
Abstract
Terpenoids account for more than 60% of all natural products, and their carbon skeletons originate from common isoprenoid units of different lengths such as geranyl pyrophosphate and farnesyl pyrophosphate. Here we characterize a metal-dependent, bifunctional isoprenyl diphosphate synthase from the leaf beetle Phaedon cochleariae by structural and functional analyses. Inter- and intramolecular cooperative effects in the homodimer strongly depend on the provided metal ions and regulate the biosynthetic flux of terpene precursors to either biological defence or physiological development. Strikingly, a unique chain length determination domain adapts to form geranyl or farnesyl pyrophosphate by altering enzyme symmetry and ligand affinity between both subunits. In addition, we identify an allosteric geranyl-pyrophosphate-specific binding site that shares similarity with end-product inhibition in human farnesyl pyrophosphate synthase. Our combined findings elucidate a deeply intertwined reaction mechanism in the P. cochleariae isoprenyl diphosphate synthase that integrates substrate, product and metal-ion concentrations to harness its dynamic potential.

Similar content being viewed by others
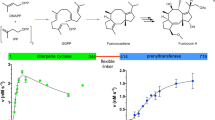

Main
Terpenes and terpenoids form the largest group of natural products, with currently over 80,000 known members, of which many display intriguing pharmaceutical properties1. Despite being chemically diverse agents of intraspecies and interspecies communication, as well as cellular integrity and proliferation, they emerge from only two isomeric building blocks, dimethylallyl pyrophosphate (DMAPP, C5) and isopentenyl pyrophosphate (IPP, C5)2. Short chain isoprenyl diphosphate synthases (scIDSs) elongate DMAPP to geranyl pyrophosphate (GPP, C10) and further to farnesyl pyrophosphate (FPP, C15) by consuming IPP in each step3. The uniform mechanism of this enzyme class requires three divalent cations that coordinate substrates at an allylic binding pocket (Al site) between the first and second aspartate-rich motif (FARM and SARM). The metal cluster provides an ionizing force that facilitates the dissociation of the pyrophosphate moiety (PPAl) once IPP binds to basic residues at the proximal homoallylic site (HAl; Fig. 1). The terminal double bond of IPPHAl attacks the nascent carbocation and connects the C5 precursor with the prenyl unit. Ultimately, the released inorganic pyrophosphate (PPi) deprotonates the extended metabolite at C2 and forms the trans double bond. If the chain length determination domain of the enzyme provides access to the generated allylic product, the elongation reaction repeats4.

a, Catalysis is uniform in this class of enzymes and starts with a prenyl substrate (DMAPP or GPP) bound to the allylic site (Al). A trinuclear, divalent metal cluster surrounds the pyrophosphate moiety (PPAl) and facilitates ionization of the ester bond (I). Basic residues of the homoallylic site (HAl) bind IPP, which attacks the nascent carbocation at C1 of the prenyl moiety (II). The generated inorganic pyrophosphate (PPi) deprotonates at C2 (III) of the intermediate and restores the allylic character of the elongated metabolite (GPP or FPP). b, In P. cochleariae, GPP is the main product of PcIDS1 in the presence of HMIs and precursor of the defensive monoterpene (C10) chrysomelidial. FPP formation is driven by Mg2+ and essential for sesquiterpene hormones (C15) during larval development. c, Active site architecture of scIDS enzymes shown for PcIDS1 in complex with DMAPPAl and IPPHAl. The first and second aspartate-rich DDxxD motifs (FARM and SARM; D, aspartate; x, any amino acid; red numbers indicate residue position) on helix D and helix H coordinate Mg2+ at positions A, B and C. Colour coding is described in the Methods.
FPP synthases (FPPSs) are distributed in all three kingdoms of life and catalyse both GPP and FPP formation, with the latter being the preferred reaction under physiological conditions. By contrast, GPP-specific synthases (GPPSs) are predominantly found in plants, but some insects, especially aphids and beetles, use GPP to yield defensive and pheromonal monoterpenes5,6,7. To date, only a single animal GPPS has been identified, in a bark beetle8, suggesting that alternative regulatory mechanisms to accumulate GPP exist within the repertoire of FPPS. Indeed, several scIDSs in insects control the biosynthesis of mono- (C10) and sesquiterpenes (C15) to meet the organism’s current needs9,10,11,12. Isoprenyl diphosphate synthase in P. cochleariae (PcIDS1, EC 2.5.1.1) produces FPP in the presence of Mg2+, but preferably forms GPP when exposed to the heavy metal ions (HMIs) Co2+ or Mn2+. Using C10 units as a building block, this leaf beetle secretes monoterpenes like chrysomeldial to protect itself against predation in its larval stadium. However, insects constantly require sesquiterpenoid hormones that drive large transformations during metamorphosis (Fig. 1b). Thus, balancing the concentrations of terpene precursors in accordance with environmental and developmental demands is an essential cellular process13.
Here we elucidate catalytic principles in PcIDS1 that either break or enforce dimer symmetry in response to HMIs. High-resolution crystal structures combined with functional and mutagenetic analyses reveal how different conformational states determine the product spectrum. In addition, a Mg2+–water cluster was identified that regulates the elongation reaction by reducing the affinity for IPP. We demonstrate that Mg2+ deactivates one of both active sites in PcIDS1 during FPP formation, whereas HMIs recruit GPP to a specific, allosteric pocket similar to end-product inhibition in the human FPP synthase14. Overall, our combined findings provide insights into the biosynthetic flux of terpene precursors towards biological defence or physiological development.
Results
Compared to well-studied scIDS enzymes that specifically produce C10, C15 or C20 isoprene precursors, the product chain length of PcIDS1 depends heavily on the provided metal cofactor. This synthase yields 96% GPP and only 4% FPP in the presence of Mn2+ or Co2+, whereas 18% GPP and 82% FPP form with Mg2+ (ref. 10). To elucidate the HMI-dependent molecular processes that suppress FPP accumulation, PcIDS1 was recombinantly expressed and purified (Supplementary Fig. 1). Protein stability was optimal in MgCl2 buffers and tolerated the addition of MnCl2, while CoCl2 facilitated precipitation. Next, we characterized the denaturation temperatures of different PcIDS1:metal:ligand complexes by thermal shift assays (Fig. 2a and Supplementary Fig. 2). In the absence of ligands, the stability of wild-type protein (41 °C) was not affected by Mn2+. For the allylic substrate DMAPP, the temperature shift in the presence of Mn2+ (Δ10 °C) was much stronger compared to Mg2+ (Δ3 °C). This discrepancy correlates with the higher affinity of PcIDS1 for DMAPP with Mn2+ (ref. 10), which might allow DMAPP to occupy the HAl site at high concentrations15. A similar metal dependency was observed with IPP (Δ14 °C for Mn2+, Δ7 °C for Mg2+), further highlighting the preference of HMIs to coordinate C5 substrates. The significant change in thermal stability with either metal suggests that IPP coordinates the HAl as well as the Al site in the absence of competing ligands. However, Mn2+ had no effect on the drastically increased protein stability (Δ28 °C) with the Al-specific ligand zoledronic acid (ZOL; ref. 16). The denaturation temperature, however, rose further when IPP was added (Δ37 °C), supposing that the inhibitor and substrate cooperatively interact with the Al site as well as the HAl site. For GPP, the product of the first elongation, the distinct thermal shift (Δ17 °C) was indifferent to the metal species, and FPP only had a moderate influence on protein stability (Δ5 °C for Mn2+, Δ7 °C for Mg2+). Therefore, our thermal shift assay results provide valuable information on ligand affinity to elucidate metal-dependent processes in PcIDS1 at the molecular level.

a, The fluorescence signal of recombinant PcIDS1 (6.25 µM) was recorded between 30 and 90 °C in the absence of (apo) and together with 200 µM of respective ligands. The graph displays the time-based derivative in relative units. The mean melting temperature is annotated in degrees Celsius next to the curves observed in MgCl2 buffer alone (5 mM, green) and with MnCl2 (1 mM, purple). b,c, Relative changes in thermal stability (compared to wild-type protein) for different protein mutants in the presence of DMAPP and IPP. d, ZOL binding stability in variants of the metal-coordinating SARM residue D319. Bars indicate the average value of three measurements (shown as dots); whiskers represent the relative error of the mean between mutant and wild-type protein. A detailed data analysis is in Supplementary Fig. 2.
Catalytic principles of PcIDS1
To obtain molecular insights into PcIDS1, we first determined the apo crystal structure in the presence of Mg2+ (Protein Data Bank (PDB) 8A6U). The enzyme adopts the conserved scIDS topology (best distance matrix alignment (DALI) hit17, PDB 3EWG, FPPS from Trypanosoma brucei18, Z-score (similarity of equivalent Cα-Cα distances among two proteins) 36.1, 30% sequence identity) with a dimer interface of 2,000 Å2 (ref. 19). Each subunit (subA and subB) consists of 13 helices (αA to αJ and α1 to α3; Extended Data Fig. 1a). In both chains, the N terminus and C terminus as well as the loops between αD–αE (183–201) and αH–α1 (329–332) are disordered. The FARM region (179–183) is partially resolved, while D319 of the SARM (319–323) coordinates a pentahydrated Mg2+ at site B (Mg2+B).
Next, we determined a Mg2+-bound IPP complex (PDB 8A6V) that shows significant discrepancies between the two subunits. SubA displays an open state and contains a single IPP molecule at the Al site with a 5 Å distance to Mg2+B, mimicking the native substrate DMAPP (Fig. 3a). D179 and D183 of the FARM rearrange and coordinate PPAl in a staggered conformation by Mg2+A and Mg2+C. Surprisingly, the electron density map depicts an additional pentahydrated metal site (Mg2+D) at the β-phosphate (Pβ) with a major impact on catalysis (as decribed in the following section). Binding of IPPAl introduces a strong hydrogen bond between R188 and Pβ that reorders the residues of αD–αE into a defined substrate loop. Notably, R184 and F202 in this region engage in cation–π interactions with their equivalents in subB, indicating intersubunit cooperativity between the Al sites (Fig. 4a).

Snapshots of the active site during catalysis and in complex with inhibitors. Metal ions are drawn in black (Mg2+) and pink (Mn2+), H-bonds are illustrated as black dots. Omit electron density maps are shown in green (FO–FC, 3.0 σ), grey (2FO–FC, 1.0 σ), pink (ano Mn, 10.0 σ) and blue (ano Br, 10.0 σ). a, PcIDS1Mg:IPP (PDB 8A6V); IPP imitates the native DMAPPAl moiety with a fourth Mg2+ (site D) coordinating Pβ. b, PcIDS1Mg:IPP:IPP (PDB 8A6V); a second IPP molecule binds to basic residues at the homoallylic site (HAl). Mg2+B is pushed towards PPAl, which adapts an eclipsed conformation to depart as PPi. c, PcIDS1Mg:ZOL:IPP (PDB 8A7C); the bisphosphonate inhibitor ZOL imitates PPAl coordination in the closed state. d, PcIDS1Mg:GPP (PDB 8A70); upon prenyl elongation and PPi release, GPP binds to the allylic site (Al) in the open state. Site D is occupied by a pentahydrated Mg2+ ion. Mg2+ drives the repetition of the cycle to form FPP (left). e, PcIDS1Mg:3-Br-GPP (PDB 8A7A); the bromine atom in 3-Br-GPP prevents carbocation formation. f, PcIDS1Mn:IPP:IPP (PDB 8A6Z); both subunits form the closed state with IPPAl and IPPHAl with Mn2+ at a 100-fold excess of Mg2+. g, PcIDS1Mn:ZOL:IPP; the active site is occupied by Mn2+ at sites A through C in complex with the inhibitor ZOLAl and IPPHAl (right, PDB 8A7J). A 50-fold excess of Mg2+ reduces the anomalous signal at A and B (left, PDB 8A7K). h, PcIDS1Mn:GPP:GPP (PDB 8A73); Mn2+A and Mn2+C sequester the PPAl moiety and prevent coordination of Mg2+D. A second GPP acts as an allosteric inhibitor (right). GPP is the main product of catalysis in the presence of Mn2+ (left). Inhibition arrows are highlighted in black. Colour coding is described in the Methods. A stereo version of this figure is presented in Extended Data Fig. 3. A comprehensive overview of all structures is shown in Supplementary Figs. 3–6.

Domains undergoing distinct conformational changes during catalysis are highlighted on a surface representation of PcIDS1Mg:IPP:IPP (PDB 8A6V). DMAPP was modelled into the allylic site (DMAPPAl (mod)) to chains A (white) and B (grey; Supplementary Fig. 7b). a, DMAPPAl induces substrate loop formation (183–201, mint) and initiates a cooperative effect on the second subunit through cation–π interactions between R184 and F202 residues (purple). GPP formation can occur in both active sites simultaneously (inset green scheme). b, IPPHAl forms the closed state with R188 and R189 (mint) facing the PP moieties. D319 and metal B move towards DMAPPAl and stabilize the eclipsed PP conformation with K333 (gold). Thereby, residues 328–331 arrange into a helix and anchor the substrate loop. c, The C terminus (brown) is recruited from a dynamic state (green) by inverting Y425 to engage in π-stacking with F398. F315 selects for IPPHAl and provides cation–π interactions to R427. The terminal carboxy group of A429 is located between R427 and K133, which stabilizes PPHAl. d, In the presence of IPPHAl, Q172 breaks the hydrogen bond to E169 (green) and displaces Y280, causing a rotation of the DMAPPAl prenyl moiety towards L176. e, The C10 prenyl chain of GPP disrupts the substrate loop at the N terminus of αEB in the neighbouring subunit by pushing M178A into I205B. PcIDS1 is a single-subunit catalyst for FPP production (inset red scheme). Colour coding is described in the Methods.
However, subB forms the closed state with two IPP molecules and depicts extensive conformational changes compared to the open state (Fig. 3b and Supplementary Table 4). Here, the domain around the SARM is rearranged and pushed towards the Al site together with α3 and the N terminus of αI (Extended Data Fig. 2). The flexible region 327–336 forms a short α-helix that anchors K333 between PPAl and D319 (Fig. 4b). This induced fit moves Mg2+B by 4 Å to interact with both phosphates of IPPAl, while Mg2+D is released. Thereby, the PPAl moiety is forced into an eclipsed configuration that promotes carbocation formation. Binding of the second IPP molecule to K133, R136 and R189 at the homoallylic site recruits the C terminus (425–429; Extended Data Fig. 1b). Stabilized by cation–π interactions with R427, the F315 side chain positions IPPHAl for nucleophilic attack (Fig. 4c). These structural observations are supported by a decreased thermal resilience of the K133R, K133A and F315A variants in complex with DMAPP and IPP (Fig. 2b).
The comparison between the open (subA) and closed (subB) states reveals a feedback relation between the HAl and Al sites. Binding of IPPHAl displaces interactions between E169, Q172 and Y280, resulting in a 120° clockwise rotation of the allylic prenyl unit (Fig. 4d and Supplementary Fig. 7c). The significantly reduced stability for DMAPP and IPP in the Y280F mutant indicates catalytic relevance of this regulatory cascade (Fig. 2c). Modelling of DMAPPAl and IPPHAl into the active site shows that reorientation of the allylic substrate results in an ideal distance (3.2 Å) between its C1 atom and the homoallylic double bond, facilitating C–C bond formation.
To investigate the second elongation step from GPP to FPP, we determined the complex structure of PcIDS1Mg:GPP (PDB 8A70). In the presence of Mg2+, GPPAl binds to the metal cluster (A–D) at the Al site in subA (open state; Fig. 3d) and subB remains vacant. While GPPAl adopts the binding mode of IPPAl with C1–C5, the distal C6–C10 unit inverts the M178 side chain and generates a spacious specificity pocket that is inaccessible in the IPPAl complex. To elucidate the structural features of FPP formation in the closed state, we applied IPP together with a geranyl pyrophosphate derivative brominated at position C3 (3-Br-GPP). In this surrogate, the electrophilic bromine prevents carbocation formation and blocks catalysis20. Our crystallographic studies show that 3-Br-GPP mimics GPPAl coordination, and the halogen atom occupies a similar position as the native 3-methyl group (Fig. 3e; PDB 8A7A). However, the HAl site remains vacant as 3-Br-GPP competes with IPPHAl binding by stabilizing the open state through a polar interaction between Y280 and Br (Supplementary Fig. 4f). In the absence of experimental data, we modelled the active GPPAl:IPPHAl complex (Supplementary Fig. 7d). To reach the closed state, GPPAl must undergo a similar reorientation as that of DMAPPAl, suggesting that GPP and FPP syntheses follow the same catalytic process. Yet, the relationship between structure and product distribution remains unresolved.
Dimer symmetry governs product distribution
Structural analysis of PcIDS1Mg:GPP (PDB 8A70) reveals an asymmetric complex in which the ligand is bound only to subA. This broken dimer symmetry originates from the C10 prenyl moiety that flips the side chain of M178 in subA and displaces I205 in the adjacent subB (Fig. 4e). The rearrangements disrupt the substrate loop and destabilize the Al and HAl sites of subB (Extended Data Fig. 4a). Interestingly, the structure of the F315A variant reveals that GPPAl still binds to both subunits in response to a modified catalytic network (PDB 8A74; Extended Data Fig. 4b). While GPPAl follows the described binding mode in subA, the orientation of M178 in subB forces the prenyl moiety of the ligand into a kinked conformation. This unphysiological state is stabilized by the metal cluster, and the substrate loop in subB is only defined at the FARM region. Although this asymmetric complex displays an affinity for ligands at the Al site, IPPHAl cannot coordinate to subB unless GPPAl dissociates from subA. In consequence, PcIDS1 acts as a single-subunit catalyst during FPP formation.
On the other hand, the PcIDS1Mg complex with IPPAl (PDB 8A6V) displays intersubunit cooperativity between the two substrate loops. However, IPPHAl is bound only to one subunit and does not prove that catalysis can take place simultaneously in both active sites. To obtain a symmetric complex with IPPHAl in both subunits, a strong ligand for the Al site like ZOL is required. This inhibitor significantly increases the thermal stability of all PcIDS1 variants apart from D319A, which prevents coordination of Mg2+B (Fig. 2 and Supplementary Figs. 2 and 4h). These results indicate that the PcIDS1:ZOL complex favours the closed state and induces formation of the HAl site, which is confirmed by the 9 °C shift upon addition of IPP. Indeed, the structure of PcIDS1Mg:ZOL:IPP (PDB 8A7C) displays symmetry between subA and subB and mimics the active site prior to GPP synthesis (Fig. 3c). The protonated imidazole group of ZOLAl imitates the nascent carbocation of DMAPPAl, while the central hydroxymethyl group constrains the bisphosphonate to a fixed conformation. With four negative charges, bisphosphonate resembles the eclipsed PPAl moiety and engages with all metal sites to induce IPPHAl binding in its native orientation (Supplementary Fig. 5b). Thus, the binding strength of C5 ligands at the Al site correlates with the affinity for IPPHAl and promotes catalysis in both subunits.
Mn2+ ions increase affinity for the allylic ligand
To determine the binding mode of HMIs, we solved the crystal structure of PcIDS1Mn:ZOL:IPP in a buffer containing a mixture of MgCl2 and MnCl2 (5:1 stoichiometry; PDB 8A7J). The subunits are identical to PcIDS1Mg:ZOL:IPP (PDB 8A7C) apart from the anomalous Mn signal for all three metal positions (Fig. 3g, left and Supplementary Table 4). The influence of environmental HMI concentrations was evaluated by a second structure in the presence of 50-fold Mg2+ excess over Mn2+ (PDB 8A7K). Despite the abundance of Mg2+, sites A and B still harbour Mn2+ (Fig. 3g, right), confirming the preference of the enzyme for ligands coordinated by HMIs. Unlike Mn2+A and Mn2+B, which rest in a rigid, bidentate coordination with the phosphonate groups of ZOLAl, position C can be selected through interactions with the FARM during substrate binding. Accordingly, Mn2+C remains the dominant metal species even at high Mg2+ concentrations to enhance the ionization of Pα during catalysis and stabilize the nascent carbocation.
However, the enforced closed state through ZOLAl coordination does not provide information about the metal-dependent interplay between the Al and HAl sites. Therefore, we applied IPP as a surrogate for DMAPPAl to evaluate Mn2+ binding in a native context and solved the structure of PcIDS1Mn:IPP:IPP (PDB 8A6Z). The complex forms a symmetric dimer with IPPAl and IPPHAl in the closed state and is similar to subB of PcIDS1Mg:IPP:IPP (PDB 8A6V; Supplementary Fig. 3f). Remarkably, the three metal positions depict a uniform anomalous density for Mn2+ despite a 100-fold Mg2+ excess in the crystallization buffer (Fig. 3f). Thus, in the absence of IPPHAl, the staggered configuration of PPAl selects for Mn2+A and Mn2+C. Once IPP coordinates the HAl site, the high affinity of Mn2+ replaces Mg2+B and stabilizes the eclipsed orientation of PPAl, driving PPi release.
So far, the described Mn2+-bound complexes all form symmetric homodimers with ligands at both Al and HAl sites. To determine whether HMIs are also recruited to asymmetric arrangements like those in PcIDS1Mg:IPP or PcIDS1Mg:GPP, we solved the structure of PcIDS1Mn:GPP (PDB 8A73). With both subunits in the open state, GPPAl is bound only to subA. While Mn2+A and Mn2+C coordinate GPPAl, the pentameric water cluster at site B favours Mg2+ (Fig. 3h). Interestingly, the complex lacks electron density for a metal at site D, as observed in PcIDS1Mg:GPP (PDB 8A70). Apparently, the stronger Lewis acidity of Mn2+A and Mn2+C reduces the charge density of PPAl and prevents coordination of Mg2+D. Without this auxiliary metal cluster, Pβ moves 1.0 Å towards the SARM and interacts with the hydration sphere of Mg2+B (Supplementary Fig. 8). These rearrangements align the subunit for IPPHAl coordination and facilitate the transition to the closed state. In consequence, HMIs enhance the intramolecular effect that forms the HAl site when a ligand is bound to Al. This cooperativity stabilizes both HAl sites during GPP synthesis but is limited to subA upon elongation to FPP. Therefore, HMIs counteract FPP formation by recruiting IPPHAl to subB and displacing GPPAl from subA.
Mn2+ recruits GPP to an allosteric pocket
The structure of PcIDS1Mn:GPP (PDB 8A73) contains well-defined electron density for a second GPP molecule (GPPAllo; Fig. 3h) that is absent in the Mg2+-bound complex. Its prenyl moiety protrudes into a 7-Å-wide and 15-Å-deep channel between αC and αG as well as αJ in subA. V286 and I420 form the bottom of this specificity pocket and act as a molecular ruler, whereas F282 and F315 engage in π-stacking with both allyl groups. Notably, the PP moiety of this allosteric ligand coordinates with residues of the HAl site. While R136 contacts both phosphates, Pβ is further stabilized by the αC helix dipole and forms a salt bridge with R189 of the substrate loop. GPPAllo keeps the subunit in the open conformation, but the C terminus is directed towards the HAl site, and residues 328–331 form the helical turn as seen in PcIDS1Mn:IPP:IPP (PDB 8A6Z). These findings indicate that IPPHAl binding and GPPAllo binding are mutually exclusive, and both depend on the intramolecular effect of HMIs. To elucidate the architecture of the allosteric site in the closed state, we determined the structure of PcIDS1Mg:ZOL:GPP (PDB 8A7L). In this complex, ZOLAl displaces GPPAl, and the structural rearrangements prevent formation of the allosteric specificity pocket (Supplementary Fig. 6c). However, GPP still binds to the HAl site by inverting its PP moiety and exposing the prenyl unit towards the protein surface. Taken together, GPPAllo represents a competitive inhibitor to IPPHAl that stabilizes the open state in the presence of HMIs.
Discussion
Our study reveals how PcIDS1 couples metal coordination and dimer symmetry with an allosteric site to fine-tune product distribution during the life cycle of P. cochleariae. In this enzyme, diversified regulatory domains evolved from a common scIDS topology to steer the biosynthetic flux of defensive monoterpene or hormonal sesquiterpene precursors. Exogenous metal cofactors shift the product distribution from FPP to GPP. Compared with the abundance of Mg2+ in the larval fat body tissue, Mn2+ and Co2+ are found at a ratio of 1:300 and 1:20,000, respectively10. HMIs compensate these low intracellular concentrations with their strong Lewis acidity and engage in stable substrate complexes that bind to PcIDS1 (refs. 21,22). Since HMIs accelerate GPP and FPP formation to the same extent, they exploit additional mechanisms to alter product distribution10,22,23,24,25.
Symmetry is a common regulatory principle in scIDS enzymes that use the dimer interface to form selective binding sites for their allylic substrates. While heterodimeric GPPSs with a single active site have been identified26, PcIDS1 enters the reaction as a symmetric homodimer. For GPP synthesis, DMAPPAl promotes intersubunit cooperativity and induces structural rearrangements that enable IPPHAl binding. Compared to the apo enzyme, the thermal resilience of PcIDS1:DMAPP is increased, but an even stronger effect is seen for IPP (Fig. 2a). Combined with crystallographic studies, these findings prove a competitive inhibition of IPPAl at high concentrations by binding to the open state as well as the closed state10,27,28. In addition, with either IPPHAl or DMAPPHAl, HMIs abolish Mg2+D coordination at PPAl and enhance GPP formation (Fig. 5a). This metal-dependent cross-talk between the Al and HAl sites results in improved protein stability with both C5 ligands and agrees with previous kinetic studies10.

The two subunits of the homodimer (subA and subB) are depicted in either the open (rhombic) or closed (rectangle) state. Substrate loops are drawn in black with unstructured regions as grey dashes. a, PcIDS1 catalyses the reaction of DMAPP and IPP to GPP in both subunits at the same time (symmetric, pink scheme). HMIs accelerate the transition to the closed state, while Mg2+ slows down catalysis. b, The enzyme procedurally converts GPP and IPP to FPP in a single subunit (asymmetric, green scheme). HMIs counteract GPPAl coordination, while Mg2+ stabilizes the open state. c, Mg2+D represents a modulator of IPPHAl binding during GPP (left) and FPP (right) synthesis. d, In the presence of HMIs, GPPAllo binds to both substrate loops and prevents GPP accumulation (left). GPPAllo binds only to subA (right), and C5 ligands that coordinate to subB displace GPPAl from subA to counteract FPP formation (dashed arrow). An extended reaction network is provided in Supplementary Fig. 11.
Once GPP is formed, it can either dissociate to enter monoterpene biosynthesis or proceed to the Al site for further elongation. Binding of GPPAl, however, breaks the dimer symmetry and restricts catalysis to one active site for FPP synthesis (Fig. 5b). Key to this asymmetry is the unique chain length determination domain sequence SLxxM (located at −5 to −1 upstream of the FARM; S, serine; L, leucine; M, methionine; x, any amino acid). So far, characterized scIDSs harbour aromatic sidechains at −5 and −4 that mediate substrate selectivity, and a small residue at −1 (refs. 4,29,30,31,32). In PcIDS1, S174 of the chain length determination domain motif provides stability at the dimer interface, and L175 forms a hydrophobic patch that guides GPPAl towards M178. An induced flip of the methionine side chain restricts GPPAl to one active site at a time. Such a regulatory connection between distant active sites of homodimeric enzymes was recently described for the human transketolase and represents a fundamental principle of positive or negative cooperativity33. Notably, methionine at −1 is conserved in a mosquito scIDS with similar metal-dependent product regulation to PcIDS1 (ref. 11).
The binding mode of GPPAl involves significant hydrophobic contacts that stabilize the complex and are independent from the metal cluster at Al. During FPP formation in the presence of Mg2+, the additional metal site D, however, decelerates the reaction rate10 and selectively recruits IPPHAl only to the catalytic subunit (Fig. 5c). HMIs, on the other hand, put the asymmetric GPPAl complex at a disadvantage by simultaneously increasing the affinity of C5 ligands to all Al and HAl sites. In consequence, IPPHAl binding to the disrupted subunit counteracts GPPAl coordination (Fig. 5d). This results in a 20-fold decreased GPP affinity when IPP is applied with HMIs during FPP formation10. Thus, HMIs will always prefer the symmetric state with two stabilized substrate loops and accelerate GPP synthesis under physiological conditions.
To prevent GPP accumulation, HMIs fine-tune PcIDS1 activity by generating an allosteric binding site (Fig. 5d). The C10 prenyl unit of GPPAllo protrudes into a hydrophobic channel, while the PPAllo moiety competes with IPPHAl. This coordination turns the product GPP into an allosteric inhibitor and slows down catalysis by trapping the subunit in a transition state (Supplementary Fig. 9). A similar regulation has been characterized for a C15-specific binding site in human FPPS (HsFPPS) that mediates end-product inhibition14. However, the human FPPAllo does not interact with the substrate loop and protrudes into a 4-Å-deeper cavity by inverting the prenyl moiety. This orientation is stabilized by L344, whereas the corresponding I420 in PcIDS1 tailors the allosteric binding channel to GPP (Supplementary Fig. 10). When this amino acid is replaced with alanine, the size of the cavity increases, but neither FPPAllo, nor GPPAllo, nor Mn2+ at the Al site are observed in the mutant structure (Supplementary Fig. 6d; PDB 8A7R). These findings highlight that the conserved architecture of allo sites has diverged into distinct roles during evolution. GPPAllo in PcIDS1 is strictly dependent on the allylic metal cluster that induces formation of the HAl site and features a unique mode of action.
Our present study highlights an intricate reaction network for PcIDS1 that combines inter- and intramolecular effects with allosteric regulation. As indicated by previous kinetic assays, HMIs attain a high affinity for DMAPPAl (Michaelis constant (Km) = 11.6 µM) and IPPHAl (Km = 0.84 µM) through cooperative symmetry between the subunits10. However, if DMAPP and IPP deplete during GPP accumulation, GPPAllo could promote allosteric feedback and throttle the impact of HMIs on the reaction rate. PcIDS1 catalyses the first elongation step significantly slower with Mg2+ alone (25% of maximum velocity of the enzymatic reaction (vmax)) and instead displays higher affinity for GPPAl (Km = 1.2 µM)10. Our crystal structures highlight that Mg2+ stabilizes an asymmetric complex with half-site reactivity to produce FPP, the precursor of sesquiterpene hormones. In vivo, an influx of exogenous HMI cofactors counteracts this second reaction and guides synthesis back to GPP (Supplementary Fig. 11). Thus, the dynamic nature of PcIDS1 allows P. cochleariae to fine-tune its terpene metabolism in response to environmental trigger factors during all life stages.
Methods
Bacterial culture
The gene of PcIDS1 (GenBank AGE89831.1), which lacks the N-terminal signal peptide (residues 1 to 85)10, was cloned into a pET-28b expression vector modified to encode an N-terminal His6-SUMO tag. Site-directed mutagenesis (Supplementary Table 1) was performed with the QuikChange II kit (Agilent Technologies) according to the manufacturer’s instructions and verified by Sanger sequencing (GATC Biotech). Escherichia coli K12 BL21(DE3) cells were transformed by electroporation and grown in glass shake flasks containing 2 l lysogenic broth (50 mg l–1 kanamycin) at 37 °C. After reaching an optical density measured at a wavelength of 600 nm (OD600) of 0.6, flasks were stored at 4 °C for 30 minutes before adding 1 mM isopropylthiogalactoside (final concentration) to induce protein expression, which occurred overnight at 20 °C. Cell pellets were collected by centrifugation, washed once with 0.9% (w/v) NaCl and stored at −20 °C.
Protein purification
E. coli pellets of 10 g were dissolved in 50 ml buffer A (100 mM Tris/HCl pH 7.5, 500 mM NaCl, 10% (v/v) glycerol, 20 mM imidazole, 10 mM 2-mercaptoethanol (β-ME), 5 mM MgCl2) and solubilized by sonication (Branson Digital Sonifier 250). After centrifugation (40,000g, 4 °C, 30 min), the supernatant was applied (5 ml min−1) to a 5 ml HisTrap HP column with an ÄKTA Pure system (GE Healthcare), previously equilibrated with buffer A. Buffer A containing 5% buffer B (buffer A with 500 mM imidazole) was used to wash the column until the absorbance signal (280 nm) returned to the baseline. The protein of interest was eluted by a linear gradient (5–100% buffer B) within 50 ml total volume. Fractions containing sufficient concentrations of pure protein were pooled and spiked with 0.5 mg SUMO protease (Ulp1 from Saccharomyces cerevisiae) and dialysed overnight at 4 °C against 5 l buffer C (20 mM Tris/HCl pH 7.5, 100 mM NaCl, 10% (v/v) glycerol, 1 mM β-ME, 5 mM MgCl2). HisTrap affinity chromatography was repeated, and the flow-through was concentrated to 2 ml using Amicon Ultra-15 centrifugal filters. Centrifugation (20,000g, 4 °C, 10 min) removed residual protein aggregates, and the supernatant was used for size exclusion chromatography with a HiLoad Superdex 200 16/60 column in buffer D (buffer C containing 1 mM tris(2-carboxyethyl)phosphine (TCEP) instead of β-ME) at 1.5 ml min−1. High purity peak fractions were pooled, concentrated to at least 40 mg ml−1 (1 mM) and either used immediately or stored at −80 °C.
Thermal shift assay
A 96-well transparent polymerase chain reaction plate was prepared with 17 µl buffer in each well (final concentrations, 100 mM MES buffer pH 7.0, 100 mM NaCl, 10% (v/v) glycerol, 5 mM MgCl2, 2.5 mM TCEP) both with and without addition of 1 mM MnCl2. Ligand molecules (in 1 µl H2O) were added to achieve final concentrations of 25 and 200 µM, respectively. In the presence of ZOL, IPP and GPP were applied at 100 µM. Some 1 µl of a 0.25 mg ml−1 protein–buffer solution (6.25 µM final concentration) was added using a Phoenix nano-dispenser system (Art Robbins Instruments), followed by 1 µl of an aqueous 1:40 dilution of Sypro Orange Stain (Merck). Plates were sealed, carefully shaken and centrifuged (1,000g, 4 °C, 5 min) to remove air bubbles. All samples were equilibrated to 4 °C in a CFX96 reverse transcriptase-polymerase chain reaction (RT-PCR) system (Biorad), and emission was detected while the temperature increased by 0.5 °C in 10 s intervals to 95 °C. Melting temperatures were derived from the inflection points of the fluorescence signal with CFX Maestro (v.4.1) and analysed (in triplicate) with OriginPro 2020 (v.9.7.0.185).
Protein crystallization and structure determination
Crystals of PcIDS1 wild-type and mutant proteins were grown using sitting drop vapour diffusion at 20 °C. Protein samples were applied at 20 mg ml–1 in buffer D (with/without 1 mM MnCl2) and mixed with reservoir solution by an Oryx4 system (Douglas Instruments). Droplet compositions including the applied ligand molecules (2 mM, 1 mM for ZOL) are denoted in Supplementary Table 2. Diffracting crystals grew within a few weeks and were cryoprotected by adding 1 µl of mother liquor containing 30% (v/v) glycerol prior to vitrification in liquid nitrogen.
Native datasets were collected at beamline X06SA (Swiss Light Source, Paul Scherrer Institute, Villigen, Switzerland) with synchrotron radiation (wavelength λ = 1.0 Å). Anomalous scattering was additionally recorded at 1.89 Å (Mn) or 0.92 Å (Br) for crystals grown in the presence of MnCl2 and 3-Br-GPP, respectively. Diffraction intensity data were evaluated by the XDS program and processed with XSCALE (Supplementary Table 3)34. Resolution limits were chosen to meet the following criteria: signal to noise ratio I/σ(I) > 2.0, statistic for the precision of the measurements of each unique reflection Rmerge < 70% and redundancy of >3.0 (Supplementary Table 3). The initial structure of PcIDS1 was solved in space group P21 (PDB 8A6U). The structure of FPPS from Gallus gallus (PDB 1UBX; ref. 4) was used for molecular replacement by Patterson search calculations with Phaser35. All other PcIDS1 structures were determined using the apo structure. Models were built, refined and completed with cofactors and ligands in COOT36 with intermittent, restrained refinements using REFMAC5 (ref. 37). Water molecules were positioned by ARP/wARP solvent38. TLS (Translation/Libration/Screw) and restrained refinements yielded adequate model parameters (Supplementary Table 3) as well as root-mean-square deviation (r.m.s.d.) values of bond lengths and angles (validated by PROCHECK39). To identify Mn and Br positions in the protein–ligand complex, anomalous difference Patterson maps were calculated with fast Fourier transformation40. The crystal structures were deposited in the RCSB Protein Data Bank (Supplementary Table 3).
Figure illustration
To systematically characterize substrate coordination in PcIDS1, figures were prepared with Molscript/Bobscript41. Protein residues (grey, marked by a one-letter code) and ligands at the allylic (Al, cyan), homoallylic (HAl, purple) and allosteric (Allo, teal) sites are shown as sticks. Mg2+ ions (black), Mn2+ ions (pink) and water molecules (red) are depicted as balls with cluster interactions represented by black dashes. Structural superpositions with PyMol (ref. 42) highlight discrepancies at the active site (coloured and grey). Unless noted otherwise, all electron density maps are FO–FC (green or blue mesh, contoured to 3.0 σ) or anomalous signal maps (pink for Mn and blue mesh for Br, 10.0 σ) with respective ligands omitted prior to phasing. The 2FO–FC maps around Mg2+ ions are contoured to 1.0 σ and coloured grey. Calculations combine the observed diffraction data, FO, with the diffraction data calculated from the atomic model, FC; σ represents the density value above the average value.
Annotations
The r.m.s.d. values were calculated for the Cα backbone with Top3D (ref. 43) and are annotated in Supplementary Table 4.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
References
Buckingham, J. Natural Products Desk Reference (CRC Press, 2017).
Hillier, S. G. & Lathe, R. Terpenes, hormones and life: isoprene rule revisited. J. Endocrinol. 242, R9–R22 (2019).
Wang, K. Isoprenyl diphosphate synthases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1529, 33–48 (2000).
Tarshis, L. C., Proteau, P. J., Kellogg, B. A., Sacchettini, J. C. & Poulter, C. D. Regulation of product chain length by isoprenyl diphosphate synthases. Proc. Natl Acad. Sci. USA 93, 15018–15023 (1996).
Cusson, M. et al. Biosynthesis and release of juvenile hormone and its precursors in insects and crustaceans: the search for a unifying arthropod endocrinology. Insect Biochem. 21, 1–6 (1991).
Kislow, C. J. & Edwards, L. J. Repellent odour in aphids. Nature. 235, 108–109 (1972).
Weibel, D. B. et al. Iridoid biosynthesis in staphylinid rove beetles (Coleoptera: Staphylinidae, Philonthinae). Insect Biochem. Mol. Biol. 31, 583–591 (2001).
Gilg, A. B., Bearfield, J. C., Tittiger, C., Welch, W. H. & Blomquist, G. J. Isolation and functional expression of an animal geranyl diphosphate synthase and its role in bark beetle pheromone biosynthesis. Proc. Natl Acad. Sci. USA 102, 9760–9765 (2005).
Vandermoten, S. et al. Characterization of a novel aphid prenyltransferase displaying dual geranyl/farnesyl diphosphate synthase activity. FEBS Lett. 582, 1928–1934 (2008).
Frick, S. et al. Metal ions control product specificity of isoprenyl diphosphate synthases in the insect terpenoid pathway. Proc. Natl Acad. Sci. USA 110, 4194–4199 (2013).
Rivera-Perez, C., Nyati, P. & Noriega, F. G. A corpora allata farnesyl diphosphate synthase in mosquitoes displaying a metal ion dependent substrate specificity. Insect Biochem. Mol. Biol. 64, 44–50 (2015).
Vandermoten, S. et al. Structural features conferring dual geranyl/farnesyl diphosphate synthase activity to an aphid prenyltransferase. Insect Biochem. Mol. Biol. 39, 707–716 (2009).
Nagel, R., Schmidt, A. & Peters, R. J. Isoprenyl diphosphate synthases: the chain length determining step in terpene biosynthesis. Planta 249, 9–20 (2019).
Park, J., Zielinski, M., Magder, A., Tsantrizos, Y. S. & Berghuis, A. M. Human farnesyl pyrophosphate synthase is allosterically inhibited by its own product. Nat. Commun. 8, 14132 (2017).
Gabelli, S. B. et al. Structure and mechanism of the farnesyl diphosphate synthase from Trypanosoma cruzi: implications for drug design. Proteins 62, 80–88 (2006).
Ling, Y., Li, Z.-H., Miranda, K., Oldfield, E. & Moreno, S. N. J. The farnesyl-diphosphate/geranylgeranyl-diphosphate synthase of Toxoplasma gondii is a bifunctional enzyme and a molecular target of bisphosphonates. J. Biol. Chem. 282, 30804–30816 (2007).
Holm, L. Using Dali for protein structure comparison. Methods Mol. Biol. 2112, 29–42 (2020).
Mao, J. et al. Solid-state NMR, crystallographic, and computational investigation of bisphosphonates and farnesyl diphosphate synthase-bisphosphonate complexes. J. Am. Chem. Soc. 128, 14485–14497 (2006).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Vattekkatte, A., Gatto, N., Schulze, E., Brandt, W. & Boland, W. Inhibition of a multiproduct terpene synthase from Medicago truncatula by 3-bromoprenyl diphosphates. Org. Biomol. Chem. 13, 4776–4784 (2015).
Trachtman, M., Markham, G. D., Glusker, J. P., George, P. & Bock, C. W. Interactions of metal ions with water: ab initio molecular orbital studies of structure, bonding enthalpies, vibrational frequencies and charge distributions. 1. Monohydrates. Inorg. Chem. 37, 4421–4431 (1998).
Bock, C. W., Katz, A. K., Markham, G. D. & Glusker, J. P. Manganese as a replacement for magnesium and zinc: functional comparison of the divalent ions. J. Am. Chem. Soc. 121, 7360–7372 (1999).
Sissi, C. & Palumbo, M. Effects of magnesium and related divalent metal ions in topoisomerase structure and function. Nucleic Acids Res. 37, 702–711 (2009).
Dismukes, G. C. Manganese enzymes with binuclear active sites. Chem. Rev. 96, 2909–2926 (1996).
Irving, H. & Williams, R. J. P. Order of stability of metal complexes. Nature 162, 746–747 (1948).
Chang, T.-H. et al. Structure of a heterotetrameric geranyl pyrophosphate synthase from mint (Mentha piperita) reveals intersubunit regulation. Plant Cell 22, 454–467 (2010).
Reed, B. C. & Rilling, H. C. Substrate binding of avian liver prenyltransferase. Biochemistry. 15, 3739–3745 (1976).
Barnard, G. F. & Popják, G. Human liver prenyltransferase and its characterization. Biochim. Biophys. Acta Enzymol. 661, 87–99 (1981).
Wang, K. & Ohnuma, S. Chain-length determination mechanism of isoprenyl diphosphate synthases and implications for molecular evolution. Trends Biochem. Sci. 24, 445–451 (1999).
Vandermoten, S., Haubruge, E. & Cusson, M. New insights into short-chain prenyltransferases: structural features, evolutionary history and potential for selective inhibition. Cell. Mol. Life Sci. 66, 3685–3695 (2009).
Stanley Fernandez, S. M., Kellogg, B. A. & Poulter, C. D. Farnesyl diphosphate synthase. Altering the catalytic site to select for geranyl diphosphate activity. Biochemistry 39, 15316–15321 (2000).
Narita, K., Ohnuma, S. & Nishino, T. Protein design of geranyl diphosphate synthase. Structural features that define the product specificities of prenyltransferases. J. Biochem. 126, 566–571 (1999).
Dai, S. et al. Low-barrier hydrogen bonds in enzyme cooperativity. Nature 573, 609–613 (2019).
Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 (2011).
Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 (2008).
Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. PROCHECK. J. Appl. Crystallogr. 26, 283–291 (1993).
Read, R. J. & Schierbeek, A. J. A phased translation function. J. Appl. Crystallogr. 21, 490–495 (1988).
Kraulis, P. J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24, 946–950 (1991).
Delano, W. L. The PyMOL Molecular Graphics System (Schrödinger, 2002).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011).
Acknowledgements
This research was supported by the German Research Foundation (DFG, grant no. 1861/5-2) and the Max Planck Society. We thank E. M. Huber for valuable input during manuscript preparation and the staff of beamline X06SA at the Paul Scherrer Institute, SLS, Villigen (Switzerland) for assistance during data collection.
Funding
Open access funding provided by Max Planck Society.
Author information
Authors and Affiliations
Contributions
F.E., A.V., W.B. and M.G. designed, conducted and analysed the experiments. F.E. and M.G. draughted the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemistry thanks Kai Tittmann and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Topology of PcIDS1.
a, Cartoon representation of PcIDS1 with α-helices shown as cylinders in a spectrum from blue to red (N- to C-terminus). Helices are labelled from A-H, 1-3 and I-J. Sticks and balls representations of the natural ligands highlight their respective binding mode. b, Topology of the arrangement in a single subunit. The Cα chain trace is indicated by arrows. Residues coordinating ligands in the allylic (Al, cyan), homoallylic (HAl, purple) and allosteric (Allo, teal) site are connected to their respective position in the enzyme and coloured according to the nature of the interaction. An asterisk indicates a residue from the adjacent subunit. The C-terminus that orders upon binding of IPPHAl or GPPAllo is highlighted in red.
Extended Data Fig. 2 Structural plasticity of PcIDS1 during catalysis.
Surface representations of PcIDS1 subunits in complex with different ligands. The apo structure (1, PDB ID 8A6U) features a deep central cleft surrounded by disordered regions (highlighted in red). Upon binding of DMAPPAl, the substrate loop between αD-E folds around the pyrophosphate (PP) moiety (2, PDB ID 8A6V, subA). Areas undergoing significant structural changes are shown in light green and residues disordered in the apo structure in dark green. The binding of IPPHAl (3, PDB ID 8A6V, subB) shifts the peripheral domain around the SARM towards the active centre (light pink) and orders the region between αH-1 as well as the C-terminus (dark pink). The presence of heavy metal ions (HMI) stabilises the accommodation of an allosteric GPP molecule (4, PDB ID 8A73) that interferes with IPPHAl binding. The C-terminus adapts a conformation as observed in the closed state. In absence of HMIs or upon release of GPPAllo, the PcIDS1:GPP complex (5, PDB ID 8A70) can either form FPP or start a new reaction cycle. Colour coding is described in Materials and Methods.
Extended Data Fig. 3 Stereo views of PcIDS1 structures.
a-j. All representations according to Fig. 3.
Extended Data Fig. 4 GPPAl-binding disrupts the substrate loop and HAl site in the neighbouring subunit.
a, Superposition of PcIDS1Mg:GPP (coloured) and PcIDS1Mg:IPP:IPP (white, DMAPP* in the figure): The C10 prenyl chain of GPPAl disrupts substrate loop formation in the neighbouring subunit by displacing M178A and I205B, which disorders the N-terminus of αEB. While GPP synthesis can take place synchronistically (top left scheme), the reaction to FPP can only occur successively (bottom right). b, PcIDS1_F315AMg:GPP: In presence of GPPAl in subA (green), IPPHAl coordination is prevented in subB (grey). The FARM at the disrupted substrate loop retains affinity for allylic ligands. GPPAl adopts a non-physiological, kinked conformation in subB to avoid steric clashes (orange dashes) with M178. c, Superposition of PcIDS1Mg:GPP (grey) with subA and subB of PcIDS1_F315AMg:GPP (cyan and teal): In absence of the phenyl sidechain, major rearrangements are observed for the regulatory network between R136, E169, Q172 and Y280. The kinked conformation of the GPPAl moiety in F315A_B displaces L176 with its 3-methyl group (arrows). However, C5 ligands would not interfere with M178 and L176 and could still bind to subB in presence of GPPAl. Colour coding is described in Materials and Methods.
Supplementary information
Supplementary Information
Supplementary Figs. 1–11, Tables 1–4, references and additional source data.
Supplementary Data 1
Source data for Supplementary Fig. 1a.
Supplementary Data 2
Source data analysis for Supplementary Fig. 2.
Supplementary Data 3
Source data for Supplementary Fig. 2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ecker, F., Vattekkatte, A., Boland, W. et al. Metal-dependent enzyme symmetry guides the biosynthetic flux of terpene precursors. Nat. Chem. 15, 1188–1195 (2023). https://doi.org/10.1038/s41557-023-01235-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41557-023-01235-9
