally focused by the objective into the cryostat. The emission from the sample passed the cryostat window, the objective, the glass, a collimator, a fibre, a collimator again and finally focused into the spectrograph.
fs-TA measurement
The fs-TA spectroscopy was conducted with a HELIOS fs-TA spectrometer (Ultrafast Systems). A portion of the 800 nm fundamental output from the regenerative amplifier (Coherent Libra, 1 kHz, 50 fs) was used to pump an optical parametric amplifier (Coherent OPerA Solo). The 650 nm pump pulses were generated from an optical parametric amplifier. Another portion of the 800 nm fundamental beam was focused into a 10 mm sapphire crystal to generate a broadband NIR continuum (800–1,600 nm) that was used as the probe pulse. The probe beam was passed through an 850 nm long-pass filter to eliminate the residual 800 nm fundamental components to prevent strong secondary photoexcitation of the sample. The signals were collected using a complementary metal–oxide–semiconductor sensor within the spectrometer.
Hall measurements
The Hall measurements were performed in a chip with van der Pauw geometry. The four bottom contact electrodes were deposited with Cr and Au. The CsSnI3 films were encapsulated with PMMA to avoid degradation. The chips were placed perpendicular to the magnetic field changing from –1.2 to 1.2 T. The applied current and voltage were recorded by Keithley 2400 and Nanovoltmeter model 2182A instruments, respectively. All the measurements were performed in air at room temperature.
Structural, morphological and chemical characterizations
The XRD measurements were conducted using an X-ray diffractometer (PANalytical X’Pert Pro) with an X-ray tube (Cu Kα, λ = 1.5406 Å). The attenuated total reflectance Fourier transform infrared measurements were performed on a PIKE MIRacle attenuated total reflectance accessory with a diamond prism in a Vertex 70 spectrometer (Bruker) using a DLATGS detector at room temperature. The spectra were acquired at a 2 cm–1 resolution. SEM was conducted using a Philips XL30 FEG SEM instrument under a voltage of 8 kV. Proton (1H) NMR (500 MHz) and carbon (13C) NMR (126 MHz) were recorded on a Varian 500 MHz spectrometer at room temperature. Dried DMSO-d6 was used as the solvent and to calibrate the NMR isotropic chemical shifts. XPS (monochromatized Al Kα, hν = 1,486.6 eV) spectra were recorded with a Scienta 200 hemispherical analyser and calibrated by referencing to the Fermi level and Au4f7/2 peak position of the Ar+-ion sputter-clean gold foil.
Device fabrication and characterization
ITO-coated glass substrates were cleaned by sonication in deionized water and then in organic solvents (acetone followed by ethanol), and then dried under a nitrogen flow. The substrates were cleaned using an ultraviolet–plasma treatment to enrich the ITO surface with oxygen and increase the ITO work function. The PEDOT:PSS hole injection layer was spin coated at 4,000 r.p.m. (acceleration speed, 4,000 r.p.m. s–1) for 40 s on ITO followed by annealing in air at 150 °C for 30 min, and then CsSnI3 films were deposited on PEDOT:PSS in a nitrogen-filled glovebox. Finally, the substrates were transferred to a vacuum chamber, where TPBi (55 nm) and LiF/Al electrodes (1/150 nm) were deposited using a thermal evaporation system through a shadow mask under a high vacuum of 1 × 10−6 torr. The device active area was 7.25 mm2 as defined by the overlapping area of the ITO and Al electrodes. The performance of the LEDs was measured in a nitrogen-filled glovebox at room temperature. A Keithley 2400 source meter and a fibre integration sphere (FOIS-1) coupled with a QE Pro spectrometer (Ocean Optics) were used for the LED measurements. It should be noted that our LED setup was programmed to measure the EL spectra ranging from 350 to 930 nm. Therefore, the full-range EL spectra up to 1,100 nm were then obtained from another spectrometer (Shamrock 303i; the detector is an Andor Newton electron-multiplying charge-coupled device) with a wide emission detection range driven by a Keithley 2400 source meter, and the performance is, thus, calibrated by the comparison of the EL spectra measured in the LED setup and from Shamrock 303i. The LED stability was measured either in the glovebox in a small confined space (without a nitrogen flow) or in a home-made nitrogen-filled box at room temperature with nitrogen flow, as specified in the figures. The devices were driven by a Keithley 2400 source meter at a constant current and the light emission signals were measured using a commercial photodiode.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Medintz, I. L., Uyeda, H. T., Goldman, E. R. & Mattoussi, H. Quantum dot bioconjugates for imaging, labelling and sensing. Nat. Mater. 4, 435–446 (2005).
Konstantatos, G. et al. Ultrasensitive solution-cast quantum dot photodetectors. Nature 442, 180–183 (2006).
Kim, S. et al. Near-infrared fluorescent type II quantum dots for sentinel lymph node mapping. Nat. Biotechnol. 22, 93–97 (2004).
Sun, L. et al. Bright infrared quantum-dot light-emitting diodes through inter-dot spacing control. Nat. Nanotechnol. 7, 369–373 (2012).
Yoon, J. et al. GaAs photovoltaics and optoelectronics using releasable multilayer epitaxial assemblies. Nature 465, 329–333 (2010).
Walther, T., Cullis, A., Norris, D. & Hopkinson, M. Nature of the Stranski-Krastanow transition during epitaxy of InGaAs on GaAs. Phys. Rev. Lett. 86, 2381 (2001).
Tuong, Ly,K. et al. Near-infrared organic light-emitting diodes with very high external quantum efficiency and radiance. Nat. Photon. 11, 63–68 (2017).
Cao, Y. et al. Perovskite light-emitting diodes based on spontaneously formed submicrometre-scale structures. Nature 562, 249–253 (2018).
Xu, W. et al. Rational molecular passivation for high-performance perovskite light-emitting diodes. Nat. Photon. 13, 418–424 (2019).
Wei, Y. C. et al. Overcoming the energy gap law in near-infrared OLEDs by exciton-vibration decoupling. Nat. Photon. 14, 570–577 (2020).
Gong, X. et al. Highly efficient quantum dot near-infrared light-emitting diodes. Nat. Photon. 10, 253–257 (2016).
Pradhan, S. et al. High-efficiency colloidal quantum dot infrared light-emitting diodes via engineering at the supra-nanocrystalline level. Nat. Nanotechnol. 14, 72–79 (2019).
Gao, L. et al. Efficient near-infrared light-emitting diodes based on quantum dots in layered perovskite. Nat. Photon. 14, 227–233 (2020).
Vasilopoulou, M. et al. Efficient colloidal quantum dot light-emitting diodes operating in the second near-infrared biological window. Nat. Photon. 14, 50–56 (2020).
Dong, Y. et al. Bipolar-shell resurfacing for blue leds based on strongly confined perovskite quantum dots. Nat. Nanotechnol. 15, 668–674 (2020).
Wang, Y.-K. et al. All-inorganic quantum-dot LEDs based on a phase-stabilized α-CsPbI3 perovskite. Angew. Chem. Int. Ed. 60, 16164–16170 (2020).
Wang, Y.-K. et al. Chelating-agent-assisted control of CsPbBr3 quantum well growth enables stable blue perovskite emitters. Nat. Commun. 11, 3674 (2020).
Zheng, X. et al. Chlorine vacancy passivation in mixed halide perovskite quantum dots by organic pseudohalides enables efficient Rec. 2020 blue light-emitting diodes. ACS Energy Lett. 5, 793–798 (2020).
Wang, Z. et al. Multifunctional p-type carbon quantum dots: a novel hole injection layer for high-performance perovskite light-emitting diodes with significantly enhanced stability. Adv. Opt. Mater. 7, 1901299 (2019).
Qiu, W. et al. Mixed lead–tin halide perovskites for efficient and wavelength-tunable near-infrared light-emitting diodes. Adv. Mater. 31, 1806105 (2019).
Yu, H. Q. et al. Alkalis-doping of mixed tin-lead perovskites for efficient near-infrared light-emitting diodes. Sci. Bull. 67, 54–60 (2022).
Lu, J. X. et al. Dendritic CsSnI3 for efficient and flexible near-infrared perovskite light-emitting diodes. Adv. Mater. 33, 2104414 (2021).
Yuan, F. et al. Color-pure red light-emitting diodes based on two-dimensional lead-free perovskites. Sci. Adv. 6, eabb0253 (2020).
Wang, K. et al. Lead-free organic-perovskite hybrid quantum wells for highly stable light-emitting diodes. ACS Nano 15, 6316–6325 (2021).
Liang, H. et al. High color purity lead-free perovskite light-emitting diodes via Sn stabilization. Adv. Sci. 7, 1903213 (2020).
Lin, R. et al. Monolithic all-perovskite tandem solar cells with 24.8% efficiency exploiting comproportionation to suppress Sn(ii) oxidation in precursor ink. Nat. Energy. 4, 864–873 (2019).
Lin, J. et al. Harnessing dielectric confinement on tin perovskites to achieve emission quantum yield up to 21%. J. Am. Chem. Soc. 141, 10324–10330 (2019).
Yakunin, S. et al. High-resolution remote thermometry and thermography using luminescent low-dimensional tin-halide perovskites. Nat. Mater. 18, 846–852 (2019).
Hong, W. et al. Efficient low-temperature solution-processed lead-free perovskite infrared light-emitting diodes. Adv. Mater. 28, 8029–8036 (2016).
Xue, S. et al. Advances in electrically driven light-emitting diodes based on lead-free metal halides. Chem. Commun. 59, 1116–1124 (2023).
Wei, Q. et al. Chances and challenges for tin perovskites. Trends Chem. 4, 1–4 (2022).
Chung, I. et al. CsSnI3: semiconductor or metal? High electrical conductivity and strong near-infrared photoluminescence from a single material. High hole mobility and phase-transitions. J. Am. Chem. Soc. 134, 8579–8587 (2012).
Ricciarelli, D. et al. Instability of tin iodide perovskites: bulk p‑doping versus surface tin oxidation. ACS Energy Lett. 5, 2787–2795 (2020).
Li, B. et al. Tin-based defects and passivation strategies in tin-related perovskite solar cells. ACS Energy Lett. 5, 3752–3772 (2020).
Barrigón, E. et al. Synthesis and applications of III–V nanowires. Chem. Rev. 119, 9170–9220 (2019).
Tsintzos, S. et al. A GaAs polariton light-emitting diode operating near room temperature. Nature 453, 372–375 (2008).
Feldmann, S. et al. Photodoping through local charge carrier accumulation in alloyed hybrid perovskites for highly efficient luminescence. Nat. Photon. 14, 123–128 (2020).
Treglia, A. et al. Effect of electronic doping and traps on carrier dynamics in tin halide perovskites. Mater. Horiz. 9, 1763–1773 (2022).
Zhang, X. et al. First-principles simulation of carrier recombination mechanisms in halide perovskites. Adv. Eng. Mater. 10, 1902830 (2020).
Meng, X. et al. Crystallization kinetics modulation of FASnI3 films with pre-nucleation clusters for efficient lead-free perovskite solar cells. Angew. Chem. Int. Ed. 60, 3693–3698 (2021).
Wang, S. et al. High-performance perovskite solar cells with large grain-size obtained by using the Lewis acid-base adduct of thiourea. Sol. RRL 2, 1800034 (2018).
Guo, Q. et al. Passivation of the grain boundaries of CH3NH3PbI3 using carbon quantum dots for highly efficient perovskite solar cells with excellent environmental stability. Nanoscale 11, 115–124 (2019).
Ye, T. et al. Ambient-air-stable lead-free CsSnI3 solar cells with greater than 7.5% efficiency. J. Am. Chem. Soc. 143, 4319–4328 (2021).
Milot, R. et al. The effects of doping density and temperature on the optoelectronic properties of formamidinium tin triiodide thin films. Adv. Mater. 30, 1804506 (2018).
Bubnova, O. et al. Semi-metallic polymers. Nat. Mater. 13, 190–194 (2014).
Shen, J. et al. Unexpectedly strong Auger recombination in halide perovskites. Adv. Energy Mater. 8, 1801027 (2018).
Martani, S. et al. Defect engineering to achieve photostable wide bandgap metal halide perovskites. ACS Energy Lett. 8, 2801–2808 (2023).
Acknowledgements
We thank B. Guan and L. Liang from the Institute of Chemistry, Chinese Academy of Sciences, for the cross-sectional SEM measurement; H. Zhang for help with the EL, PL and low-temperature-dependent PL spectra measurements; Z. Hu and J. Zhang for the NMR spectra measurement; C. Wang for the XPS and ultraviolet photoelectron spectroscopy measurements; H. Lei for the SEM characterization; C. Kuang for the Fourier transform infrared spectra measurement; and L. Sui for help with the carrier recombination dynamic simulations. F.Y. acknowledges financial support from the National Key Research and Development Program of China (2023YFB3611800), National Natural Science Foundation of China (22302012), National Science Fund for Excellent Young Scholars (Overseas) and Special Funds for Introducing Talents from Beijing Normal University (312200502508). F.G. acknowledges support from Knut and Alice Wallenberg Foundation (Dnr. KAW 2019.0082), the European Research Council Consolidator Grant (LEAP, 101045098), the Swedish Research Council Vetenskapsrådet (grant 2020-03564) and the Swedish Government Strategic Research Area in Materials Science on Functional Materials at Linköping University (faculty grant SFO-Mat-LiU no. 2009-00971). I.A.A. is a Wallenberg Academy Scholar (grant no. KAW-2018.0194). S.I.S. acknowledges support from the Swedish Research Council (VR) (project no. 2019-05551) and the ERC (synergy grant FASTCORR project 854843). T.C.S. and J.W.M.L. acknowledge support from the Singapore Ministry of Education under its MOE Tier 2 grant (MOE-T2EP50120-0004) and the National Research Foundation (NRF) Singapore under its NRF Investigatorship (NRF-NRFI2018-04). A.P., G.F. and A.T. acknowledge funding from the European Union’s Horizon 2020 research and innovation programme through the ERC project SOPHY under grant agreement no. 771528, the MSCA-ITN SMART-X under grant agreement no. 860553 and the Marie Skłodowska-Curie grant agreement no. 956270. The computations were enabled by resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) and the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Center (NSC) and Center for High Performance Computing (PDC), partially funded by the Swedish Research Council through grant agreement nos. 2022-06725 and 2018-05973.
Funding
Open access funding provided by Linköping University.
Author information
Authors and Affiliations
Contributions
F.G. and F.Y. conceived the idea and designed the experiments. F.Y. prepared the samples and carried out the device fabrication and measurements. F.Y., G.F., A.T. and A.P. conducted the carrier recombination dynamic simulations and PLQY measurement. T.L. helped with the device stability and Hall effect measurement. J.W.M.L. and T.C.S. performed the fs-TA measurements. U.S., J.K., S.I.S. and I.A.A. conducted the theoretical calculations. F.Y. wrote the first draft of the paper. All authors discussed the results and contributed to the paper.
Corresponding author
Ethics declarations
Competing interests
F.Y. and F.G. are filing a patent application related to this work.
Peer review
Peer review information
Nature Photonics thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–34, Table 1 and Discussion.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yuan, F., Folpini, G., Liu, T. et al. Bright and stable near-infrared lead-free perovskite light-emitting diodes. Nat. Photon. 18, 170–176 (2024). https://doi.org/10.1038/s41566-023-01351-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41566-023-01351-5
