
Abstract
Understanding the contribution of immune mechanisms to Parkinson’s disease pathogenesis is an important challenge, potentially of major therapeutic implications. To further elucidate the involvement of peripheral immune cells, we studied epigenome-wide DNA methylation in isolated populations of CD14+ monocytes, CD19+ B cells, CD4+ T cells, and CD8+ T cells from Parkinson’s disease patients and healthy control participants. We included 25 patients with a maximum five years of disease duration and 25 controls, and isolated four immune cell populations from each fresh blood sample. Epigenome-wide DNA methylation profiles were generated from 186 samples using the Illumina MethylationEpic array and association with disease status was tested using linear regression models. We identified six differentially methylated CpGs in CD14+ monocytes and one in CD8 + T cells. Four differentially methylated regions were identified in monocytes, including a region upstream of RAB32, a gene that has been linked to LRRK2. Methylation upstream of RAB32 correlated negatively with mRNA expression, and RAB32 expression was upregulated in Parkinson’s disease both in our samples and in summary statistics from a previous study. Our epigenome-wide association study of early Parkinson’s disease provides evidence for methylation changes across different peripheral immune cell types, highlighting monocytes and the RAB32 locus. The findings were predominantly cell-type-specific, demonstrating the value of isolating purified cell populations for genomic studies.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder, for which there is currently no effective disease-modifying therapy. Over the last decades, a growing body of evidence has demonstrated the important role of the immune system in PD pathogenesis, yet the specific mechanisms involved and the interrelations between microglial and peripheral immune processes are incompletely understood1,2. The prospect of immunomodulation is currently one of the most promising strategies for disease modification in PD3, and an improved understanding of the contribution of different classes of leukocytes may be pivotal for further progress.
Highlighting the relevance of the adaptive immune system, T cells have been shown to infiltrate the substantia nigra4,5 and recognize peptides of the neuropathological hallmark protein α-synuclein presented by human leukocyte antigen (HLA) class II proteins6. Levels of α-synuclein autoantibodies have also been studied with mixed results7, and genome-wide association studies (GWAS) have consistently reported a significant PD risk signal in the HLA region on chromosome 68,9. However, studies integrating genetic association statistics with cell-specific data have primarily linked genetic PD risk to innate immunity and the myeloid lineage10,11,12,13, a finding that is also mirrored in Alzheimer’s disease (AD)14. In line with these observations, a recent transcriptomic study of CD14+ monocytes reported widespread alterations in PD15.
Epigenetics influence complex disease by mechanisms of gene regulation that are partially dynamic over time, cell-type-specific, and shaped by both genetic and environmental factors16. A growing body of evidence implicates epigenetic mechanisms in neurodegeneration and PD pathogenesis17,18,19. DNA methylation at CpG dinucleotides is the most studied epigenetic modification in the context of complex disease. Hypothesis-free epigenome-wide association studies (EWAS) have been performed in a small series of post-mortem human brain tissue comparing PD to control brains20,21,22 and a larger study of bulk cortical tissue from our group investigated Lewy body pathology as outcome, including donors with both PD and dementia with Lewy bodies23. Two PD EWAS have studied isolated cortical neurons using either flow cytometry24 or magnetic-activated cell separation25. A number of PD studies have investigated differential methylation in whole blood26,27,28,29,30. Sample sizes of whole blood PD EWAS have been gradually increasing, and the largest study to date included more than 2000 participants and reported a hypermethylated CpG site in PD near SLC7A11, a gene involved in glutamate signaling30. Methylation analyses in blood may be particularly interesting as a tool to characterize immune mechanisms in PD, and a recent EWAS study highlighted immune dysregulation both in PD generally and specifically in patients with depression29. These are promising findings, yet the use of whole blood for differential methylation analyses entails major caveats. Whole-blood EWAS typically applies an algorithm to adjust for estimated cell-type composition, but essentially measures the noisy, joint effect of methylation across all cell types in the sample, limiting both the power to detect cell-type-specific signals and the interpretation of positive findings. Furthermore, most studies have included patients many years from PD diagnosis, when downstream effects of the disease and its treatment may have a major impact on methylation.
To further elucidate the contribution of different classes of immune cells and identify cell-type-specific alterations in gene regulation, we performed epigenome-wide DNA methylation profiling of purified cell populations representing major peripheral cell types of the innate and adaptive immune system, namely CD14+ monocytes, CD19+ B cells, CD4+ T helper cells and CD8+ cytotoxic T cells in 25 patients with early PD and 25 healthy controls. We nominate differentially methylated CpGs and regions, with monocytes showing the highest number of significant findings.
Results
Patients were diagnosed based on Movement Disorders Society criteria31 and had a maximum disease duration of five years from motor onset at the time of blood draw. Eight out of 25 PD patients were included at the time of diagnosis and had not used any medication for PD. Mean disease duration from the time of PD diagnosis was 1.2 years and mean age at diagnosis was 55. Controls had no immunological nor neurological disease and no parkinsonism in first-degree family members. Demographics are presented in Table 1.
Visualizing overall methylation patterns across cell types
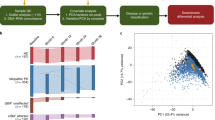
After quality control and filtering (see “Methods” and Supplementary Fig. 1), the final normalized methylation dataset included 482,470 probes and 186 samples. As expected, a multidimensional scaling plot demonstrates that the overall methylation pattern separates the CD14+ monocytes from the three classes of lymphocytes (Fig. 1). Within the lymphocyte group, CD4+, and CD8+ T cells cluster closer to each other than to CD19+ B cells.

The plot visualizes the overall similarities in methylation profiles across CD14+ monocytes, CD19+ B cells, CD4+ T cells and CD8+ T cells. It was generated based on the 100,000 most variable probes using the plotting function implemented in the minfi package.
Power analyses and distribution of P values
Power analyses using pwrEWAS32 indicated that for a study with 25 patients and 25 controls, the difference in methylation level would need to be 0.1 SD in order for statistical power to be 80% at a false discovery rate 0.05. This corresponds to a larger effect size than many differential methylation signals reported in complex disease EWAS. We therefore expected statistical power to be marginal, although the use of purified cell populations could limit the variance and improve power substantially compared to whole-blood EWAS of similar sample size.
Following association testing in each cell type by linear regression, including sex, age, and the first surrogate variable as covariates (see “Methods”), we assessed the distribution of P values by quantile–quantile plots and P value histograms33 (Supplementary Figs. 2 and 3). Assuming an adequate model and true association with disease for a subset of probes, we would expect a test-statistic inflation parameter λ close to 1 and a uniform distribution of P values with an enrichment in the lower end representing the true signals. This pattern was seen most clearly in CD14+ monocytes (λ = 1.03), with a slight overabundance of low P values also in CD8 + T cells.
In particular CD4 + T cells showed skewed P value distributions which could indicate poor modeling of data33. Previous EWAS studies in both PD25 and Alzheimer’s disease34 have reported striking sex differences, arguing for sex-stratified analysis of methylation data rather than merely inclusion of sex as a covariate in linear regression. We considered our sample size too small to attempt the identification of specific differentially methylated CpGs in sex-stratified analyses, yet we performed linear regression split by sex to evaluate P value distributions. Interestingly, sex-stratified regression improved the pattern of P value distributions for both CD8 + T cells and CD19 + B cells, but not for CD14+ monocytes or CD4 + T cells. This may indicate that sex-stratified analysis is warranted for specific cell types and should be explored in future larger studies.
Differentially methylated CpGs
Association testing in linear regression models including sex, age, and the first surrogate variable as covariates (see “Methods”) identified a total of seven signals passing a Bonferroni-corrected significance threshold of P < 2.25 × 10-8 (Table 2), including six signals from four different genomic loci in CD14+ monocytes and one signal in CD8+ T cells. No significant signals were detected in CD4+ T cells or CD19+ B cells. Manhattan plots for the cell types with significant findings are shown in Fig. 2.

The figure shows Manhattan plots from linear regression for CD14+ monocytes and CD8+ T cells. Loci with one or more significant probes are annotated with the name of the gene closest to the top associated probe.
To further assess whether these association signals were cell-type-specific or shared, we extracted summary statistics from the seven significant probes for all cell types (Supplementary Table 1). A nominal association (P < 0.05) was seen in at least one other cell type for four out of the seven probes, but no probe showed association at P < 0.05 across all cell types. Associations in additional cell types were generally much weaker than the top cell type, and only one passed a Bonferroni-corrected threshold for a total of 21 candidate tests (cg11473614, genome-wide significant in monocytes, P = 0.0021 in CD19+ B cells). Figure 3 illustrates the methylation patterns of a highly monocyte-specific probe versus a probe showing a similar trend across cell types. Violin plots of uncorrected beta values for the remaining significant probes are shown in Supplementary Fig. 4. We noted an unusual distribution of methylation levels for two of the probes, cg18523915 and cg14704780, showing particularly high variability in healthy controls. This might indicate that some unknown external factor is having a major impact on methylation at these CpGs, potentially confounding our association results. Cg18523915 has been reported to be significantly associated with outcome of COVID-19 in two recent EWAS studies of whole blood35 and peripheral blood mononuclear cells (PBMCs)36, yet blood sampling in our study was performed prior to the COVID-19 pandemic.

The figure shows unadjusted methylation beta values across groups for probes cg12134806 and cg11473614, illustrating the contrast between a cell-type-specific and shared pattern of differential methylation. Cg12134806 shows minimal variation for all other cell types except CD14+ monocytes. Cg11473614 reached genome-wide significance in CD14+ monocytes, yet with similar directions of effect for CD19+ B cells (P = 0.0021) and CD18+ T cells (P = 0.020). White bars represent group means. Single asterisk (*) denotes P value < 0.05 and double asterisks (**) denotes P value < 2.25 × 10–8 in linear regression analysis, including sex, age, and one surrogate variable as covariates.
We compared our results to the two largest whole-blood EWAS published in refs. 30 and Henderson-Smith et al. (Supplementary Table 2). The two significant probes reported by Vallerga et al. showed no evidence of association in any cell type in our data. Conversely, we looked up the seven significant probes from the present study in whole-blood summary statistics and found that three are present in the Vallerga et al. data, yet none showed evidence of association with PD in whole-blood data. Out of the seven probes reported as significantly associated with PD in whole blood by Henderson-Smith et al., the two top probes showed associations at P < 0.05 with one cell type each (cg06889422 in CD4+ T cells, P = 0.042, and cg16133681 in CD14+ monocytes, P value = 0.042), both with direction of effect consistent with the whole-blood study (Supplementary Table 2). None of the previously reported probes showed association in any of the four immune cell types when adjusting for 36 independent lookups.
We further compared the genomic location of the seven significant probes to the top signals reported in the largest PD GWAS to date9. One CpG probe, cg14704780 annotated to TSBP1, is located less than 300 kb from the top GWAS SNP of the HLA locus, rs112485576. No other CpG probe fell within a 1-Mb window of any GWAS top SNP. We assessed methylation quantitative trait loci (mQTL) associated with cg14704780, cg20636526 or rs112485576 in whole blood using the ARIES mQTL database37, but found no data to support a significant association between the GWAS SNP and differential methylation at these particular CpGs.
Differentially methylated regions and gene ontology enrichment in CD14+ monocytes
P value histograms and individual CpG association results clearly suggested that our experiment had the best potential to detect true signals in CD14+ monocytes, and we prioritized this cell type for analysis of differentially methylated regions. The DMRcate package estimates a smoothed test statistic across probes within a given window and compares this to the expected statistic, using the Benjamini–Hochberg false discovery rate method for calling individually associated probes. A more liberal significance threshold for this initial step than the Bonferroni correction used in the individual CpG association analysis has been recommended38, as the method requires multiple associated probes in the same region, which is also more likely to be biologically relevant than single associated probes. The method identified four differentially methylated regions at P < 0.05 based on Fisher’s multiple comparison statistic, all in CD14+ monocytes (Table 3). Among the significant individual CpGs in monocytes were three hypomethylated probes located near LINC03062 on chromosome 9 and 1 hypermethylated probe annotated to TSBP1. These were also represented in the two most significant regions (chr9: 92,276,541–92,277,266 and chr6: 32,305,106–32,305,145) identified by DMRcate. The next region at chr6: 146,863,357–146,863,680 is located upstream of RAB32 and includes three hypomethylated probes that did not reach individual significance at the Bonferroni-corrected threshold. Gene ontology pathway analysis using gometh (see “Methods”) did not identify any significantly enriched GO or KEGG pathways, which was not unexpected given the limited number of significant probes identified for each cell type.
Comparison with monocyte gene expression and qPCR analysis of RAB32
A predominance of significant findings in monocytes is in line with previous research implicating the myeloid lineage in PD. A recent study investigated monocyte gene expression in 135 PD patients and 95 controls and identified 300 differentially expressed genes15. Comparison with our results is not straightforward, as differential methylation analysis identifies regulatory genomic loci rather than genes. Mindful of this caveat, we performed a lookup of the closest gene to each of the differentially methylated probes and regions in monocytes (ten different genes in total) in the summary statistics from differential gene expression provided by Navarro et al. Applying Bonferroni correction for ten independent tests, RAB32 was the only differentially expressed gene (P value = 0.0012), upregulated in PD. We also compared the genes proximal to differentially methylated loci in monocytes to 29 genes previously highlighted in a transcriptome-wide association study (TWAS) using gene expression models from peripheral monocytes but found no overlap with our ten monocyte signals39.
To further assess whether differential methylation near RAB32 was also reflected on the mRNA level in our monocyte samples, we performed qPCR (see “Methods”). In line with the results from Navarro et al., we found that RAB32 was upregulated in PD compared to controls (P value = 0.0020, effect size of disease status on expression Z score (SD) = 0.93 (0.28)). We further observed a strong negative correlation between methylation and RAB32 expression (Pearson’s r2 = −0.43, P value = 0.0045) (Fig. 4) Taken together, these results indicate that PD patients show hypomethylation upstream of RAB32 combined with increased RAB32 expression in monocytes.

The plot illustrates the correlation between cg05420134 methylation beta values and RAB32 mRNA expression assessed by quantitative PCR. PD patient data points are shown as triangles (red) and control data points as circles (green). Expression values are calculated as relative to the mean. The regression line indicates the coefficient (SD) = − 1.30 (0.43).
Exploring the association between differentially methylated probes and dopaminergic treatment
DNA methylation may be altered by medical treatment, and previous work has indicated that this may be relevant in PD40. Aiming to explore the likelihood that observed differences in methylation may be driven by treatment effects, we assessed the association between all differentially methylated loci and dopaminergic medication as a binary (treatment-naïve versus on PD medication) variable. No association passed adjustment for multiple testing, but we observed P value < 0.05 for the probe in the TSBP1 locus of hypomethylation with dopaminergic treatment (coefficient = −0.60 (0.20), P value = 0.0062 for association between cg14704780 and treatment). Interestingly, this indicates that methylation levels differed more from controls in treatment-naïve patients than in patients receiving medication, a pattern also reported previously for SNCA promoter methylation in whole blood40. We note however, that the level of motor symptoms as assessed by the Unified Parkinson’s Disease Rating Scale (UPDRS) III was significantly higher in the patients who had not yet started PD treatment (Wilcoxon rank sum test P value = 0.02). The association between 14704780 and dopaminergic treatment was weakened by including UPDRS III in the model, but not eliminated (P value = 0.014).
Discussion
There is an urgent need to elucidate the immune mechanisms that contribute to PD pathogenesis. We performed a hypothesis-free methylome-wide association study of purified immune cell populations in early PD, identifying differential methylation across ten different genomic loci in CD14+ monocytes, and one in CD8+ T cells. Our findings indicate dysregulation of immune cells in PD, showing cell-type-specific changes that would not be readily detectable by whole-blood methylation analyses. We included PD patients with less than five years disease duration from diagnosis, 1.2 years on average, indicating that these methylation changes are present early in the disease course. Of the four different immune cell types studied, CD14+ monocytes showed by far the strongest evidence of differential methylation, both as individually significant probes and differentially methylated regions identified by DMRcate. An overrepresentation of low P values at the tail of an otherwise uniform distribution was seen only in CD14+ monocytes, which might potentially reflect more true disease-associated methylation changes in this cell type. These observations are in line with an increasing body of evidence implicating the myeloid lineage in neurodegeneration generally10,11, as well as in PD specifically13,15,39.
A differentially methylated region with three individually significant probes was identified in monocyte data near the long noncoding RNA LINC03062, alternative name UNQ6494, on chromosome 9. This transcript has been linked to immune infiltration in endometrial carcinoma41 and survival in lung adenocarcinoma42, but potential roles in neuroinflammation are unknown. The region is located ~50 kb downstream of its closest coding gene, GADD45G, which has functions related to DNA damage response and cellular stress signaling43.
We also identified a hypomethylated region in monocytes upstream of RAB32 and found by qPCR that RAB32 monocyte expression both correlated negatively with methylation in this region and was increased in PD patients relative to controls. Independent evidence supporting a role for this gene was found in summary statistics from a recently published transcriptomic study, also indicating increased expression in PD monocytes15. Interestingly, RAB32 encodes a small Rab GTPase that has been demonstrated to directly interact with and regulate leucine-rich repeat kinase 2, LRRK244,45,46. Coding mutations in LRRK2 cause autosomal dominant PD47,48 and noncoding common variants in the same locus are associated with sporadic PD in GWAS9. Several lines of evidence link the pathogenic mechanisms of LRRK2 to cells of the myeloid lineage, potentially involving both microglia49,50 and monocytes15,51. Our findings thus provide further support for RAB32 as implicated in PD pathogenesis through interaction with LRRK2 in myeloid cells.
Comparing the methylation patterns of significant probes across all cell types revealed that the differential methylation was largely cell-type-specific. Furthermore, we observed very limited overlap between our own findings and results from recent PD EWAS of whole blood28,30. It is well established that PD patients have increased levels of granulocytes and decreased levels of lymphocytes compared to healthy controls52,53, and although algorithms for cell-type deconvolution are used, there is always the risk that cell composition may be affecting the results. Even if the adjustment for cell composition is accurate, the methylation measured in whole blood will be a mixture reflecting all cell types, quantitatively dominated by granulocytes. Our findings indicate that isolation of homogenous cell populations is essential to identify disease-relevant methylation changes in less abundant cell types. Fluorescence-activated cell sorting (FACS) is an alternative to isolation with magnetic beads used here, and we acknowledge that processing a subset of samples with both methods would have allowed for further methodological validation of this step. Of note, RNA sequencing is increasingly being applied at single-cell resolution, yet this methodology is generally not yet available for DNA methylation analysis.
Our study has several limitations. Most importantly, there is a lack of independent replication of the results. The findings must therefore be interpreted with caution until they can be corroborated by similar methylation analyses from other cohorts. In our attempt to find independent support for 11 genes nominated in monocytes by comparison with summary statistics from a monocyte differential expression analysis reported in a recent PD study15, RAB32 was the only gene passing an adjusted significance threshold. This strengthens our report of hypomethylation upstream of RAB32 yet limits the interpretability of other signals. It is worth noting however, that simple one-to-one relationships between methylation and gene expression are not the rule in analyses incorporating both types of data54.
DNA methylation studies cannot differentiate between disease causes and downstream effects, the latter being less relevant for translational research aiming toward novel disease-modifying therapies. We included patients in an early phase of the disease, including nearly one-third de novo patients who had not started medication for PD. Nevertheless, we cannot exclude that treatment effects or other disease-related effects without causal relevance may have contributed to the observed methylation changes. We observed a trend towards association with medication for probes annotated to TSBP1, which were also located close to the established PD GWAS signal in the HLA region, but where treatment seemed to associate with a normalization of methylation values.
Importantly, a wide range of lifestyle and environmental factors may differ across groups of PD patients and healthy controls, potentially influencing DNA methylation. Such exposure-driven methylation differences may be mediators of PD risk, but could also be by-products of risk-associated exposures with no causal role or downstream effects of living with PD. Our study did not include the rich exposure data that would be required to dissect these complex relationships. This limitation is a major general caveat for most current complex disease EWAS, however. As highlighted in a recent review, improved integration of exposure variables is highly warranted in future epigenetic studies of PD55.
The included number of participants (25 patients and 25 controls) was modest, although we analyzed a total of 186 methylomes across four cell types. Power analysis indicated that our design was underpowered for smaller effect sizes, which we recognize as a major limitation of our study. P value histograms suggested that the data would have been better modeled in sex-stratified analysis for CD8+ T cells and CD19+ B cells, yet our study was too small to identify significant signals using this design. Power was also limited for pathway analyses, and larger studies are needed to further characterize methylation changes across the studied cell types. Furthermore, this study was performed in an ethnically homogeneous European population. This is a strength with respect to the marginal statistical power, yet do not know to what degree disease-associated methylation changes tend to be population-specific. As with genetic association studies, we acknowledge that more research in underrepresented populations is urgently needed.
In conclusion, we performed a methylome-wide association study of purified CD14+ monocyte, CD19+ B cell, CD4+ T-cell and CD8+ T-cell populations in PD, identifying differential methylation predominantly in CD14+ monocytes, highlighting the RAB32 locus in particular. Our findings shed further light on immune mechanisms in PD and warrant further studies of these cell types in order to clarify their role in pathogenesis and identify potential targets of immune-modifying therapy.
Methods
Subjects
The project was approved by the Regional Committee for Medical and Health Research Ethics, Norway. Participants were recruited at the Department of Neurology, Oslo University Hospital and gave written informed consent. A diagnosis of PD was made based on the Movement Disorder Society criteria31, yet levodopa response was not required for inclusion of treatment-naïve patients. Subjects with immunological disorders were excluded, and a clinical blood screen was performed to confirm no gross abnormalities in leukocyte counts at the time of blood draw.
Isolation of immune cell types
Peripheral blood mononuclear cells (PBMC) were isolated from whole blood immediately after venipuncture by Lymphoprep (Stemcell Technologies, Vancouver, Canada) and density gradient centrifugation. Next, CD8+, CD14+ and CD19+ cells were isolated by positive selection with EasySepTM Human CD8/CD14/CD19 Positive Selection Kit II (Stemcell Technologies, Vancouver, Canada). CD4+ cells were isolated by negative selection with EasySepTM Human CD4 Isolation Kit (Stemcell Technologies, Vancouver, Canada). All procedures followed the standard protocol from the kit manufacturer. Flow cytometry was performed as a quality control the next day to ensure a purity of >90% for each isolated cell population.
Power analysis
We estimated power using the online tool pwrEWAS32. The number of CpGs tested was set to 500,000, target CpGs to 50 and false discovery rate to 0.05. Based on these parameters, a study of 25 patients and 25 controls was estimated to have 80% power to detect a differential methylation corresponding to 0.1 SD. Assuming a replication scenario where 10 candidate CpGs are tested, out of which 5 are truly associated, the required effect size for 80% power was 0.028 SD.
DNA methylation analyses, quality control, and data normalization
DNA from the four cell types was isolated with QiAamp DNA Mini Kit (Qiagen, Hilden, Germany) using the standard protocol from the manufacturer. DNA isolation yield varied across samples. From a total of 50 participants, a sufficient amount of DNA was available for 49 CD14+ samples, 50 CD19+ samples, 49 CD8+ samples, and 38 CD4+ samples. Insufficient DNA yield occurred primarily for CD4+ T cells, at a similar rate in patients (5/25) and controls (7/25). 500ng DNA from each sample was bisulfite treated and assessed using the Illumina Infinium MethylationEPIC BeadChip (Illumina, San Diego, CA). Samples of different disease status and cell type were deliberately intermixed on plates and arrays in a balanced design. From four of the samples, we included an identical technical DNA replicate in the experiment for quality check purposes. The MethylationEPIC array targets ~850,000 CpGs across the regulatory human genome, including both promoter regions and distal enhancers.
Data processing was performed using R 4.0.3. Raw signal intensity data were imported into R before applying a series of quality checks and filtering steps implemented in the minfi56 and wateRmelon57 R packages. Quality control, normalization and evaluation of potential batch effects were performed jointly on the full experiment dataset including all cell types. One sample where >1% of sites had a detection P value greater than 0.05 was filtered out using the pfilter function in the wateRmelon package. CpG sites with a beadcount <3 in 5% of samples or detection P value > 0.05 in 1% of samples were also filtered out. Sex chromosome CpGs were used to estimate sample sex and one sample failing sex check was removed. All remaining data were of high quality as evaluated by inspection of bisulfite conversion and other control probe metrics, outlier detection by the wateRmelon outlyx function and assessment of median signal intensities as evaluated by the minfi getQC function. From a broad range of available normalization methods, we chose functional normalization (funnorm) implemented in the minfi package, as this method is recommended when large differences in methylation patterns are expected, including data from different cell types58. We note that although assessment of differential methylation within each cell type was the main aim of the study, appropriate evaluation of potential batch effects required reliable comparison also across the different cell types.
The normalized MethylSet data object was mapped to the genome and probes on sex-chromosomes, probe sequences overlapping with known SNPs in the MethylationEPIC annotation and previously reported cross-reactive probes59 were filtered out. Taking advantage of the technical replicates we used the CpGFilter package to compute the intra-class correlation coefficient (ICC), which characterizes the relative contribution of the biological variability to the total variability for each probe60. Probes with low ICC have large measurement errors, making them unsuitable for statistical association testing in complex disorders, and a considerable proportion of probes may thus be filtered out, reducing the multiple testing burden. We filtered out probes with ICC < 0.2. Furthermore, we filtered out constitutively methylated or unmethylated probes with mean beta values < 0.025 or >0.975. The wateRmelon pwod function was used separately on data from each cell type to filter out outlier values lying more than four times the interquartile range from the mean, assumed to result from rare SNP artifacts.
RAB32 expression analysis with quantitative PCR
RNA was extracted from CD14+ monocytes using the RNeasy Mini Kit (Qiagen, Hilden, Germany), with adequate yield for 23 PD and 20 control samples, followed by cDNA synthesis using SuperScript IV VILO Master Mix with ezDNase (Invitrogen, Waltham, MA). Quantitative PCR (qPCR) was performed using TaqMan gene expression assays on a ViiA7 instrument (Applied Biosystems, Waltham, MA) with standard settings as recommended by the manufacturer. Based on previous literature61,62, we evaluated ACTB, RPL37A and B2M as reference genes and selected RPL37A and B2M as the most suitable based on a strong pairwise correlation (Pearson’s R2 = 0.77, P value = 1.06e-9) compatible with stable expression. The geometric mean of the cycle threshold (CT) value for these genes was used as normalization factor to estimate the relative expression of RAB32 using the comparative CT method63.
Statistical analyses
Methylation array experiments are prone to technical batch effects. We evaluated experiment plate and chip position as potential covariates but found that these showed no association with top five principal components. To adjust for potentially unknown batch effects, we estimated surrogate variables (SVs) using the sva package64. We applied the “Leek” method to determine the number of SVs to include in the model65, which identified 1 SV as appropriate. Methylation beta values were logit transformed into M values. To identify differentially methylated sites (CpGs), linear regression was performed using the limma package66, using the makeContrasts function to define the contrast of interest between PD and control samples of each cell type. We included sex, age, and the first SV as covariates in the model: M ~ disease status + age + sex + SV1. We used the Bonferroni-corrected significance threshold recommended by Mansell et al.67 (P < 9 × 10–8) divided by four cell types (P < 2.25 × 10–8) to adjust for multiple testing. To assess differentially methylated regions, we used the contrast matrix generated for linear regression as input for the R package DMRcate with default parameters68. Finally, we assessed gene ontology enrichment of significant CpGs using the gometh function implemented in the missMethyl R package69. In CD14+ monocytes we tested both Gene Ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways for CpGs passing significance thresholds of P value < 10–7, as well as a more liberal threshold of P value < 10–4. We explored the association between significant probes and dopaminergic treatment using linear regression, comparing treated (N = 17) to untreated (N = 8) patients with sex, age and one the first surrogate variable as covariates.
The correlation between RAB32 expression and adjacent CpGs was investigated using Pearson’s product-moment correlation. Linear regression with sex and age as covariates was used to assess the association between RAB32 expression and disease status. After the identification of differentially methylated CpGs and regions, we assessed the potential association of these probes with PD medication and clinical outcomes using linear or logistic regression models as appropriate, with sex and age as covariates.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The individual-level raw data generated in the current study are not publicly available as open sharing was not included in the informed consent signed by the participants. Data will be provided from the corresponding author for noncommercial PD research use provided a collaboration and data transfer agreement has been signed and approved by the institutional Data Protection Office. Full summary statistics will be made publicly available at https://github.com/lpihlstrom.
Code availability
The underlying software code for this study will be made fully available on GitHub at https://github.com/lpihlstrom.
References
Tan, E. K. et al. Parkinson disease and the immune system—associations, mechanisms and therapeutics. Nat. Rev. Neurol. 16, 303–318 (2020).
Zhu, B., Yin, D., Zhao, H. & Zhang, L. The immunology of Parkinson’s disease. Semin. Immunopathol. 44, 659–672 (2022).
Vijiaratnam, N., Simuni, T., Bandmann, O., Morris, H. R. & Foltynie, T. Progress towards therapies for disease modification in Parkinson’s disease. Lancet Neurol. 20, 559–572 (2021).
McGeer, P. L., Itagaki, S., Akiyama, H. & McGeer, E. G. Rate of cell death in parkinsonism indicates active neuropathological process. Ann. Neurol. 24, 574–576 (1988).
Galiano-Landeira, J., Torra, A., Vila, M. & Bove, J. CD8 T cell nigral infiltration precedes synucleinopathy in early stages of Parkinson’s disease. Brain 143, 3717–3733 (2020).
Sulzer, D. et al. T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 546, 656–661 (2017).
Scott, K. M., Kouli, A., Yeoh, S. L., Clatworthy, M. R. & Williams-Gray, C. H. A systematic review and meta-analysis of alpha synuclein auto-antibodies in Parkinson’s disease. Front. Neurol. 9, 815 (2018).
Hamza, T. H. et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet. 42, 781–785 (2010).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
Raj, T. et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 344, 519–523 (2014).
Gagliano, S. A. et al. Genomics implicates adaptive and innate immunity in Alzheimer’s and Parkinson’s diseases. Ann. Clin. Transl. Neurol. 3, 924–933 (2016).
Lu, Q. et al. Systematic tissue-specific functional annotation of the human genome highlights immune-related DNA elements for late-onset Alzheimer’s disease. PLoS Genet. 13, e1006933 (2017).
Andersen, M. S. et al. Heritability enrichment implicates microglia in Parkinson’s disease pathogenesis. Ann. Neurol. 89, 942–951 (2021).
Tansey, K. E., Cameron, D. & Hill, M. J. Genetic risk for Alzheimer’s disease is concentrated in specific macrophage and microglial transcriptional networks. Genome Med. 10, 14 (2018).
Navarro, E. et al. Dysregulation of mitochondrial and proteolysosomal genes in Parkinson’s disease myeloid cells. Nat. Aging 1, 850–863 (2021).
Cavalli, G. & Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 571, 489–499 (2019).
Hwang, J. Y., Aromolaran, K. A. & Zukin, R. S. The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat. Rev. Neurosci. 18, 347–361 (2017).
Song, H. et al. Epigenetic modification in Parkinson’s disease. Front. Cell Dev. Biol. 11, 1123621 (2023).
Pavlou, M. A. S. & Outeiro, T. F. Epigenetics in Parkinson’s disease. Adv. Exp. Med. Biol. 978, 363–390 (2017).
Kaut, O. et al. Epigenome-wide analysis of DNA methylation in Parkinson’s disease cortex. Life 12, 502 (2022).
Young, J. I. et al. Genome-wide brain DNA methylation analysis suggests epigenetic reprogramming in Parkinson disease. Neurol. Genet. 5, e342 (2019).
Masliah, E., Dumaop, W., Galasko, D. & Desplats, P. Distinctive patterns of DNA methylation associated with Parkinson disease: identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 8, 1030–1038 (2013).
Pihlstrom, L. et al. Epigenome-wide association study of human frontal cortex identifies differential methylation in Lewy body pathology. Nat. Commun. 13, 4932 (2022).
Marshall, L. L. et al. Epigenomic analysis of Parkinson’s disease neurons identifies Tet2 loss as neuroprotective. Nat. Neurosci. 23, 1203–1214 (2020).
Kochmanski, J., Kuhn, N. C. & Bernstein, A. I. Parkinson’s disease-associated, sex-specific changes in DNA methylation at PARK7 (DJ-1), SLC17A6 (VGLUT2), PTPRN2 (IA-2beta), and NR4A2 (NURR1) in cortical neurons. NPJ Parkinsons Dis. 8, 120 (2022).
Moore, K., McKnight, A. J., Craig, D. & O’Neill, F. Epigenome-wide association study for Parkinson’s disease. Neuromol. Med 16, 845–855 (2014).
Chuang, Y. H. et al. Parkinson’s disease is associated with DNA methylation levels in human blood and saliva. Genome Med. 9, 76 (2017).
Henderson-Smith, A. et al. DNA methylation changes associated with Parkinson’s disease progression: outcomes from the first longitudinal genome-wide methylation analysis in blood. Epigenetics 14, 365–382 (2019).
Paul, K. C. et al. Immune system disruptions implicated in whole blood epigenome-wide association study of depression among Parkinson’s disease patients. Brain Behav. Immun. Health 26, 100530 (2022).
Vallerga, C. L. et al. Analysis of DNA methylation associates the cystine-glutamate antiporter SLC7A11 with risk of Parkinson’s disease. Nat. Commun. 11, 1238 (2020).
Postuma, R. B. et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 30, 1591–1601 (2015).
Graw, S., Henn, R., Thompson, J. A. & Koestler, D. C. pwrEWAS: a user-friendly tool for comprehensive power estimation for epigenome wide association studies (EWAS). BMC Bioinforma. 20, 218 (2019).
Breheny, P., Stromberg, A. & Lambert, J. P-value histograms: inference and diagnostics. High Throughput 7, 23 (2018).
Zhang, L. et al. Sex-specific DNA methylation differences in Alzheimer’s disease pathology. Acta Neuropathol. Commun. 9, 77 (2021).
Castro de Moura, M. et al. Epigenome-wide association study of COVID-19 severity with respiratory failure. EBioMedicine 66, 103339 (2021).
Bradic, M. et al. DNA methylation predicts the outcome of COVID-19 patients with acute respiratory distress syndrome. J. Transl. Med. 20, 526 (2022).
Gaunt, T. R. et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 17, 61 (2016).
Campagna, M. P. et al. Epigenome-wide association studies: current knowledge, strategies and recommendations. Clin. Epigenet. 13, 214 (2021).
Li, Y. I., Wong, G., Humphrey, J. & Raj, T. Prioritizing Parkinson’s disease genes using population-scale transcriptomic data. Nat. Commun. 10, 994 (2019).
Schmitt, I. et al. L-dopa increases alpha-synuclein DNA methylation in Parkinson’s disease patients in vivo and in vitro. Mov. Disord. 30, 1794–1801 (2015).
Gu, C. et al. The IFN-gamma-related long non-coding RNA signature predicts prognosis and indicates immune microenvironment infiltration in uterine corpus endometrial carcinoma. Front. Oncol. 12, 955979 (2022).
Sui, J. et al. Integrated analysis of long non-coding RNAassociated ceRNA network reveals potential lncRNA biomarkers in human lung adenocarcinoma. Int. J. Oncol. 49, 2023–2036 (2016).
Humayun, A. & Fornace, A. J. Jr. GADD45 in stress signaling, cell cycle control, and apoptosis. Adv. Exp. Med. Biol. 1360, 1–22 (2022).
Gao, Y. et al. The emerging role of Rab GTPases in the pathogenesis of Parkinson’s disease. Mov. Disord. 33, 196–207 (2018).
Waschbusch, D. et al. LRRK2 transport is regulated by its novel interacting partner Rab32. PLoS ONE 9, e111632 (2014).
McGrath, E., Waschbusch, D., Baker, B. M. & Khan, A. R. LRRK2 binds to the Rab32 subfamily in a GTP-dependent manner via its armadillo domain. Small GTPases 12, 133–146 (2021).
Zimprich, A. et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 (2004).
Paisan-Ruiz, C. et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600 (2004).
Langston, R. G. et al. Association of a common genetic variant with Parkinson’s disease is mediated by microglia. Sci. Transl. Med. 14, eabp8869 (2022).
Kim, C. et al. LRRK2 mediates microglial neurotoxicity via NFATc2 in rodent models of synucleinopathies. Sci. Transl. Med. 12, eaay0399 (2020).
Xu, E. et al. Pathological alpha-synuclein recruits LRRK2 expressing pro-inflammatory monocytes to the brain. Mol. Neurodegener. 17, 7 (2022).
Stevens, C. H. et al. Reduced T helper and B lymphocytes in Parkinson’s disease. J. Neuroimmunol. 252, 95–99 (2012).
Horvath, S. & Ritz, B. R. Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging 7, 1130–1142 (2015).
Hannon, E., Weedon, M., Bray, N., O’Donovan, M. & Mill, J. Pleiotropic effects of trait-associated genetic variation on DNA methylation: utility for refining GWAS loci. Am. J. Hum. Genet. 100, 954–959 (2017).
Tsalenchuk, M., Gentleman, S. M. & Marzi, S. J. Linking environmental risk factors with epigenetic mechanisms in Parkinson’s disease. NPJ Parkinsons Dis. 9, 123 (2023).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Pidsley, R. et al. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics 14, 293 (2013).
Fortin, J. P. et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 15, 503 (2014).
Chen, Y. A. et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 8, 203–209 (2013).
Chen, J. et al. CpGFilter: model-based CpG probe filtering with replicates for epigenome-wide association studies. Bioinformatics 32, 469–471 (2016).
Piehler, A. P., Grimholt, R. M., Ovstebo, R. & Berg, J. P. Gene expression results in lipopolysaccharide-stimulated monocytes depend significantly on the choice of reference genes. BMC Immunol. 11, 21 (2010).
Maess, M. B., Sendelbach, S. & Lorkowski, S. Selection of reliable reference genes during THP-1 monocyte differentiation into macrophages. BMC Mol. Biol. 11, 90 (2010).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 (2008).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Leek, J. T. Asymptotic conditional singular value decomposition for high-dimensional genomic data. Biometrics 67, 344–352 (2011).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Mansell, G. et al. Guidance for DNA methylation studies: statistical insights from the Illumina EPIC array. BMC Genomics 20, 366 (2019).
Peters, T. J. et al. De novo identification of differentially methylated regions in the human genome. Epigenet. Chromatin 8, 6 (2015).
Phipson, B., Maksimovic, J. & Oshlack, A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics 32, 286–288 (2016).
Acknowledgements
This work was funded by a grant from the South-Eastern Regional Health Authority, Norway. L.P. was also supported by the Norwegian Health Association. C.B. is supported by Alzheimer’s Research UK (ARUK-RF2019B-005) and the Multiple System Atrophy Trust.
Author information
Authors and Affiliations
Contributions
L.P. contributed to the conception and design of the study. M.S.A., I.S.L., I.S.B., C.C., C.B., M.T., and L.P. contributed to the acquisition, analysis, and/or interpretation of the data. M.S.A. and L.P. contributed to the drafting of the manuscript and figures. All authors contributed to the critical revision of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Andersen, M.S., Leikfoss, I.S., Brorson, I.S. et al. Epigenome-wide association study of peripheral immune cell populations in Parkinson’s disease. npj Parkinsons Dis. 9, 149 (2023). https://doi.org/10.1038/s41531-023-00594-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-023-00594-x
