
Abstract
Modern data mining methods have demonstrated effectiveness in comprehending and predicting materials properties. An essential component in the process of materials discovery is to know which material(s) will possess desirable properties. For many materials properties, performing experiments and density functional theory computations are costly and time-consuming. Hence, it is challenging to build accurate predictive models for such properties using conventional data mining methods due to the small amount of available data. Here we present a framework for materials property prediction tasks using structure information that leverages graph neural network-based architecture along with deep-transfer-learning techniques to drastically improve the model’s predictive ability on diverse materials (3D/2D, inorganic/organic, computational/experimental) data. We evaluated the proposed framework in cross-property and cross-materials class scenarios using 115 datasets to find that transfer learning models outperform the models trained from scratch in 104 cases, i.e., ≈90%, with additional benefits in performance for extrapolation problems. We believe the proposed framework can be widely useful in accelerating materials discovery in materials science.
Similar content being viewed by others
Introduction
Accurate materials property prediction using crystal structure occupies a primary and often critical role in materials science, particularly when screening through a near-infinite space of candidate materials for desirable materials performance. Upon identification of a candidate material, one has to go through either a series of hands-on experiments or intensive density functional theory (DFT) calculations which can take hours to days to even months depending on the complexity of the system. Hence, the ability to accurately predict the properties of interest of the material prior to synthesis can be extremely useful to prioritize available resources for simulations and experiments, which can significantly accelerate the process of materials exploration and discovery. Owing to significant advances in materials theory1,2,3 and computational power, it has become possible to compute several materials properties of a compound using DFT. This has led to the creation of large DFT databases4,5, which when combined with various advanced data mining techniques have extensively contributed to enhanced property prediction models6,7,8,9,10,11,12,13 and catalyzed the development of the field of materials informatics14,15,16,17,18,19,20.
Since the size of data available for training the model has a significant impact on the quality of the predictive models21,22,23, reliable and accurate models are still limited to a few selected materials properties that are relatively easy to compute. Several works have attempted to improve the performance of the model for small datasets24,25,26,27,28. However, the quality of the prediction for these studies rely on the materials property-specific feature engineering performed prior to training the model, making it less applicable for generalized use across various properties. Alternatively, transfer learning (TL), an advanced data mining technique is often applied for scarce data problems which utilizes the knowledge learned from a large collection of historical data29,30,31,32,33,34,35. For instance, it can use the knowledge of a model for a given property trained on a large DFT dataset to build a model of the same property but on a small experimental dataset. However, the absence of a large collection of historical data for most of the materials properties prohibits the broad application of this same-property transfer learning, i.e., where both source and target properties are the same. Gupta et al.36,37,38 attempt to address this by introducing cross-property transfer learning, which allowed training models on target properties for which corresponding big source datasets may not be readily available. However, the models were confined to only taking composition as input. Although composition-only based predictive models can be helpful for screening and identifying potential material candidates without the need for structure as an input, they are by design not capable of distinguishing between structure polymorphs of a given composition, which would end up being duplicates in the data, and thus would need to be removed before ML modeling. This prevents us from applying transfer learning in cases where the datasets contain large amounts of structure polymorphs, and the removal of duplicate entries might result in significantly less data available for model training. It might also prevent the implementation of cross-materials class transfer learning, thereby limiting the application of transfer learning to the same materials class only. Thus composition-based models may have limited applicability in the materials discovery process, as structure information is critical to define the material and to perform DFT computations and further experiments for validation. Further, composition-only based models could potentially have substantial errors in the predicted values as compared to ground truth, as different structure polymorphs of a given composition can have drastically different properties. These shortcomings of models trained on composition-based inputs can be mitigated by incorporating structure-based inputs, and hence structure-based modeling presents bigger opportunities than composition-based modeling to advance the discovery process in the field of materials science.
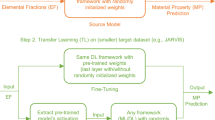
In this work, we present a framework that combines advanced data mining techniques with a structure-aware graph neural network (GNN) to improve the predictive performance of the model for materials properties with sparse data. The overall workflow of the proposed framework is shown in Fig. 1. Here, we first apply a structure-aware GNN-based deep learning architecture to capture the underlying chemistry associated with the existing large data containing crystal structure information. The resulting knowledge learned is then transferred and used during training on the sparse dataset to develop reliable and accurate target models. For simplicity, we call the large body of available data as the source dataset, the model trained on the source dataset as the source model, the sparse data as the target dataset, and the model trained on the target dataset as the target model. The transfer of information can be performed by either fine-tuning or feature extraction methods. Fine-tuning uses the weights from the pre-trained model as the preliminary weight initialization for the network, which are further refined using the target dataset. In the feature extraction method, we treat the pre-trained model as a feature extractor to extract robust features for the target dataset and use them to build the target model using representation learning. In this work, we use structure-aware GNN-based model, ALIGNN39 as the source model architecture, as it has been shown to significantly outperform several other contemporary models (SchNet40, CGCNN41, MEGNet31, DimeNet++42) on materials property prediction task across a wide variety of datasets (MP4, QM943, JARVIS5) with upto 52 solid-state and molecular properties of different data sizes using crystal structure information as the model input. Interested readers can refer to the publication39 for more details. We implement fine-tuning-based TL for ALIGNN and design a ALIGNN-based feature extractor for feature extraction-based TL using atom, bond, and angle-based features. Therefore, all the models developed in this work are structure-aware which facilitates better screening and identification of the potential material candidates, making it easier for the domain scientists to perform follow-up DFT-computations and experiments, thereby saving time and resources in the process of future materials discovery. We compare models obtained using the proposed framework with models trained from scratch (SC). Note that the proposed framework can be easily adapted to the ever-increasing datasets and ever-advancing data mining techniques to improve the models further. The significant improvements gained by using the proposed framework are expected to be useful for materials science researchers to more gainfully utilize data mining techniques to help screen and identify potential material candidates more reliably and accurately for accelerating materials discovery.

First, a data mining model (e.g., Atomistic Line Graph Neural Network (ALIGNN)39 comprised of ALIGNN layers and Graph Convolutional Network (GCN) layers is trained from scratch on a big source data set (e.g., Materials Project (MP)4) using structure files (e.g., atomic positions for the Vienna Ab initio Simulation Package (POSCAR)) to produce knowledge model. Next, the data mining model is trained on smaller target datasets (e.g., Joint Automated Repository for Various Integrated Simulations5 (JARVIS)) with different properties by using available information contained within the knowledge model to improve the predictive ability of the model further.
Results
Datasets
We use nine datasets of DFT-computed and experimental properties in this work: Materials Project (MP)4, Joint Automated Repository for Various Integrated Simulations (JARVIS) 3D with 46 properties and 2D with 32 properties5, Flla44 with three properties, Dielectric Constant (DC)45 with five properties, Piezoelectric Tensor (PT)46 with two properties, Experimental Formation Energy (EFE)47 with one property, Kingsbury Experimental Formation Energy (KEFE)48 with one property, Kingsbury Experimental Bandgap (KEB)49 with one property, and Harvard Organic Photovoltaic Dataset (HOPV)50 with 24 properties. MP dataset was downloaded from39, JARVIS-3D (https://figshare.com/collections/ALIGNN_data/5429274), JARVIS-2D (https://ndownloader.figshare.com/files/26808917) and HOPV (https://ndownloader.figshare.com/files/28814184) from their respective figshare links and the rest of the datasets were obtained using Matminer51.
A model trained on the formation energy of the MP dataset39 is used as the source model to perform fine-tuning and feature extraction-based transfer learning as formation energy has shown to lead to meaningful representations from large source datasets36, which can then be applied during the model training on the smaller target datasets to improve their predictive performance. The rest of the datasets are used to perform target model training followed by materials property prediction and evaluation. The target datasets are randomly split with a fixed random seed into training, validation, and holdout test sets in the ratio of 80:10:10. The data size for every materials property in each of the datasets are shown in Supplementary Table 1, 2 and 3, and modifications made to some of the target dataset’s materials properties to suit the model input are shown in Supplementary Table 4. We use mean absolute error (MAE) as the primary evaluation metric for all models. We also incorporate a ‘Base’ model, which always uses the average property value of all the training data provided to it as the predicted property of a test compound as a naive baseline for comparison with scratch (SC) and transfer learning (TL) methods. Note that due to the large number of materials properties investigated in this work and the limited computational resources, we do not investigate the aleatoric uncertainty caused by the random initialization of the models.
ALIGNN-based Feature Extractor
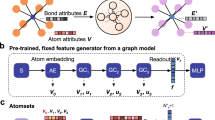
We use a structure-aware GNN-based architecture, ALIGNN39 as our base architecture for training the source models, performing transfer learning using fine-tuning method, and extracting structure-based features, as it has shown to significantly outperform other known GNN models31,40,41,42,52 for materials property prediction across a wide variety of datasets with different data sizes39 using crystal structure information as the model input. For the initial set of input features used to train ALIGNN, please refer to the publication39. To extract structure-based features from ALIGNN, we design a ALIGNN-based Feature Extractor, which is shown in Fig. 2.

Blue color indicates atom-based features, orange color indicates bond-based features and green color indicates angle-based features.
The structure file containing information on lattice geometry and the ionic positions of a compound is divided into atom, bond, and angle-based features before feeding into the ALIGNN-based Feature Extractor where we perform feature extraction. As the graph neural network (ALIGNN) used for extracting features comprises of an intricate arrangement of layers, simply extracting features from every layer would yield nearly 100 variations of possible features without any definite meaning. If each of these sets of features is used as model input to perform deep learning-based model training, it will make the entire process too costly and time-consuming. Hence, we define several analytical checkpoints, mainly after the ALIGNN layer and GCN layer, each containing two edge-gated graph convolution layers53 and one edge-gated graph convolution layer, respectively to extract features instead of extracting features from every layer in order to design a more generalized mechanism for performing feature extraction based TL, which is both meaningful as well as helps save time and resources to carry out the model training for the proposed framework. After performing feature extraction from the pre-defined analytical checkpoints, we obtain 9 sets of atom-based features, 9 sets of bond-based features and 5 sets of angle-based features, each with a different 256-vector representation of the compound. We also test the effect of features on the performance of the model by combining atom-bond and atom-bond-angle features from the same checkpoint. Moreover, as it is known that features extracted from the last layer of a given architecture are also helpful when performing transfer learning (also known as TL based on the freezing method54), we also combine the last set of atom, bond, and angle-based features (called atom-bond-angle features(last)) to see its effect on the performance. Note that we do not try all possible combinations of atom, bond and angle-based features extracted from different checkpoints in order to facilitate further generalizability of the workflow. Due to the nature of the source model architecture, all the features extracted from the feature extractor are structure aware. For a detailed explanation of the pre-processing of the structure-based features associated with the feature extractor, please refer to the methods section. Next, we perform model training using the above-defined set of features as input for the deep neural network where we use a 17-layered neural network comprising of stacks of fully connected layers and ReLU as the activation function inspired from21,22,55 as the base architecture and formation energy of JARVIS-3D dataset as the materials property for property prediction task, the results of which are shown in Table 1. In this work, we use a very basic deep neural network to perform model training on the extracted features to see the potential of the extracted features to predict the materials properties.
Table 1 shows that, in general, feature representations containing structure-aware atom-based features tend to perform better as compared to only bond or angle-based features. Moreover, the combination containing the last set of the atom, bond, and angle-based features, called atom-bond-angle features(last), performs the best among the 38 sets of features used for the analysis. Hence, for the rest of the analysis, we only atom-bond-angle features(last) as the feature set to perform feature extraction-based TL for generalizability. Moreover, we use the model with the least validation error only (among fine-tuning and atom-bond-angle features(last) based TL models) to perform model testing on the holdout test set to have a fair comparison with the SC model, i.e., both the TL and SC models look at the holdout test set only once during testing.
JARVIS-3D database
Here, we demonstrate the performance of TL models on different target materials properties in the JARVIS-3D dataset. We compare the performance of TL models with the SC models, i.e., ALIGNN trained directly on the target dataset from scratch. Table 2 presents the prediction accuracy of the best SC and best TL model on the test set for each of the 48 target properties.
Table 2 indicates that TL models outperform the SC models in 42/46 cases, i.e., in ≈91% of the cases. We observe higher percent error improvement in the TL model for materials properties with less number of data points (below ~19,000 data points). Supplementary Table 5 shows that among the TL models, fine-tuning-based TL model performed the best for 27/42 target properties, and feature extraction-based TL model performed the best for 15/42. The results illustrate the benefit of using the proposed framework even when the materials properties of the source datasets and target datasets are different using structure-based features as model input. We believe this is because the source model was able to learn and extract useful and widely applicable features during the model training on the source data.
Other DFT-based databases
In the previous section, we only used a single DFT-computed dataset to perform the model training using the proposed framework to improve the performance of the target model. However, as various DFT-computed datasets are calculated using different computational settings and can show significant discrepancies across each other56, these differences may affect the performance of the target model when applying TL. Hence, here we investigate the effect of using the same source model trained on the formation energy of MP dataset on other small DFT-based databases.
Table 3 indicates that TL models outperform the SC models in 10/10 cases, i.e., in 100% of the cases. Supplementary Table 6 shows that among the TL models, the fine-tuning-based TL model performed the best for 2/10 target properties, and feature extraction-based TL model performed the best for 8/10. It is interesting to see that on smaller DFT databases, not only the feature-extraction-based TL gives the more accurate model for a large fraction of evaluated properties, but the best TL model is also quantitatively much more accurate than the best SC model, underscoring the power of structure-aware feature-extraction based TL for small datasets.
JARVIS-2D database
In the previous sections, we used different DFT-computed datasets containing 3D materials to perform the model training using the proposed framework to improve the performance of the target model. However, there also exist a class of materials that exhibit plate-like 2D shapes whose physical and chemical properties may differ in nature from that of 3D materials. Hence, here we investigate the effect of using the same source model trained on 3D materials dataset with TL to build target models on datasets containing 2D materials. Table 4 presents the prediction accuracy of the best SC and best TL model on the test set for each of the 34 target properties in JARVIS-2D database.
Table 4 indicates that TL models outperform the SC models in 27/32 cases, i.e., in ≈84% of the cases. As most of the materials properties have a small number of data points, we observe even larger improvement in the performance of the TL model. Supplementary Table 7 shows that among the TL models, the fine-tuning-based TL model performed the best for 5/27 target properties, and feature extraction-based TL model performed the best for 22/27. The results demonstrate that our proposed framework is able to improve the performance of the predictive model even when the source model trained on 3D materials is applied to 2D materials across different materials properties.
Other materials class data
So far, we have observed the advantages of using the proposed framework on a variety of materials properties from different DFT-computed datasets of crystalline solids where TL models typically outperform SC models. However, as there are different classes of materials available, it would be interesting to see if the knowledge learned from one class of materials can be helpful in building a more accurate model on another class of materials. Hence, in this section, we explore the effectiveness of our proposed framework by applying it on datasets comprised of molecular properties.
Table 5 indicates that TL models outperform the SC models in 22/24 cases, i.e., in ≈92% of the cases. We also observe for some specific materials properties, improvement in the performance is always very little, such as scharber jsc, scharber pce, and scharber voc. It would be interesting to see if it is possible to analyze and quantify possible relations between materials properties from different materials classes which can lead to possible improvement in the performance of the target model for cross-property transfer learning scenarios in future work. Supplementary Table 8 shows that among the TL models, the fine-tuning-based TL model performed the best for 7/22 target properties, and the feature extraction-based TL model performed the best for 15/22. It is quite encouraging to observe that the proposed TL models outperform the SC models even when using properties from another materials class as the target properties for most of the cases. This shows that the ALIGNN model is able to successfully and automatically capture relevant atom, bond, and angle-based domain knowledge features from source data and effectively and appropriately apply that information for building improved predictive models for a variety of target properties on small target datasets across different materials classes using the proposed structure-aware TL framework.
Experimental data
Here, we demonstrate the performance of our proposed framework on experimental datasets with formation energy and band gap as materials properties.
Table 6 indicates that TL models outperform the SC models in 3/3 cases, i.e., in 100% of the cases. Supplementary Table 9 shows that among the TL models, the fine-tuning-based TL model performed the best for 1/3 target properties, and feature extraction-based TL model performed the best for 2/3. It is very encouraging to observe the improvement in performance not only for computational datasets but also for experimental datasets. This along with the other results demonstrates that the proposed framework can significantly and consistently help improve the prediction of the materials properties across various domains and classes, thereby potentially saving time and resources in the process of future materials discovery.
Discussion
In this paper, we presented a framework that combines structure-aware GNN architecture with advanced data-mining techniques to build a powerful source model whose information is then used to build significantly and consistently accurate target models on various materials properties from smaller datasets for enhanced materials property prediction across various domains and materials classes. To show the benefit of the proposed approach, we built source models using a structure-aware GNN-based architecture called ALIGNN on the MP dataset by using only formation energy as the source materials property. This trained model was then used to perform transfer learning on 115 different dataset-property combinations to find that the proposed framework yields highly accurate and robust models even when the source property and target property are different, which is expected to be especially useful in building predictive models for properties for which big datasets are not available. We compare the performance of the TL models with ALIGNN model trained from scratch.
To check the robustness of the proposed framework even further, we perform empirical and statistical analysis to examine the performance difference between SC and TL models. First we describe empirical analysis, where we perform training size-based and extrapolation-based analysis using formation energy as materials property (as it is one of the most studied property) from JARVIS dataset. For training size-based analysis we perform model training with different training data size using the same test set (10% of the total data size) to create a learning curve with prediction error as a function of the training set size. Figure 3 shows that TL model outperform SC model for all the training sizes for formation energy prediction.

Training curve for predicting formation energy in JARVIS dataset for different training data sizes on a fixed test set.
For extrapolation-based analysis, we divide the whole dataset into different splits, where data points corresponding to the bottom 10% of formation energy values were set aside as the ‘Extrapolation test set’, and the remaining data was divided into training, validation, and test split (as ‘Interpolation test split’). The lower values for formation energy indicate a more stable compound, and it is desirable to have a model that can predict the lower values accurately and even extrapolate. The scatter plot of the prediction error for ‘Extrapolation test set’ and ‘Interpolation test set’ is shown in Fig. 4. It shows that the best TL model (in this case, fine-tuning based TL model) performs better as compared to the best SC model for both the test splits.

Prediction error analysis with mean absolute error (MAE) as error metric for predicting formation energy in JARVIS dataset using best scratch (SC) and best transfer learning (TL) model.
Next, we perform statistical analysis where we perform uncertainty and statistical significance analysis using different materials properties. For uncertainty analysis, we perform 9-fold cross-validation (as the datasets were divided into 8:1:1 ratio) for SC and proposed TL model with the best modeling configuration using formation energy and bandgap (as they are widely studied materials properties) of JARVIS 3D, JARVIS 2D, and Experimental datasets. Supplementary Table 10 shows the distribution of performance for the models across different train/test splits, where we observe that TL outperforms SC in terms of MAE for all six cases. Additionally, to see if the observed MAE is statistically distinguishable from one another, we perform a corrected resampled t-test57 and obtain p value < 0.01 for all cases. This shows the MAE obtained using the proposed TL model is statistically distinguishable from the MAE obtained using the SC model at α=0.01. For statistical significance analysis, we estimate a one-tailed p-value to compare the test MAEs obtained on 115 target datasets (out of which TL models outperformed SC models on 104 target datasets) in order to see if the observed improvement in the accuracy of TL models over SC models is significant or not. Here, as we are dealing with different properties obtained from different datasets, whose differences in MAE may not be directly comparable58, we use the Signed Test59 to estimate the one-tailed p-value. Here, the null hypothesis is ‘TL model is not better than the SC model’ and the alternate hypothesis is ‘TL model is better than the SC model’. After performing the statistical testing using a sign test calculator60, we get the p value < 0.00001, thus rejecting the null hypothesis at α=0.01. This suggests that the difference in test MAE between SC and TL models is unlikely to have arisen by chance, and thus we can infer that in general the proposed TL models perform significantly better than SC models. Additionally, we train ALIGNN on multiple materials properties simultaneously for both the source and target models to examine its performance as compared to training the source and target models with just a single property, as performed in this study. We use the formation energy and bandgap as the materials properties where the source model is trained on the MP dataset, and the target model is trained on the JARVIS 3D dataset. Supplementary Table 11 shows the test MAE of the SC model and proposed TL model when the source and target models are trained on single and multiple materials properties. When training the model on single materials property, we observe that using the corresponding source model as well as formation energy as the source property helps improve the performance of the model. When training the model on multiple materials properties, we observe a decrease in model accuracy for formation energy and negligible difference in accuracy for bandgap. This suggests that training models on multiple materials properties simultaneously for both the source and target datasets is not beneficial for improving the accuracy of the model.
We also observe that out of 115 materials properties analyzed in our work, the SC model performed the best for 11 properties, fine-tuning-based TL model performed the best for 42 properties, and feature extraction-based TL model performed best for 62 properties (Supplementary Figure 1). We observe that in general, fine-tuning-based TL models perform better for larger target datasets, and feature extraction-based TL models perform better for smaller target datasets, which is consistent with a previous study on composition-based cross-property TL36. Additionally, we plot the percent error improvement of the TL model against the SC model as a function of dataset size with a histogram in Supplementary Figures 2 and 3 and observe larger improvement in the model accuracy for smaller datasets as compared to larger datasets. The mean ± standard deviation, 1st quartile, median, 3rd quartile, minimum and maximum percent error improvements are -11.95 ± 20.23, -15.16, -5.48, -2.54, -96.09 and 34.97, respectively. Although we only used formation energy as the source material property to train the feature extractor (source model) and a basic deep neural network to build target models using the extracted features, feature extraction-based TL was found to perform better for more number of materials properties as compared to fine-tuning based TL for small datasets. This shows the powerful ability of the feature extractor to learn relevant, robust, and versatile sets of features that can be leveraged even with relatively simple data mining techniques, thereby providing flexibility and interoperability. We also observe that transfer learning works not only for classical quantities such as Deltae (5.25%) but also for electronic properties such as bandgap (6.19%) equally well. The TL-based improvements are also mostly isotropic, e.g., improvements in Meps (x,y,z) components are similar. While some properties like PMDiEl show substantial improvements, the underlying reasons for this remain unclear. A potential future utility could involve a GNNExplainer-like tool61 for ALIGNN architecture. Hence, the proposed method can help improve the robustness and accuracy of the target model on small datasets by incorporating the rich set of hierarchical features that can be learned using the ever-increasing data and ever-improving data mining techniques. The proposed framework is thus flexible and can leverage state-of-the-art data mining techniques to improve upon the performance and can be applied to other materials properties across various domains and materials classes for which enough source data may not be available. Although transfer learning is not always effective for all kinds of materials properties with varying data sizes, we observe that the benefit of transfer learning is more for materials properties with smaller number of data points, transferring knowledge from periodic (e.g., crystalline) to non-periodic (e.g., molecular) properties, i.e., performing cross materials class transfer learning to increase the accuracy of the target model is possible when using structure-based modeling (albeit with smaller benefits), and there is larger improvement in performance for ‘extrapolation’ than ‘interpolation’ problems. Further, the proposed framework is expected to be easily adaptable to other scientific domains beyond materials science. The presented framework is conceptually easy to implement, understand, use, and build upon. For future work, it would be interesting to explore the effect on the performance of the target model when materials properties other than formation energy are used as the source material property and GNN architecture other than ALIGNN is used for training the source model. Although in the current study, we have used DFT-relaxed structures, which hold origin one way or another in experimental crystal structures, we plan to use such TL models for crystal generative models as well62 where property predictions and pre-screening with TL-performance boosted models will be useful. It would also be interesting to explore the uncertainty associated with the materials property prediction by incorporating neural network components that help perform uncertainty estimation, such as dropout within the network architecture, or by creating an ensemble model using multiple graph neural networks and/or input from multiple checkpoints. One can also explore different sets of features to train the neural network or use more sophisticated neural network architectures for the target model in a bid to boost the performance of the target model for a specific materials property.
Methods
Scratch and transfer learning models
In this work, we implement a scratch (SC) model and two types of transfer learning (TL) models. For SC models, the model training is performed directly on the small target dataset without providing the model with any form of knowledge from source data. We use the graph neural network model, ALIGNN, as the model architecture for the SC model. For TL models, we use a model pretrained on the MP dataset with formation energy as the materials property using ALIGNN as the model architecture. The TL techniques comprise of traditional fine-tuning and a feature extraction method from a graph neural network. Fine-tuning uses the weights from the pre-trained model as the preliminary weight initialization for the network (which is the same architecture as used during source model training) and is further refined using a small dataset. In the feature extraction method, we treat the pre-trained model as the feature extractor and extract atom, bond, and angle-based features from a given layer, each containing a variable number of rows depending on the number of the atom, bond, and angle information present in the input file and 256 columns as features for each row. For example, let us consider a hypothetical compound AaBbCc where a + b + c = x, number of bonds = y and number of angles = z (generally, number of angles > number of bonds > number of atoms) and we extract the features from a checkpoint. Then, the dimensions of the extracted vectors will be (x, 256) for atom-based features, (y, 256) for bond-based features, and (z, 256) for the angle-based features. In order to pre-process them into a form that can be given to the deep learning (DL) model, which takes a one-dimensional vector as input, we take the mean of all features across each column. This creates a (1, 256) vector representation for each of the structure-based features (atom, bond, and angle) for a given compound of the target dataset. The extracted feature from a given layer can then be either concatenated or used separately as an input for any DL model. For example, if we use atom based features from a given layer as the materials representation, each compound will be represented as a 256-dimensional feature vector. Similarly, for atom+bond-based features it will be a 512-dimensional feature vector, and for atom+bond+angle-based features it will be a 768-dimensional feature vector representation. For our analysis, we only use atom+bond+angle (last) as the set of features for the feature extraction-based TL. The ‘Base’ model used in this work always uses the average property value of all the training data provided to it as the predicted property of a test compound as a naive baseline for comparison with SC and TL methods.
Network settings and model architecture
ALIGNN was implemented using Pytorch and a 17-layered neural network (NN-17) was implemented using TensorFlow 2 (with Keras). Detailed configurations for the network architecture is [FC1024-Re x 4]-[FC512-Re x 3]-[FC256-Re x 3]-[FC128-Re x 3]-[FC64-Re x 2]-[FC32-Re]-FC1 where the notation [...] represents a stack of model components comprising a sequence (where FC: fully connected layer, Re: ReLU activation function). The number of layers for the neural network was decided based on the analysis performed in55, where they investigate the performance of deep learning models of different depths in model architecture and show that the error improves with the number of layers up to 17 layers, after which the accuracy stagnated. The hyperparameters used in the ALIGNN comprise of the following: Sigmoid Linear Unit (SiLU) as the base activation function, Adaptive Moment Estimation with decoupled weight decay (AdamW) as the optimizer with normalized weight decay of 10−5, mini-batch size of 64 (32 or 16 where the holdout test set is small or the size of the input files is larger than the available GPU memory), and learning rate as 0.001. We train all ALIGNN models for 300 epochs with a fixed random seed as done in the original work39. The hyperparameters used in the NN-17 comprise of the following: rectified linear activation unit (ReLU) as the base activation function after each layer (except for the last layer), Adaptive Moment Estimation (Adam) as the optimizer, mini-batch size as 64 with a learning rate of 0.0001. We used early stopping with a patience of 200 to stop the model training if the validation loss does not improve for 200 epochs to prevent overfitting. All NN-17 model training used a fixed random seed. Readers interested in in-depth hyperparameter settings for ALIGNN and NN-17 models are referred to those publications22,39,55 for details. We use mean absolute error (MAE) as the loss function as well as the primary evaluation metric for all models. We use DFT-relaxed or experimentally determined structures as input for all the models trained in this study.
Data availability
The datasets used in this paper are publicly available from the corresponding websites- MP4 from https://materialsproject.org/, JARVIS5 from https://jarvis.nist.gov, Flla44, Dielectric Constant45, Piezoelectric Tensor46, Experimental Formation Energy47, Kingsbury Experimental Formation Energy48, Kingsbury Experimental Bandgap49 from AutoMatminer63 (https://github.com/hackingmaterials/automatminer), and HOPV from https://ndownloader.figshare.com/files/28814184.
Code availability
The codes required to perform fine-tuning and feature extraction based TL used in this study is available at https://github.com/GuptaVishu2002/ALIGNNTL.
References
Roemelt, M., Maganas, D., DeBeer, S. & Neese, F. A combined dft and restricted open-shell configuration interaction method including spin-orbit coupling: Application to transition metal l-edge x-ray absorption spectroscopy. J. Chem. Phys. 138, 204101 (2013).
Curtarolo, S., Morgan, D. & Ceder, G. Accuracy of ab initio methods in predicting the crystal structures of metals: A review of 80 binary alloys. Calphad 29, 163–211 (2005).
Asta, M., Ozolins, V. & Woodward, C. A first-principles approach to modeling alloy phase equilibria. JOM 53, 16–19 (2001).
Jain, A. et al. The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Choudhary, K. et al. JARVIS: An integrated infrastructure for data-driven materials design (2020).
Morgan, D. & Jacobs, R. Opportunities and challenges for machine learning in materials science. Annu. Rev. Mater. Res. 50, 71–103 (2020).
Mannodi-Kanakkithodi, A. & Chan, M. K. Computational data-driven materials discovery. Trends Chem. 3, 79–82 (2021).
Friederich, P., Häse, F., Proppe, J. & Aspuru-Guzik, A. Machine-learned potentials for next-generation matter simulations. Nat. Mater. 20, 750–761 (2021).
Pollice, R. et al. Data-driven strategies for accelerated materials design. Acc. Chem. Res. 54, 849–860 (2021).
Westermayr, J., Gastegger, M., Schütt, K. T. & Maurer, R. J. Perspective on integrating machine learning into computational chemistry and materials science. Chem. Phys. 154, 230903 (2021).
Jha, D., Gupta, V., Liao, W.-k., Choudhary, A. & Agrawal, A. Moving closer to experimental level materials property prediction using ai. Sci. Rep. 12 (2022).
Mao, Y. et al. An ai-driven microstructure optimization framework for elastic properties of titanium beyond cubic crystal systems. Npj Comput. Mater. 9, 111 (2023).
Gupta, V. et al. Physics-based data-augmented deep learning for enhanced autogenous shrinkage prediction on experimental dataset. In Proceedings of the 2023 Fifteenth International Conference on Contemporary Computing, 188–197 (2023).
Agrawal, A. & Choudhary, A. Perspective: Materials informatics and big data: Realization of the “fourth paradigm” of science in materials science. APL Mater. 4, 053208 (2016).
Hill, J. et al. Materials science with large-scale data and informatics: unlocking new opportunities. MRS Bull. 41, 399–409 (2016).
Ward, L. & Wolverton, C. Atomistic calculations and materials informatics: A review. Curr. Opin. Solid State Mater. Sci. 21, 167–176 (2017).
Ramprasad, R., Batra, R., Pilania, G., Mannodi-Kanakkithodi, A. & Kim, C. Machine learning in materials informatics: recent applications and prospects. Npj Comput. Mater. 3, 54 (2017).
Agrawal, A. & Choudhary, A. Deep materials informatics: Applications of deep learning in materials science. MRS Commun. 9, 779–792 (2019).
Choudhary, K. et al. Large scale benchmark of materials design methods. Preprint at: https://arxiv.org/abs/2306.11688 (2023).
Gupta, V., Liao, W.-k., Choudhary, A. & Agrawal, A. Evolution of artificial intelligence for application in contemporary materials science. MRS Commun.1–10 (2023).
Jha, D. et al. Enabling deeper learning on big data for materials informatics applications. Sci. Rep. 11, 1–12 (2021).
Gupta, V., Liao, W.-k., Choudhary, A. & Agrawal, A. Brnet: Branched residual network for fast and accurate predictive modeling of materials properties. In Proceedings of the 2022 SIAM International Conference on Data Mining (SDM), 343–351 (SIAM, 2022).
Gupta, V., Peltekian, A., Liao, W.-k, Choudhary, A. & Agrawal, A. Improving deep learning model performance under parametric constraints for materials informatics applications. Sci. Rep. 13, 9128 (2023).
Seko, A. et al. Prediction of low-thermal-conductivity compounds with first-principles anharmonic lattice-dynamics calculations and bayesian optimization. Phys. Rev. Lett. 115, 205901 (2015).
Ghiringhelli, L. M., Vybiral, J., Levchenko, S. V., Draxl, C. & Scheffler, M. Big data of materials science: Critical role of the descriptor. Phys. Rev. Lett. 114, 105503 (2015).
Lee, J., Seko, A., Shitara, K., Nakayama, K. & Tanaka, I. Prediction model of band gap for inorganic compounds by combination of density functional theory calculations and machine learning techniques. Phys. Rev. B 93, 115104 (2016).
Sendek, A. D. et al. Holistic computational structure screening of more than 12000 candidates for solid lithium-ion conductor materials. Energy Environ. Sci. 10, 306–320 (2017).
Mao, Y. et al. Ai for learning deformation behavior of a material: Predicting stress-strain curves 4000x faster than simulations. In 2023 International Joint Conference on Neural Networks (IJCNN), 1–8 (IEEE, 2023).
Kaya, M. & Hajimirza, S. Using a novel transfer learning method for designing thin film solar cells with enhanced quantum efficiencies. Sci. Rep. 9, 5034 (2019).
Yamada, H. et al. Predicting materials properties with little data using shotgun transfer learning. ACS Cent. Sci. 5, 1717–1730 (2019).
Chen, C., Ye, W., Zuo, Y., Zheng, C. & Ong, S. P. Graph networks as a universal machine learning framework for molecules and crystals. Chem. Mater. 31, 3564–3572 (2019).
Feng, S. et al. A general and transferable deep learning framework for predicting phase formation in materials. Npj Comput. Mater. 7, 1–10 (2021).
Lee, J. & Asahi, R. Transfer learning for materials informatics using crystal graph convolutional neural network. Comput. Mater. Sci. 190, 110314 (2021).
McClure, Z. D. & Strachan, A. Expanding materials selection via transfer learning for high-temperature oxide selection. JOM 73, 103–115 (2021).
Dong, R., Dan, Y., Li, X. & Hu, J. Inverse design of composite metal oxide optical materials based on deep transfer learning and global optimization. Comput. Mater. Sci. 188, 110166 (2021).
Gupta, V. et al. Cross-property deep transfer learning framework for enhanced predictive analytics on small materials data. Nat. Commun. 12, 1–10 (2021).
Gupta, V. et al. Mppredictor: An artificial intelligence-driven web tool for composition-based material property prediction. J. Chem. Inf. Model. 63, 1865–1871 (2023).
Gupta, V., Liao, W.-k., Choudhary, A. & Agrawal, A. Pre-activation based representation learning to enhance predictive analytics on small materials data. In 2023 International Joint Conference on Neural Networks (IJCNN), 1–8 (IEEE, 2023).
Choudhary, K. & DeCost, B. Atomistic line graph neural network for improved materials property predictions. Npj Comput. Mater. 7, 1–8 (2021).
Schütt, K. et al. Schnet: A continuous-filter convolutional neural network for modeling quantum interactions. Adv. Neural Inf. Process. Syst. 30 (2017).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Klicpera, J., Giri, S., Margraf, J. T. & Günnemann, S. Fast and uncertainty-aware directional message passing for non-equilibrium molecules. Preprint at: https://arxiv.org/abs/2011.14115 (2020).
Ramakrishnan, R., Dral, P. O., Rupp, M. & Von Lilienfeld, O. A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 1, 1–7 (2014).
Faber, F., Lindmaa, A., von Lilienfeld, O. A. & Armiento, R. Crystal structure representations for machine learning models of formation energies. Int. J. Quantum Chem. 115, 1094–1101 (2015).
Petousis, I. et al. High-throughput screening of inorganic compounds for the discovery of novel dielectric and optical materials. Sci. Data 4, 160134 (2017).
de Jong, M., Chen, W., Geerlings, H., Asta, M. & Persson, K. A. A database to enable discovery and design of piezoelectric materials. Sci. Data 2, 150053 (2015).
Kim, G., Meschel, S. V., Nash, P. & Chen, W. Experimental formation enthalpies for intermetallic phases and other inorganic compounds. Sci. Data 4, 170162 (2017).
Wang, A. et al. A framework for quantifying uncertainty in dft energy corrections. Sci. Rep. 11, 1–10 (2021).
Zhuo, Y., Mansouri Tehrani, A. & Brgoch, J. Predicting the band gaps of inorganic solids by machine learning. J. Phys. Chem. Lett. 9, 1668–1673 (2018).
Lopez, S. A. et al. The harvard organic photovoltaic dataset. Sci. Data 3, 1–7 (2016).
Ward, L. T. et al. Matminer: An open source toolkit for materials data mining. Comput. Mater. Sci. 152, 60–69 (2018).
Qiao, Z., Welborn, M., Anandkumar, A., Manby, F. R. & Miller III, T. F. Orbnet: Deep learning for quantum chemistry using symmetry-adapted atomic-orbital features. J. Chem. Phys. 153, 124111 (2020).
Dwivedi, V. P., Joshi, C. K., Laurent, T., Bengio, Y. & Bresson, X. Benchmarking graph neural networks. Journal of Machine Learning Research, 24, 1–48. (2023).
Oquab, M., Bottou, L., Laptev, I. & Sivic, J. Learning and transferring mid-level image representations using convolutional neural networks. In Proceedings of the IEEE conference on computer vision and pattern recognition, 1717–1724 (2014).
Jha, D. et al. ElemNet: Deep learning the chemistry of materials from only elemental composition. Sci. Rep. 8, 17593 (2018).
Hegde, V. I. et al. Quantifying uncertainty in high-throughput density functional theory: A comparison of AFLOW, Materials Project, and OQMD. Phys. Rev. Mater. 7, 053805 (2023).
Nadeau, C. & Bengio, Y. Inference for the generalization error. Mach. Learn. 52, 239–281 (2003).
Demšar, J. Statistical comparisons of classifiers over multiple data sets. J. Mach. Learn. Res. 7, 1–30 (2006).
Sheskin, D. J. Handbook of parametric and nonparametric statistical procedures (Chapman and Hall/CRC, 2003).
Statistics, S. S. Sign test calculator. https://www.socscistatistics.com/tests/signtest/default.aspx (2018).
Ying, Z., Bourgeois, D., You, J., Zitnik, M. & Leskovec, J. Gnnexplainer: Generating explanations for graph neural networks. Adv. Neural Inf. Process. Syst. 32 (2019).
Wines, D., Xie, T. & Choudhary, K. Inverse design of next-generation superconductors using data-driven deep generative models. Preprint at: https://arxiv.org/abs/2304.08446 (2023).
Dunn, A., Wang, Q., Ganose, A., Dopp, D. & Jain, A. Benchmarking materials property prediction methods: the matbench test set and automatminer reference algorithm. Npj Comput. Mater. 6, 1–10 (2020).
Acknowledgements
This work was performed under the following financial assistance award 70NANB19H005 from U.S. Department of Commerce, National Institute of Standards and Technology as part of the Center for Hierarchical Materials Design (CHiMaD). Partial support is also acknowledged from NSF award CMMI-2053929, DOE award DE-SC0021399, and Northwestern Center for Nanocombinatorics. This research was supported by the Exascale Computing Project (17-SC-20-SC), a joint project of the U.S. Department of Energy’s Office of Science and National Nuclear Security Administration, responsible for delivering a capable exascale ecosystem, including software, applications, and hardware technology, to support the nation’s exascale computing imperative.
Author information
Authors and Affiliations
Contributions
V.G. designed and carried out the implementation and experiments for the ALIGNN-based deep transfer learning framework under the guidance of A.A., A.C. and W.L., K.C., B.D., F.T. and C.C. provided the necessary domain expertise for this work. V.G., A.A., K.C., B.D. and F.T. wrote the manuscript. All authors discussed the results and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41524_2023_1185_MOESM1_ESM.pdf
Supplementary Information: Structure-Aware Graph Neural Network-Based Deep Transfer Learning Framework For Enhanced Predictive Analytics On Diverse Materials Datasets
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gupta, V., Choudhary, K., DeCost, B. et al. Structure-aware graph neural network based deep transfer learning framework for enhanced predictive analytics on diverse materials datasets. npj Comput Mater 10, 1 (2024). https://doi.org/10.1038/s41524-023-01185-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-023-01185-3
