
Abstract
The complex interplay between chemistry, microstructure, and behavior of many engineering materials has been investigated predominantly by experimental methods. Parallel to the increase in computer power, advances in computational modeling methods have resulted in a level of sophistication which is comparable to that of experiments. At the continuum level, one class of such models is based on continuum thermodynamics, phase-field methods, and crystal plasticity, facilitating the account of multiple physical mechanisms (multi-physics) and their interaction during microstructure evolution. This paper reviews the status of simulation approaches and software packages in this field and gives an outlook towards promising research directions.
Similar content being viewed by others


Introduction
The diversity and complexity of materials and their behavior are rooted in the complex, generally nonlinear interplay between microstructural features such as voids, cracks, phases, grains, dislocations, and local chemical composition with each other and with environmental conditions. Designing improved materials requires an in-depth understanding of several intertwined metastable configurations and intertwined, hierarchical and self-organized networks of defects that influence each other through various interactions across multiple lengths- and timescales1. The interplay of chemistry and mechanical failure, for example, is important for the longevity of rechargeable batteries2,3; microscopic cracks which form over time as the electrodes are mechanically deformed during load cycles result in electrical disconnections and loss of capacity. There are numerous other examples where inelastic deformation features and damage are initiated or accelerated due to harsh environmental conditions such as hydrogen embrittlement4, stress corrosion cracking5, and radiation-inflicted damage6. Ni-based superalloys are another example where carefully tuned chemical composition and processing results in a complex microstructure which ultimately gives rise to the high performance of these alloys at elevated temperatures7. The number of microstructural and compositional parameters, processes, and configurations to combine to invent a new material is enormous. A complex system (either in terms of composition or microstructure) has a higher potential for exhibiting interesting, unusual, and surprising behavior compared to a simple system. This trend is observed in generations of alloys, where typically the number of alloying elements has increased in the quest to improve material properties and behavior. Most of the current technologically important materials have therefore complex chemical compositions and carefully designed microstructures, however, often with high costs for expensive alloy ingredients and reduced compatibility when subject to recycling. Superalloys8, modern Al alloys9, and semiconductors10, are examples of such systems. Improved models that consider both composition and microstructure can be employed to investigate the trade-off between these in the optimization of material properties. This opens up a theory-based design pathway into a competition between the exploitation of conserved parameters (e.g. composition) vs. non-conserved parameters (e.g. microstructure) for optimal materials, process, and performance design. Note that although the spatial distribution of composition, depending on lengthscale, can be regarded as a type of microstructure, in this work, the microstructure is defined as structural variations at small scales. These are often in form of configurational defects in the solid structure and therefore, non-conservative. Multi-scale and multi-physics phenomena are not limited to microscopic and macroscopic lengthscales. The deformation behavior of planetary crusts11 or that of polar ice caps leading to calving12,13 are specific examples of much larger-scale material behavior where multiple physical processes and lengthscales come together.
In past ages, material design and discovery have relied on trial and error methods. With the advent of the scientific method, however, such discovery has been based increasingly on hypothesis-guided experimentation. Due to the inherent complexity of the task and the vast configuration space, as well as experimental and real-world limitations, however, material design and discovery based on experiments alone is extremely difficult and time-consuming. This is one reason why computational approaches have become an important tool in material science and engineering14,15,16. Many quantities that are inherently or technologically not measurable in experiments are possible to track and investigate in simulations. In this review, we mostly focus on approaches to model solid materials, in particular metallic systems. In this case, several modeling approaches and simulation packages exist, based on a number of different physical models and mathematical methods that have been proposed to describe these phenomena. Multi-physics approaches in these systems typically involve mechanical deformation, phase transformations, thermal and compositional fluctuations, and solid-state chemical reactions. Crystal plasticity17,18 for modeling inelastic deformation at mesoscopic lengthscales, phase-field (PF) modeling for spatially-resolved moving boundary problems19, the Cahn–Hilliard model for component diffusion and spinodal decomposition phenomena20,21, as well as CALPHAD22,23 for equilibrium thermodynamic phase diagram calculations are prominent examples of models and computational means for simulation of different phenomena involving microstructure and chemistry. A recent review of multi-scale methods in material science both in terms of historical development and current status can be found in ref. 24. More topic-specific reviews on multi-scale and multi-physics material modeling and simulation are available, for example on additive manufacturing25 or microstructure-property relationships in metals26.
In addition to modeling and simulation, the development of analytical methods based on statistics, machine learning, material information theory (informatics), data science, and artificial intelligence, is providing tools for the investigation and elucidation of very large sets of complex empirical data and simulation results for material systems. Such methods offer unparalleled computational efficiency in navigating and exploring complex material property landscapes27,28. A particular attractive area of application for such methods is material science, given the extremely large sets of experimental data or simulation results generated for example via microscopy or molecular dynamics. These developments are touched on only briefly at the end of the current review; for a very recent comprehensive treatment of these in the physical sciences, the interested reader is referred to ref. 29.
The ever-increasing computational resources and the development of advanced physical models and numerical algorithms make multi-physics modeling a rapidly growing topic. At the same time, improved experimental techniques make it possible to benchmark the models. The purpose of this review is to concisely present and summarize recent advancements in continuum-based multi-physics and multi-scale modeling and simulation techniques. Figure 1 provides an overview of basic aspects of the modeling of materials with microstructure. In this review, a model is considered multi-physical if it couples at least two different fields representing two different physical phenomena. Although the multi-scale paradigm is most obvious in methods such as direct atomistic-continuum coupling30, FEM231, or FEM-FFT32, scale-bridging is also realized in atomistically-informed continuum models. In such models, the data from small-scale simulations is transferred and employed on a larger scale. The models discussed in the current review are often calibrated by atomistic data and employed for simulation at much larger lengthscales. Despite a large number of proposed models, only a few of them find further use outside of their developer community. Thus, one goal of this review is to explore possibilities for more sustainable and accessible software development that facilitates collaborative use and successive improvement. This is analogous to other fields of open-source software development, such as the widely used community codes for molecular dynamics and density functional theory. The increased computational power, availability of numerous open-source software packages, as well as emergence of data-driven approaches, has led to many research groups employing multi-physics and multi-scale methods for material simulations. Therefore, the current review is beneficial for the community as a summary of important aspects of such methods to note either as a developer or as a user.

a–c Fracture and dislocation activity at the crack tip and triple junction during loading are depicted at the atomic scale (a), the single grain scale (b), and polygrain scale (c). (d) Abstract visualization of microstructure-composition-property space at these scales. (e) Schematic of simulation scale versus the number of parameters involved based on ideas in ref. 238. The complexity of microstructure-composition-property relationships at different scales poses a tremendous challenge to determining optimal material design and discovering new materials.
The current work begins with a summary of the theoretical framework for the formulation of multi-physics models and a detailed example of the formulation of such a model. This is followed by an overview of software packages based on such models as well as a short discussion of selected technical aspects related to these. Then examples of multi-physics integration in several applications are presented. The review ends with a summary of emerging and ongoing work related to machine learning, data-driven models, and statistics.
Theoretical framework and formulation of models
General considerations and background
The purpose of this section is a brief overview of the theoretical basis for, and approaches to, the formulation of models upon which the modeling and simulation of microstructure evolution and the resulting material behavior in selected engineering materials is based. Examples of the latter include structural (e.g., lightweight metallic alloys), functional (e.g., ferroelectric, ferromagnetic ceramics), energy (i.e., battery materials), high-temperature (e.g., Ni superalloys), and reactive (e.g., oxidation, reduction) materials. Such materials are generally metallic or ceramic in character. The corresponding microstructure consists of phases, grains, grain boundaries, defects (vacancies, voids, dislocations, interfaces, cracks), and so on. Even restricted to these materials, the relevant literature on theory, methods, and models is vast, and one cannot possibly hope to do justice to it in a short review. For a modicum of completeness, consider a few selected relevant approaches and models not treated here. These include for example atomistic computational models for multiphase, multicomponent systems such as (non-kinetic) hybrid Monte Carlo molecular dynamics (e.g., ref. 33) or (kinetic) diffusive molecular dynamics (e.g., refs. 34,35,36,37). Likewise neglected in this review is the formulation of continuum balance relations and flux constitutive relations via ensemble and spatial averaging of microstructure dynamics (e.g., refs. 38,39,40,41,42,43,44,45,46,47,48,49). Not considered as well are models for heterogeneous media with random microstructure based on statistics, stochastics, and averaging (e.g., refs. 50,51), or more generally those based on micromechanics and homogenization theory (e.g., refs. 52,53,54,55). The latter theory is also employed to formulate relevant models for generalized or extended continua with microstructure micromorphic:56; see also for example refs. 57,58. Another approach to formulate such models is based on a generalization of continuum mechanics to higher-order continua with additional kinematic (i.e., microstructural) degrees of freedom (e.g.,59,60,61,62). This approach has been applied to materials as diverse as liquid crystals, polymers (e.g., ref. 63), or polar ice (e.g., ref. 64).
The recent literature offers several comprehensive reviews of topics related to those addressed in the current work. These include the multi-scale modeling and simulation of interfaces, dislocations, and dislocation field plasticity treated in detail in ref. 65. Likewise, as mentioned above,24 discusses recent developments in multi-scale modeling and simulation, in particular, based on high-performance computing and machine learning. The current treatment is restricted to continuum models for microstructure evolution and material behavior formulated in the framework of continuum thermodynamics (e.g., refs. 66,67,68,69), internal variable theory (e.g., refs. 70,71), material theory (e.g., refs. 68,72), and PF methods (e.g., refs. 19,73). Models formulated in the thermodynamic framework are by definition thermodynamically consistent (e.g., satisfy the dissipation principle identically). This is important in formulating multi-physics models in order to avoid hidden inconsistencies (e.g., negative entropy production). Such inconsistencies could arise for example when relations (e.g., partial differential equations) from different models are coupled together in a fashion independent from the rest of the model formulation, something which could lead to nonphysical results.
In the case of PF methods, the literature offers a number of comprehensive reviews74,75,76,77,78,79,80, including their application to finite-deformation interface mechanics in crystalline solids81 and to material discovery82. Historically, the PF approach is generally considered to have originated in work aimed at extending the thermodynamics of spatially-uniform (bulk) systems to spatial non-uniformity; seminal works to this end include for example van der Waals (1893) (see ref. 83), Cahn and Hilliard (CH)20, Landau84, and Allen and Cahn (AC)85. In the context of classic density functional theory (CDFT: e.g., ref. 86), these early PF models are all weakly non-local. This is in contrast to strongly non-local models formulated via the CDFT of freezing87,88, atomic density field theory (ADFT: e.g., refs. 73,89), or PF crystal (PFC: e.g., refs. 78,90,91). A number of (higher-order) weakly non-local approximations of PFC have also been developed (see for example ref. 92). For more details on ADFT and PFC, the interested reader is referred to80. In more recent times, weakly non-local PF-based models have also been formulated via non-CDFT-based approaches (e.g., Clausius–Duhem thermodynamics:93,94,95).
Generally speaking, PFs and their evolution mediate transitions between thermodynamic states of a system determined by coupled and (energetically, kinetically) competing physical mechanisms. The PFs in ADFT or PFC for example take the form of atomic density fields which are naturally lattice-periodic. In contrast, PFs in microscopic PF models are represented by non-atomic, lattice fields (e.g., lattice disregistry) whose transition regions represent defects such as dislocation cores or interfaces. A prominent example here is PF microelasticity (PFM: e.g., refs. 96,97,98). Another example of PFs is represented by symmetry-adapted (e.g., strain-based) structural (non-conservative) order parameters (e.g., refs. 99,100,101,102).
In microscopic PF models, there is no scale separation between the gradient energy (internal, material) and system (external) lengthscales. In contrast, these lengthscales are scale-separated in mesoscopic PF models. In this latter case, the gradient energy is physically well-approximated by its sharp-interface (limit) value20. Furthermore, the gradient energy lengthscale loses any physical meaning in the mesoscopic context, in which case it can be employed as a mathematical convergence parameter (e.g., sharp-interface limit103,104, Γ-convergence105) and as a numerical regularization parameter (e.g., ref. 106) for more robust numerical convergence behavior.
Example of multi-physics-based thermodynamic model formulation
In the thermodynamic context, accounting for multiple physical mechanisms (multi-physics) and their (energetic, kinetic) competition or interaction with each other during processes resulting in microstructure evolution is completely natural. Models for materials with continuum microstructure formulated in this context can be classified in particular as being non-isothermal or isothermal. For example, dynamic processes involving in particular melting or solidification and heat transport (e.g., laser-heating- or powder-fusion-based additive manufacturing:107,108) are clearly non-isothermal. As discussed in more detail below, the (physically) more general non-isothermal case is based on the free entropy (e.g., refs. 109,110,111). Under certain assumptions (e.g., gradient-free entropy independent of temperature:112), this more general formulation reduces naturally to one based on the free energy. The majority of multi-physics models for materials with continuum microstructure in the literature are restricted to isothermal conditions, and in the case of elastic behavior, to geometric and physical linearity. In this case, temperature parameterizes energetic and kinetic properties and processes. A prime example here is the pioneering work of Khachaturyan73 on the energetic coupling of elasticity and chemistry to model multicomponent, multiphase crystalline solids.
As a concrete example of the formulation of a multi-physics model, the case of a multicomponent, multiphase, solid mixture with mass exchange (e.g., due to chemical reactions) is considered here. For simplicity, memory and fluctuation effects are neglected, and local thermodynamic equilibrium is assumed (e.g., refs. 66,68). In addition, attention is restricted to thermochemoelastic solid behavior. Rate-dependent gradient inelasticity (i.e., slip, damage, fracture) can also be formulated in a (non-conservative) PF setting (e.g., refs. 113,114,115,116,117,118); again for simplicity, this is neglected here. Not treated as well, for this reason, is the case of functional materials and processes (e.g., ref. 119), or charged chemical components (e.g., ref. 120). Instead, attention is focused on non-isothermal model formulation and its specialization to the isothermal case. Further details on the former are given for example in ref. 112; more comprehensive treatment of the latter, including non-equilibrium chemo-mechanical interface modeling, can be found for example in ref. 121.
Consider a non-isothermal mixture of nc chemical components and np solid phases. Neglecting any supplies of momentum, energy, or entropy for simplicity, the balance relations
(e.g., refs. 66,68) hold for the mass of component a = 1, …, nc, linear momentum, internal energy, and entropy, respectively. Here, ϱa, ja and σa represent the densities of mass, mass flux, and mass supply-rate, respectively, of component a. Mixture fields include the motion χ, as well as the densities of mass \(\varrho =\mathop{\sum }\nolimits_{a = 1}^{{n}_{{{{\rm{c}}}}}}{\varrho }_{a}\), momentum flux P (i.e., first Piola–Kirchhoff stress), internal energy ε, heat flux q, entropy η, entropy flux h, and entropy production-rate π. In the context of (1), the corresponding form
for the generalized Gibbs relation (e.g., refs. 66,68,122) is central to the formulation of thermodynamically consistent constitutive relations. Here, κa is the component entropic chemical potential (having units of entropy per unit mass, i.e. JK−1 Kg−1 in SI), and θ the mixture absolute temperature.
For inhomogeneous thermochemoelastic behavior, the constitutive forms
hold for the internal energy and entropy densities, respectively, with \({{{\mathbf{\varrho }}}}:= ({\varrho }_{1},\ldots ,{\varrho }_{{n}_{{{{\rm{c}}}}}})\) and \({{{\boldsymbol{\phi }}}}:= ({\phi }_{1},\ldots ,{\phi }_{{n}_{{{{\rm{p}}}}}})\), where ϕα represent the (structural) order parameter associated with phase α = 1, …, np. To model for example anisotropic interface energy and faceting (e.g., refs. 123,124), or lower-symmetry crystalline systems (e.g., ref. 125), ε and η would depend on even higher-order gradients of ϱ and ϕ (again for simplicity, such possibilities are neglected here). In particular, (3) determine in turn the thermochemoelastic (dependent) constitutive relations
for the absolute temperature and stress, respectively. The residual local deformation FR (dimensionless) is due to microstructure evolution resulting in stress relaxation. It represents a geometrically nonlinear generalization of the stress-free strain73 and (elastic) energy reduction. The geometrically exact kinematic constitutive form
for the evolution of FR depends on that of ϱ and ϕ, mediated by the material properties Nϱ a (having units of volume per unit mass, i.e., m3 kg−1 in SI) and Nϕ α (dimensionless). Alternative to (5) is the split FR = FCFS with \({\dot{{{{\bf{F}}}}}}_{{{{\rm{C}}}}}=\mathop{\sum }\nolimits_{a = 1}^{{n}_{{{{\rm{c}}}}}}{\dot{\varrho }}_{a}{{{{\bf{N}}}}}_{{{{\boldsymbol{\varrho }}}}a}\,{{{{\bf{F}}}}}_{{{{\rm{C}}}}}\) and \({\dot{{{{\bf{F}}}}}}_{{{{\rm{S}}}}}=\mathop{\sum }\nolimits_{\alpha = 1}^{{n}_{{{{\rm{p}}}}}}{\dot{\phi }}_{\alpha }{{{{\bf{N}}}}}_{{{{\boldsymbol{\phi }}}}\alpha }\,{{{{\bf{F}}}}}_{{{{\rm{S}}}}}\). Given (2)–(5), one obtains the forms
for the entropy production-rate density and entropy flux density (e.g., ref. 126), respectively, via the generalized Cahn–Hilliard (CH) form
for the entropic chemical potential in the case of inhomogeneous thermochemoelasticity. Here, φ is the negative free entropy density, i.e.,
with ψ the free energy density. Note that (6)2 reduces to its homogeneous form \({{{\bf{h}}}}={\theta }^{-1}{{{\bf{q}}}}-\mathop{\sum }\nolimits_{a = 1}^{{n}_{{{{\rm{c}}}}}}{\kappa }_{a}{{{{\bf{j}}}}}_{a}\) (e.g., ref. 66) when the generalized no-flux boundary conditions
are assumed on the boundary of any (referential) mixture region with (outward) unit normal n. These are relevant to purely bulk (i.e., system-size independent: e.g., ref. 20) inhomogeneous behavior. From the residual form (6)1 of π, one obtains the thermodynamic flux-force relations
in terms of the thermal conductivity K, component (mass) mobility Mab, component (mass) exchange coefficient kab, and phase mobility mαβ. For simplicity, the cross-coupling (off-diagonal) contributions to the general forms (e.g., refs. 66,69) of the flux-force relations are neglected in (10). Substituting (10) into (6)1 yields a form for the entropy production-rate density π which is (in general quasi-) quadratic in the driving forces and linear in the kinetic properties. As usual, requiring these latter properties to be nonnegative definite is then sufficient to insure π ⩾ 0 in all processes and so nonnegative entropy production.
In summary, the system
of evolution-field relations and coupled partial differential equations (PDEs) is obtained for ϱa, χ, ϕα, θ (recall \(\varrho =\mathop{\sum }\nolimits_{a = 1}^{{n}_{{{{\rm{c}}}}}}{\varrho }_{a}\)) from (1)1−3, (4) and (7)–(10). In particular, note that the temperature relation (11)4 is based on (9). The last three terms on the right-hand side of (11)4 determine the volumetric heating rate. Once specific model forms for ε, η, Nϱ a, Nϱ a, K, Mab, kab, and mαβ, are given, initial-boundary-value problems based on the system (11) and corresponding boundary conditions can be formulated and numerically solved to simulate microstructure evolution and the corresponding material behavior in multicomponent, multiphase systems undergoing thermochemomechanical loading. As mentioned above, a current and prominent example of the latter is laser-heating- or powder-fusion-based additive manufacturing107,108.
Consider next a few special cases of the above formulation employed in the literature. For example, assume further that the mixture is closed with respect to mass. In this case, the additional constraints \({{{{\bf{j}}}}}_{{n}_{{{{\rm{c}}}}}}=-\mathop{\sum }\nolimits_{a = 1}^{{n}_{{{{\rm{c}}}}}-1}{{{{\bf{j}}}}}_{a}\) and \({\sigma }_{{n}_{{{{\rm{c}}}}}}=-\mathop{\sum }\nolimits_{a = 1}^{{n}_{{{{\rm{c}}}}}-1}{\sigma }_{a}\) apply, \(\varrho =\mathop{\sum }\nolimits_{a = 1}^{{n}_{{{{\rm{c}}}}}}{\varrho }_{a}\) is constant via (1)1, and \({\varrho }_{{n}_{{{{\rm{c}}}}}}=\varrho -\mathop{\sum }\nolimits_{a = 1}^{{n}_{{{{\rm{c}}}}}-1}{\varrho }_{a}\) is no longer independent. In this context, it makes sense to introduce the mass concentration ca ≔ ϱa/ϱ such that \({c}_{{n}_{{{{\rm{c}}}}}}=1-\mathop{\sum }\nolimits_{a = 1}^{{n}_{{{{\rm{c}}}}}-1}{c}_{a}\) and \({\dot{\varrho }}_{a}=\varrho {\dot{c}}_{a}\). Likewise, the term \(\mathop{\sum }\nolimits_{a = 1}^{{n}_{{{{\rm{c}}}}}}{\kappa }_{a}(\varrho {\dot{c}}_{a}+{{{\rm{div}}}}{{{{\bf{j}}}}}_{a}-{\sigma }_{a})=\mathop{\sum }\nolimits_{a = 1}^{{n}_{{{{\rm{c}}}}}-1}({\kappa }_{a}-{\kappa }_{{n}_{{{{\rm{c}}}}}})\,[\varrho {\dot{c}}_{a}+{{{\rm{div}}}}{{{{\bf{j}}}}}_{a}-{\sigma }_{a}]\) in the Gibbs relation (2) depends only on \({\kappa }_{a}-{\kappa }_{{n}_{{{{\rm{c}}}}}}\). Further, the dependence of ε and η on ϱ and ∇ ϱ in (3) reduces to one on \({{{\bf{c}}}}:= ({c}_{1},\ldots ,{c}_{{n}_{{{{\rm{c}}}}}-1})\) and ∇ c. The remaining model relations reduce accordingly. Another special case takes the form of the so-called multiphase-field (MPF) approach (e.g., ref. 127; broad generalizations of this are considered in ref. 128). This is based on the constraint \(\mathop{\sum }\nolimits_{\alpha = 1}^{{n}_{{{{\rm{p}}}}}}{\phi }_{\alpha }=1\). This holds for example if (i) each ϕα is interpreted as a phase volume density (i.e., the ratio of the phase volume to total mixture volume) and (ii) the mixture is occupied only by the phases. In this case, ϕβ = 1 − ∑α≠βϕα is dependent for arbitrary β. In addition, \({\delta }_{{\phi }_{\alpha }}\varphi -{\delta }_{{\phi }_{\beta }}\varphi +({\partial }_{{{{{\bf{F}}}}}_{{{{\rm{R}}}}}}\varphi ){{{{\bf{F}}}}}_{{{{\rm{R}}}}}^{{{{\rm{T}}}}}\cdot ({{{{\bf{N}}}}}_{{{{\boldsymbol{\phi }}}}\alpha }-{{{{\bf{N}}}}}_{{{{\boldsymbol{\phi }}}}\beta })\) becomes the thermodynamic force conjugate to \({\dot{\phi }}_{\alpha }\) in (6)1 for π, in particular at the α-β interface. Further details on MPF can be found for example in ref. 76.
Lastly, consider the isothermal special case. For example, in this case, (2) reduces to \(\theta \pi -{{{\rm{div}}}}\theta {{{\bf{h}}}}=\mathop{\sum }\nolimits_{a = 1}^{{n}_{{{{\rm{c}}}}}}{\mu }_{a}(\varrho {\dot{c}}_{a}+{{{\rm{div}}}}{{{{\bf{j}}}}}_{a}-{\sigma }_{a})+{{{\bf{P}}}}\cdot \nabla \dot{{{{\boldsymbol{\chi }}}}}-{{{\rm{div}}}}{{{\bf{q}}}}-\dot{\psi }\) for the generalized Gibbs relation in terms of the dissipation-rate density θπ, energetic chemical potential μa = θκa, and free energy density ψ from (8)2. The constitutive form ψ = ψ(θ, ∇ χ, FR, ϱ, ∇ ϱ, ϕ, ∇ ϕ) for the latter is determined by (3), in which case \({\mu }_{a}={\delta }_{{\varrho }_{a}}\psi +{\partial }_{{{{{\bf{F}}}}}_{{{{\rm{R}}}}}}\psi \cdot {{{{\bf{N}}}}}_{{{{\mathbf{\varrho }}}}a}\) holds via (7). The remaining non-isothermal model relations reduce accordingly. A number of further assumptions and issues are addressed in the context of isothermal conditions in the literature, e.g., the existence of a dissipation potential (e.g., ref. 129), rate-variational formulation of initial-boundary-value problems (e.g., refs. 115,121,130,131,132), or the consequences of thermodynamic extremum principles (e.g., ref. 133,134,135,136). A detailed treatment and discussion of these goes beyond the scope of the current review. As mentioned above, the vast majority of PF models for multicomponent, multiphase solid systems in the literature are isothermal. These include for example models for single-component systems containing (i) structural phases (e.g., ref. 106), (ii) twin phases (e.g., ref. 137), (iii) equilibrium phase interfaces (e.g., refs. 138,139,140,141,142), (iv) damage, fracture phases (e.g., refs. 118,143,144,145) or (v) structural phases with dislocations (e.g., ref. 99). Equilibrium phase interfaces in two-phase, two-component chemoelastic systems have been treated for example in ref. 146. A non-mechanical treatment of non-equilibrium phase interfaces in multiphase, multicomponent systems based on the MPF approach discussed above is given for example by ref. 147. A chemo-mechanical treatment of non-equilibrium phase interfaces in multicomponent, multiphase systems can be found in ref. 121. Yet another area of application of the above model formulation in the isothermal case is the chemo-mechanical modeling of solute segregation to mechanical defects (e.g., dislocations, stacking faults:148,149) and its effect on spinodal decomposition, precipitation, and second-phase formation at defects (i.e., defect thermodynamics: e.g., refs. 150,151; see also for example ref. 152). Although the current treatment excludes electromagnetic effects, much of the above treatment still applies to functional materials (e.g., refs. 153,154,155,156) and battery materials (e.g., Li-ion:157,158,159,160). This holds true as well for electrochemical-mechanical systems (e.g., batteries, fuel cells) and processes (e.g., oxidation, reduction) containing charged chemical components or species (e.g., ref. 120). Further aspects of such isothermal models, as well as their numerical implementation, and incorporation into software packages, are discussed in the rest of this review.
Numerical methods and software packages
To make the presented theories and models usable, they need to be implemented in computer code and coupled to reliable and robust numerical solvers. To this end, many software packages have been developed for material modeling at different lengthscales or with multiple coupled physical phenomena. In this section, software and tools that are suitable for the multi-scale, multi-physics simulations, in particular for metallic systems, are discussed and three established packages are presented in more detail. These three packages are selected to illustrate the diversity of approaches and numerical methods that are employed in the field of continuum multi-physics modeling.
OpenPhase
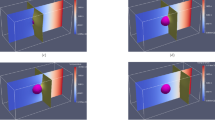
OpenPhase is open-source software, which is developed in general for the simulation of microstructure formation or phase transformation in complex systems161. The software is distributed as a library under the GNU General Public License (GPLv3). It has been used for solving physical phenomena in materials such as solidification, grain growth, creep, recrystallization, foam formation, and solid-solid transformation76,162,163,164,165,166,167,168,169. The software is based on the MPF approach76,127, where each phase/domain is defined by a PF parameter. The evolution of each phase is captured by solving the respective PF as well as those which are present in its interface. Through the coupling of the MPF approach with different fields, such as chemical, mechanical, and even magnetic fields, the software is used for the simulation of multi-physics problems for different applications. One application of OpenPhase for modeling and simulation of metallic foams as an advanced material is shown in Fig. 2. For this specific example, a model is developed by coupling the MPF approach with fluid dynamics162. Using MPF provides the possibility to assign the material parameters (such as mass, interface energy) of each bubble separately. Thus, it allows the introduction of an energy barrier (called disjoining pressure) between bubbles arising from the presence of additives which prevents bubble coalescence and stabilizes the foam formation, see Fig. 2162. However, if the liquid film between two bubbles falls below a threshold and the force pushing two bubbles towards each other exceeds the disjoining pressure, coalescence becomes inevitable. In Fig. 2a–c, for an initial setup including bubble nuclei, the final foam structures are different as a result of using different disjoining pressures (it is highest for Fig. 2a and lowest for Fig. 2c). A 3D simulation of foam and the summary of the model are depicted in Fig. 2d, e, respectively.

Microstructure formation in metallic foams (coupling of MPF with fluid dynamics)162. a–c Formation of foam structures from an identical distribution of bubble nuclei with different disjoining pressures. From a–c the disjoining pressure between bubbles is lowered and thus the number of merged/coalesced bubbles increases. Therefore, in spite of identical bubble nuclei distribution, different structures are formed. d An example of a 3D simulation of foam formation162. e A schematic representation of the coupling between MPF and fluid dynamics for modeling of microstructure formation in metallic foams162.
DAMASK
The Düsseldorf Advanced Material Simulation Kit (DAMASK)170, open-source, multi-physics software including crystal plasticity, is a simulation platform with the capability and flexibility to perform and analyze complex multi-field simulations. It is distributed with pre- and post-processing tools as a standalone software package under the GNU General Public License (GPLv3). Its modular structure allows solving of different field equations in a fully coupled way with a staggered approach, using Fast Fourier Transform (FFT) and Finite Element (FE) based solvers provided by the PETSc numerical library21,118,171. The pre- and post-processing of DAMASK facilitates coupling with other platforms such as DREAM.3D172, MTEX173, Neper174, Gmsh175, and Paraview176. The integration with other platforms allows for complex workflows. A few multi-physics example simulations using DAMASK are presented and discussed below.
MOOSE
The Multi-physics Object-Oriented Simulation Environment (MOOSE) is an open-source, parallel finite element framework that focuses on coupled multi-physics simulations in a flexible manner177. The framework includes a variety of plug-in infrastructures, which makes running or post-processing a multi-physics problem convenient. All MOOSE-based applications (apps) and community-developed physics modules are libraries that can be easily linked under the LGPL v2 license. Therefore, this design allows users to focus on their scientific endeavors without having to manually implement the intricacies of modern parallel computing. In MOOSE, the implicit method is used to solve equations from different apps in a fully coupled fashion. Multi-physics and multi-scale (FEM2) simulations can be easily set up by the MultiApp and Transfer system in MOOSE, as illustrated in Fig. 3. Furthermore, parallel solvers like PETSc171 are automatically included in this framework. As a result, users can run the simulations on a workstation or a cluster with multiple cores/CPUs with ease.

a Chemo-mechanical analysis for the lithium concentration distribution (top row) of a LiNixMnyCozO2 (NMC) particle, and the crack patterns of NMC (bottom row), with the influence of grain boundary interaction239. b The electro-mechanical analysis of lithium-ion battery cell performance under different discharge conditions178. c The workflow of multi-apps coupling. Each app is responsible for its own physical model, the data transfer between different apps is implemented by the transfer interface.
The apps in MOOSE can be used to simulate different physical problems. For instance, during the charge and discharge process of lithium-ion batteries (LIBs), the insertion of lithium not only involves the diffusion process of chemical species but also volume changes as well as mechanical deformation. Therefore, as mentioned in178,179, one can easily define the system’s free energy, as well as its derivative i.e., stress and chemical potential, with the consideration of both the mechanical deformation and diffusion in the ChemoMechanics App. The governing equations can also be defined within this module by using the Kernel from MOOSE.
Other packages
Besides those mentioned above, there is a large number of other open-source packages that could be employed to tackle multi-physics and multi-scale microstructure problems or certain aspects of it. Among them are AMITEX_FFTP for crystal plasticity and PF180, Prisms-Plasticity181 and Prisms-phase-field182 for plasticity and PF modeling, respectively, FiPy183 a finite volume partial differential equations (PDE) solver with python, FEniCS184 a computing platform for solving PDEs, and MicroSim185 to name a few. It should be noted that there is also commercially available software for multi-physics simulations such as COMSOL Multi-physics186, ANSYS187, and ABAQUS188. Although these are often powerful tools for multi-physics simulations, the black-box nature of the closed source codes will make their use limited in new model development and is often targeted to users only. In addition, many research groups have their in-house codes of which several ones are shared within projects or on request. A list of important packages and their features is presented in Table 1.
Challenges in multi-physics simulation development
System versus microstructure lengthscales
Materials containing multiple phases and chemical components consist of individual single crystals/grains/phases having distinct material properties (e.g., elastic stiffness) and fields (e.g., deformation gradient, component concentration, temperature) which vary spatially within each grain and across grain boundaries. In the extreme case, the average grain size may be of the same order of magnitude as that of the polycrystal/mixture. In this case, there is no separation of lengthscales, and the question arises as to how single-crystal- /grain- /phase-based properties and fields determine the corresponding properties and fields of the polycrystal/multicomponent, multiphase mixture as a whole.
As it turns out, this issue is related to the treatment of contrasts in phase properties and fields at phase/grain interfaces. Based on mechanical equilibrium and kinematic compatibility at interfaces, the case of equilibrium phase contrasts at elastic phase interfaces has been treated in the purely mechanical case for example by refs. 139,140,141,142. In the more general chemo-mechanical case, equilibrium phase contrasts have been treated in this way by ref. 146, and non-equilibrium (i.e., evolving) phase contrasts by ref. 121.
At sufficiently large mixture lengthscales, the assumption of scale separation becomes justified, in which case (e.g., statistical, volume) averaging of phase properties and fields is relevant. In the extreme, this takes the form of homogenization assumptions for phase properties and fields. In the classic (bulk) mechanical case53, for example, these include uniform phase (local) deformation (Voigt–Taylor), or uniform phase stress (Reuss-Sachs), and their generalizations (e.g., laminate theory:68). Analogous assumptions in the bulk chemical case are uniform phase chemical concentrations189 or uniform phase chemical potentials190. For more details, the interested reader is referred to ref. 121.
Related to these issues in the PF context is a classic challenge facing PF modeling, i.e., resolving (i) physical mechanisms over multiple lengthscales (i.e., interface and system) and (ii) physical processes over much longer timescales than dictated by interface kinetics. On the physical and mathematical side, methods which have been developed to deal with this include sharp-interface scaling20,103, thin-interface asymptotic analysis (e.g.,19,Appendix A]), Hamilton–Jacobi- and level-set-based rescaling191, or most recently the so-called sharp PF method192. On the numerical side, adaptive mesh refinement, or multigrid methods, are employed.
Model parameter identification
Multi-physics simulations take into account several different physical phenomena. It is, therefore, no surprise that they depend on a large number of model parameters. Determining material-specific values for these parameters is often a significant hurdle for the application of such models. Typically models without reliable parametrization are not useful for practical applications. In the extreme case of CALPHAD, the assessment of parameters is the more involved step in comparison to the development of the model itself. Consequently, open databases with contributions from the scientific community have been established193. However, the fragmentation of models prohibits a unified notation and exchange of parameters for full-field models. Therefore, common repositories, as opposed to the area of crystal structures194,195,196 or interatomic potentials197, have not been established. It remains therefore an open question how expensive parameter fitting198 can be avoided by sharing data and model implementations between different researchers.
Reproducibility
Coupled multi-physics simulations have predictive capabilities, i.e. they can explain non-trivial phenomena199. The ability to obtain surprising results comes, however, at a high cost: It is non-trivial to validate whether simulation results reflect reality or whether they are caused by a faulty implementation or nonphysical parameterization. Furthermore, multi-physics models are complex and many papers lack important information like employed numerical methods, tolerances, constitutive parameters, and sometimes even spatial or temporal discretization. Without making the implementation open, the model cannot be used by other researchers. One can of course try to re-implement it, but that is a lot of work and there is no guarantee that one can find solutions for the missing information. Often making the source code and the data available also does not help since one needs to maintain and support software packages with helpful documentation and guides. As a consequence, the FAIR principles for findable, accessible, interoperable, and reusable data management200 are of utmost importance for multi-physics simulations. This holds not only for the used software, which needs to be tested, documented, and versioned but also for the employed datasets. Using public repositories based on Git, e.g. on GitLab and GitHub, is today the de-facto standard for collaborative development of open-source software for scientific applications.
Examples of multi-physics integration
There is a vast trove of literature on multi-physics integration in material science that could be generally categorized into investigation of structural phases79,106, mechanical phase interfaces138,139,142,201, damage118, fracture145, polarization phases153,156, confined systems thermodynamics, defect thermodynamics148,150,151,202,203, chemical liquid and solid phases189,190, chemo-mechanical solid phases and phase interfaces121,146, as well as electrochemical-mechanical systems (e.g., batteries, fuel cells) and processes containing charged components or species120. A few of these cases are explored in more detail in this section.
Thermo-mechanical coupling is a case that is often not considered in material modeling. However, solving mechanical boundary-value problems only, completely ignoring the effect of other interacting fields is no longer sufficient for the state-of-the-art modeling of the evolution of microstructures in high-strength materials. For example, the effect of plastic dissipation on the evolving microstructure is generally ignored when performing a crystal plasticity simulation. It is, however, known that the dissipation leads to an increase in the temperature of the material, which couples back to the mechanics in the form of thermal lattice expansion, modified elastic constants, and faster dislocation velocity, affecting the overall stress response, as shown in Fig. 4a. Another example of coupling the mechanics and thermal solvers is the microstructural evolution of twinning-induced plasticity (TWIP) steel. In Fig. 4b, the total twin volume fraction shows variation when the effect of plastic dissipation is taken into account. A schematic of the thermo-mechanical coupling in DAMASK is shown in Fig. 4c.

a-left Effect of plastic dissipation on the overall stress-strain response in a uniaxial tension test on bcc-iron starting at room temperature, including the effect of varying temperature on the components of the elastic stiffness tensor and the effect of lattice expansion due to increase in temperature. Simulation using the dislotwin constitutive model198,240 where only plastic slip was considered, as implemented in DAMASK170. a-right Deviation in temperature for each material point from the final average temperature of the simulation box. b-left Varying twin volume fraction (ftw) for TWIP steel assuming both isothermal response and conduction of the dissipated plastic heat causing a change in stacking fault energy and thermal lattice expansion. Simulation using the dislotwin constitutive model240 as implemented in DAMASK170. b-right Final temperature in the case of conduction. c Schematics of thermo-mechanical coupling in DAMASK.
It is widely acknowledged that the interaction of the solute diffusion and elastoplastic deformation can strongly affect the microstructural evolution and the subsequent performance in many engineering materials149,204,205. Integration of the PF, Cahn–Hilliard, and crystal plasticity models enables the investigation of the complex role of plastic relaxation in chemo-mechanically coupled processes21,23,205,206,207,208,209, such as the rafting behavior in Ni-based superalloys149,210,211,212,213,214, lithiation of a phase-separating electrode material205,215, or the hydrogen dissolution and permeation in the metallic components for hydrogen storage and distribution207,216. The influence of the misfit stress, localized creep strain, and anisotropy of plasticity on the rafting behavior of \({\gamma }^{\prime}\) precipitates in Ni-based and Co-based superalloys has been widely studied at varying lengthscales through coupling PF models with various plasticity theories, such as at the scale of individual dislocations within the PF theory149,151,217, dislocation-density-based crystal plasticity model211,212,213,214, and the phenomenological mesoscale plasticity model204,210. In these applications, the multicomponent engineering alloys were generally simplified to binary or ternary simple alloys, due to the limitation of the incorporation of the complex chemical-free energies into the PF model. Recently, a chemical potential-based PF model has been developed based on a semi-analytical inversion of the thermodynamic relations21,23, which enables the direct incorporation of the CALPHAD-based chemical-free energies into the PF methods and can thus facilitate the application of the coupled CALPHAD/phase-field/crystal plasticity model in many multicomponent engineering alloys.
Figure 5a–c presents some exemplary applications of the multi-physics model developed in DAMASK in the investigation of the chemo-mechanically coupled ternary spinodal decomposition process21, grain boundary microchemistry, and precipitation in Al-Zn-Mg-Cu alloys23, and dislocation-twin interactions in Mg alloys208,209. Figure 5d shows the general framework of the finite strain and multicomponent chemo-mechanics model developed in DAMASK21,170. A phenomenological or dislocation-density-based crystal plasticity model was usually employed to predict the heterogeneous distribution of stress, strain, and dislocation activities. The mechanical model was implicitly coupled to an MPF model and a Cahn–Hillard model to describe the concurrent phase transformations, e.g., precipitation, twinning and martensite transformation, and multicomponent solute diffusion during many engineering materials processes.

a Influence of the plastic deformation on the ternary spinodal decomposition process, showing the morphology of the decomposition regions, the hydrostatic stress, and the plastic strain21. b CALPHAD-informed PF modeling of the grain boundary segregation and precipitation evolution in an Al-Zn-Mg-Cu alloy23. c Studying the dislocation slip and twinning interaction in the Mg polycrystal with a coupled crystal plasticity--PF model, showing the twinning microstructure and the spatial distribution of basal dislocations208,209. d Schematic of the finite-strain chemo-mechanical model in DAMASK21,170.
Hydrogen embrittlement and its effect on the cracking of materials has been modeled by coupling a PF damage model with stress-assisted hydrogen diffusion and mechanics218,219,220. The hydrogen effect is accounted for by incorporating the atomic concentration of H in the model, which alters the crack resistance of the material. It has been shown that increasing the hydrogen content significantly decreases the mechanical response of a notched plate218, which is a direct result of the lowered crack resistance in the presence of hydrogen. Accumulation of the hydrogen occurs in the crack tip due to locally large stresses as shown in ref. 218.
Emerging avenues and outlook
Although the simulation methods for multi-physics and multi-scale problems have been continuously improved as summarized above, the complexity of realistic material science problems can still easily overwhelm even the most cutting-edge approaches, both in terms of computation time and model identification. Each important physical phenomenon added to a given problem introduces not only a new set of parameters for that specific model but also the possible dependency of the parameters of the previously included models on the parameters of the new one. Quantifying the intertwined model-parameter dependencies is not an easy task when the system is complex. Model-free and data-driven computational approaches221,222 along with machine-learning methods28 are emerging as either an alternative or an augmenting tool for material simulations. The recently published works can roughly be organized into three categories: data transfer between scales (such as interatomic potentials based on density functional theory223, various forms of homogenization or averaged material-response prediction224,225), solving or accelerating the solution of PDEs226,227, and utilizing a physics-informed neural network for better prediction of material processes228.
In the field of accelerated solution of PDEs, one approach is linearization within the explicit time-stepping setting. The associated linear problems have quadratic complexity if attempted to be solved from scratch, and the memory requirements for matrix-based methods are overwhelming for fine-microstructure studies. Iterative matrix-free solvers (such as MF-FEM) prove to be more efficient if initialization is available, for which a common approach is a prior solution on a coarser grid combined with interpolation (MultiGrid). This strategy, although beneficial in some cases, becomes challenging for high-contrast material phases and fails upon the emergence of critical fractal-like self-organization. For both scenarios, stabilization and acceleration of the approach are achieved by the introduction of intermediate homogenization loops. An example of this approach for the former case is shown in refs. 229,230. The newly-introduced adaptive smoothening prolongation-based multigrid solver (AMG) designed for ill-conditioned nonlinear problems of structural mechanics – which is a broader problem-space in a part of which fractal-like patterns can emerge—shows robust performance both for benchmark models and real-world calculations (such as fine-microstructure composite-made mechanical tools), a key feature employed in the approach being adaptive factored sparse approximate inverse231,232,233. A more general study of conditioning the AMG for elliptic PDEs is conducted in ref. 234. There, the idea of intermediate stabilizing homogenization is addressed as proper identification of stable coarsening degrees of freedom, and a generalization of the Ruge-Stüben algorithm is employed. The aforementioned ill-conditioning of the standard MG approach is especially apparent in elliptic boundary-value problems with stochastic coefficients, where indeed fractal patterns can emerge and stable intermediate coarsening steps are all the more necessary. Construction of MG algorithms for such problems seems to possibly benefit from considering the ideas explored in ref. 235.
A noteworthy recent work236 combines the PDE-solution-acceleration effort with the concept of data transfer between scales. There, a finite element procedure is discussed in which instead of the full displacement-based second-order PDE formulation, the problem of plastic deformation in nonlinear materials is treated as a mixed stress-displacement first-order system. Mesh-independence and stable convergence with the increasing resolution are obtained by using a variational-multi-scale stabilization technique, based on the decomposition of the fields into their finite element component and a subscale. The subscale-corrections to the fields are expanded on a basis orthogonal to that of the upper-scale fields (in the Galerkin sense). This work demonstrates an important feature of multiple-scale methods: separation between scales is obtained by preventing resonances between the scales, which ensures the overall stabilization of the scheme. The said separation is achieved by coarse-graining (averaging) over the subscale before adding its correction. Similarly, when acknowledging a physically-distinguished subscale (rather than a numerically-constructed one), as in ref. 237, where material properties are derived from interatomic potentials, statistical averaging is implemented. For example, ref. 237 is using the Gauss approximation potential framework. Interestingly enough, in deriving material properties from interatomic potentials, often machine-learning techniques are used (as in ref. 237), which are based on low-order smooth interpolation, which naturally introduces the coarse-graining stabilizing effect as aforementioned.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Beyerlein, I. J. et al. Alloy design for mechanical properties: conquering the length scales. MRS Bull. 44, 257–265 (2019).
Zhao, Y. et al. A review on modeling of electro-chemo-mechanics in lithium-ion batteries. J. Power Sources 413, 259–283 (2019).
Grey, C. P. & Hall, D. S. Prospects for lithium-ion batteries and beyond—a 2030 vision. Nat. Commun. 11, 6279 (2020).
Dwivedi, S. K. & Vishwakarma, M. Hydrogen embrittlement in different materials: a review. Int. J. Hydrog. Energy 43, 21603–21616 (2018).
Donahue, J. R., Lass, A. B. & Burns, J. T. The interaction of corrosion fatigue and stress-corrosion cracking in a precipitation-hardened martensitic stainless steel. npj Mater. Degrad. 1, 11 (2017).
Nordlund, K. et al. Primary radiation damage: a review of current understanding and models. J. Nucl. Mater. 512, 450–479 (2018).
Kontis, P., Kostka, A., Raabe, D. & Gault, B. Influence of composition and precipitation evolution on damage at grain boundaries in a crept polycrystalline ni-based superalloy. Acta Mater. 166, 158–167 (2019).
Wu, X. et al. Unveiling the re effect in ni-based single crystal superalloys. Nat. Commun. 11, 389 (2020).
Georgantzia, E., Gkantou, M. & Kamaris, G. S. Aluminium alloys as structural material: a review of research. Eng. Struct. 227, 111372 (2021).
Zhang, J., Tse, K., Wong, M., Zhang, Y. & Zhu, J. A brief review of co-doping. Front. Phys. 11, 117405 (2016).
Knoll, M., Tommasi, A., Logé, R. E. & Signorelli, J. W. A multiscale approach to model the anisotropic deformation of lithospheric plates, Geochem. Geophys. Geosyst. 10, Q08009 (2009). https://doi.org/10.1029/2009GC002423.
Faria, S. H., Weikusat, I. & Azuma, N. The microstructure of polar ice. part ii: state of the art. J. Struct. Geol. 61, 21–49 (2014).
Montagnat, M. et al. Multiscale modeling of ice deformation behavior. J. Struct. Geol. 61, 78–108 (2014).
The Minerals Metals & Materials Society (TMS). Modeling Across Scales: A Roadmapping Study for Connecting Materials Models and Simulations Across Length and Time Scales (TMS, 2015).
Diehl, M. Review and outlook: mechanical, thermodynamic, and kinetic continuum modeling of metallic materials at the grain scale. MRS Commun. 7, 735–746 (2017).
Hong, S. et al. Reducing time to discovery: materials and molecular modeling, imaging, informatics, and integration. ACS Nano 15, 3971–3995 (2021).
Roters, F., Eisenlohr, P., Bieler, T. R. & Raabe, D.Crystal Plasticity Finite Element Methods: In Materials Science and Engineering (Wiley, 2011).
Clayton, J. D. Nonlinear Mechanics of Crystals (Springer, 2011).
Provatas, N. & Elder, K. Phase Field Methods in Material Science and Engineering (Wiley, 2010).
Cahn, J. W. & Hilliard, J. E. Free energy of a non-uniform system. I. Interfacial energy. J. Chem. Phys. 28, 258–267 (1958).
Shanthraj, P., Liu, C., Akbarian, A., Svendsen, B. & Raabe, D. Multi-component chemo-mechanics based on transport relations for the chemical potential. Comput. Methods Appl. Mech. Eng. 365, 113029 (2020).
Kattner, U. R. The calphad method and its role in material and process development. Tecnol. Metal. Mater. Min. 13, 3–15 (2016).
Liu, C. et al. CALPHAD-informed phase-field modeling of grain boundary microchemistry and precipitation in al-zn-mg-cu alloys. Acta Mater. 214, 116966 (2021).
Radhakrishnan, R. A survey of multiscale modeling: foundations, historical milestones, current status, and future prospects. AIChE J. 67, e17026 (2021).
Bayat, M., Dong, W., Thorborg, J., To, A. C. & Hattel, J. H. A review of multi-scale and multi-physics simulations of metal additive manufacturing processes with focus on modeling strategies. Addit. Manuf. 47, 102278 (2021).
Geers, M. & Yvonnet, J. Multiscale modeling of microstructure–property relations. MRS Bull. 41, 610–616 (2016).
Batra, R. Accurate machine learning in materials science facilitated by using diverse data sources. Nature 589, 524–525 (2021).
Schmidt, J., Marques, M. R. G., Botti, S. & Marques, M. A. L. Recent advances and applications of machine learning in solid-state materials science. npj Comput. Mater. 5, 83 (2019).
Kalinin, S. V. et al. Handbook on Big Data and Machine Learning in the Physical Sciences (World Scientific, 2020).
Curtin, W. A. & Miller, R. E. A perspective on atomistic-continuum multiscale modeling. Model. Simul. Mater. Sci. Eng. 25, 071004 (2017).
Schröder, J. in Plasticity and Beyond: Microstructures, Crystal-Plasticity and Phase Transitions (eds Schröder, J. & Hackl, K.) Ch. 1 (Springer, 2014).
Kochmann, J., Wulfinghoff, S., Reese, S., Mianroodi, J. R. & Svendsen, B. Two-scale FE?FFT- and phase-field-based computational modeling of bulk microstructural evolution and macroscopic material behavior. Comput. Methods Appl. Mech. Eng. 305, 89–110 (2016).
Sadigh, B. et al. Scalable parallel Monte Carlo algorithm for atomistic simulations of precipitation in alloys. Phys. Rev. B 85, 184203 (2012).
Li, J. et al. Diffusive molecular dynamics and its application to nanoindentation and sintering. Phys. Rev. B 84, 054103 (2011).
Dontsova, E., Rottler, J. & Sinclair, C. W. Solute-defect interactions in Al-Mg alloys from diffusive variational Gaussian calculations. Phys. Rev. B 90, 174102 (2014).
Venturini, G., Wang, K., Romero, I., Ariza, M. & Ortiz, M. Atomistic long-term simulation of heat and mass transport. J. Mech. Phys. Solids 73, 242–268 (2014).
Mendez, J. P. & Ponga, M. MXE: A package for simulating long-term diffusive mass transport phenomena in nanoscale systems. Comput. Phys. Commun. 260, 107315 (2021).
Irving, J. H. & Kirkwood, J. G. The statistical mechanical theory of transport processes. iv. the equations of hydrodynamics. J. Chem. Phys. 18, 817–829 (1950).
Russakoff, G. A derivation of the macroscopic Maxwell equations. Am. J. Phys. 38, 1188–1195 (1970).
Hardy, R. Formulas for determining local properties in molecular dynamics simulations: shock waves. J. Chem. Phys. 76, 622–628 (1982).
Pitteri, M. On a statistical-kinetic model for generalized continua. Arch. Ration. Mech. Anal. 111, 99–120 (1990).
Svendsen, B. In Kinetic and Continuum Theories of Granular and Porous Media (eds Hutter, K. & Wilmanski, K.) Ch. 5 (Springer, 1999).
Chen, Y. Reformulation of microscopic balance equations for multiscale materials modeling. J. Chem. Phys. 130, 134706 (2009).
Chen, Y., Zimmerman, J., Krivtsov, A. & McDowell, D. L. Assessment of atomistic coarse-graining methods. Int. J. Solids Struct. 49, 1337–1349 (2011).
Admal, N. C. & Tadmor, E. B. A unified interpretation of stress in molecular systems. J. Elast. 100, 63–143 (2010).
Lehoucq, R. B. & Lilienfeld-Toal, A. V. Translation of Walter Noll’s “derivation of the fundamental equations of continuum thermodynamics from statistical mechanics". J. Elast. 100, 5–24 (2010).
Lehoucq, R. B. & Sears, M. P. Statistical mechanical foundation of the peridynamic nonlocal continuum theory: energy and momentum conservation laws. Phys. Rev. B 84, 031112 (2011).
Chen, Y. & Diaz, A. Physical foundation and consistent formulation of atomic-level fluxes in transport processes. Phys. Rev. E 98, 052113 (2018).
Svendsen, B. Constitutive relations for polar continua based on statistical mechanics and spatial averaging. Proc. R. Soc. Lond. A Math. Phys. Sci. 476, 20190407 (2020).
Torquato, S. Random Heterogeneous Materials (Springer, 2002).
Ostoja-Starzweski, M. Microstructural Randomness and Scaling in Mechanics of Materials (Chapman & Hall/CRC, 2008).
Mura, T. Micromechanics of Defects in Solids (Springer, 1987).
Nemat-Nasser, S. & Hori, M. Micromechanics: Overall Properties of Heterogeneous Materials (Elsevier, 1999).
Li, S. & Wang, G. Introduction to Micromechanics and Nanomechanics (World Scientific, 2008).
Alleman, C., Luscher, D., Bronkhorst, C. & Ghosh, S. Distribution-enhanced homogenization framework for heterogeneous elasto-plastic problem. J. Mech. Phys. Solids 85, 176–202 (2015).
Biswas, R. & Poh, L. H. A micromorphic computational homogenization framework for heterogeneous materials. J. Mech. Phys. Solids 102, 187–208 (2017).
Eringen, A. C. Mechanics of micromorphic materials. In Proceedings of the 11th Congress of Applied Mechanic 131–138 (Springer, 1964).
Forest, S. Micromorphic approach for gradient elasticity, viscoplasticity, and damage. J Eng. Mech. 135, 117–131 (2009).
Capriz, G. Continua with Microstructure (Springer, 1989).
Blenk, S. & Muschik, W. Orientational balances for nematic liquid crystals. J. Non-Equilb. Thermody. 16, 67–87 (1991).
Papenfuss, C. Theory of liquid crystals as an example of mesoscopic continuum mechanics. Comput. Mater. Sci. 19, 45–52 (2000).
Svendsen, B. On the continuum modeling of materials with kinematic structure. Acta Mater. 152, 49–80 (2001).
Faria, S. H. Mixtures with continuous diversity: general theory and application to polymer solutions. Contin. Mech. Thermodyn. 13, 91–121 (2001).
Placidi, L. & Hutter, K. Thermodynamics of polycrystalline materials treated by the theory of mixtures with continuous diversity. Contin. Mech. Thermodyn. 17, 409–451 (2006).
McDowell, D. L. In Mesoscale Models: From Micro-Physics to Macro-Interpretation (eds Zbib, H., Forest, S. & Mesarovic, S) (Springer, 2019).
Groot, S. R. d. & Mazur, P. Non-equilibrium Thermodynamics (Dover Publications, 1984).
Müller, I. Thermodynamics (Pitman, 1985).
Šilhavý, M. The Mechanics and Thermodynamics of Continuous Media (Springer, 1997).
Balluffi, R. W., Allen, S. M. & Carter, W. C. Kinetics of Materials (John Wiley, 2005).
Maugin, G. A. The Thermomechanics of Plasticity and Fracture (Cambridge Univ. Press, 1992).
McDowell, D. L. Internal State Variable Theory. Handbook of Materials Modeling: Methods (eds Yip, Sidney) 1151–1169 (Springer Netherlands, Dordrecht 2005). https://doi.org/10.1007/978-1-4020-3286-8_58.
Truesdell, C. & Noll, W. The Non-Linear Field Theories of Mechanics (Springer, 1965).
Khachaturyan, A. G. Theory of Structural Transformations in Solids (Wiley, 1983).
Chen, L.-Q. Phase-field model for microstructure evolution. Annu. Rev. Mater. Res. 32, 113–140 (2002).
Moelans, N., Blanpain, B. & Wollants, P. An introduction to phase-field modeling of microstructure evolution. CALPHAD 32, 268–294 (2008).
Steinbach, I. Phase-field models in material science. Model. Simul. Mater. Sci. Eng. 17, 073001 (2009).
Steinbach, I. Phase-field model for microstructure evolution at the mesoscopic scale. Annu. Rev. Mater. Res. 43, 89–107 (2013).
Emmerich, H. et al. Phase-field-crystal models for condensed matter dynamics on atomic length and diffusive time scales: an overview. Adv. Phys. 61, 665–743 (2012).
Levitas, V. I. Phase transformations, fracture, and other structural changes in inelastic materials. Int. J. Plast. 140, 102914 (2021).
Chen, L.-Q. & Zhao, Y. From classical thermodynamics to phase-field method. Prog. Mater. Sci. 124, 100868 (2022).
Clayton, J. D. Mesoscale models of interface mechanics in crystalline solids: a review. J. Mater. Sci. 53, 5515–5545 (2018).
Tonks, M. R. & Aagesen, L. K. The phase field method: mesoscale simulation aiding material discovery. Annu. Rev. Mater. Res. 49, 79–102 (2019).
Rowlison, J. S. Translation of J. D. van der Waals’ “The thermodynamic theory of capillarity under the hypothesis of a continuous variation of density" (1893). J. Stat. Phys. 20, 197–243 (1979).
Landau, L. D. & Lifshitz, E. M. Statistical Physics Vol. 5 (Pergamon Press, 1963).
Allen, S. M. & Cahn, J. W. A macroscopic theory for antiphase boundary motion and its application to antiphase domain coarsening. Acta metall. 27, 1085–1095 (1979).
Engel, E. & Dreizler, R. M. Density Functional Theory (Springer, 2011).
Ramakrishnan, T. V. & Yussouff, M. First principles order-parameter theory of freezing. Phys. Rev. B 19, 2775–2794 (1979).
Singh, Y. Density functional theory of freezing and properties of the ordered phase. Phys. Rep. 207, 251–444 (1991).
Jin, Y. M. & Khachaturyan, A. G. Atomic density function theory and modeling of microstructure evolution at the atomic scale. J. Appl. Phys. 100, 013519 (2006).
Elder, K. R. & Grant, M. Modeling elastic and plastic deformations in nonequilibrium processing using phase field crystals. Phys. Rev. E 70, 051605 (2004).
Hütter, M. & Svendsen, B. Formulation of strongly non-local, non-isothermal dynamics for heterogeneous solids based on the GENERIC with application to phase-field modeling. Mater. Theory 1, 2–20 (2017).
Elder, K. R., Provatas, N., Berry, J., Stefanovic, P. & Grant, M. Phase-field crystal modeling and classical density functional theory of freezing. Phys. Rev. B 75, 064107 (2007).
Fried, E. & Gurtin, M. E. Continuum theory of thermally induced phase transitions based on an order parameter. Phys. D: Nonlinear Phenom. 68, 326–343 (1993).
Fried, E. Continua described by a microstructural field. Z. Angew. Math. Phys. 47, 168–175 (1996).
Gurtin, M. E. Generalized Ginzburg-Landau and Cahn-Hilliard equations based on a microforce balance. Phys. D Nonlinear Phenom. 92, 178–192 (1996).
Wang, Y. U., Jin, Y. M., Cutiño, A. M. & Khachaturyan, A. G. Nanoscale phase field microelasticity theory of dislocations: model and 3D simulations. Acta Mater. 49, 1847–1857 (2001).
Wang, Y. & Li, J. Phase field modeling of defects and deformation. Acta Mater. 58, 1212–1235 (2010).
Mianroodi, J. R. & Svendsen, B. Atomistically determined phase field modeling of dislocation dissociation, stacking fault formation, dislocation slip, and reactions in fcc systems. J. Mech. Phys. Solids 77, 109–122 (2015).
Gröger, R., Marchand, B. & Lookman, T. Dislocations via incompatibilities in phase-field models of microstructure evolution. Phys. Rev. B 94, 054105 (2016).
Rudraraju, S., der Ven, A. V. & Garikipati, K. Mechanochemical spinodal decomposition: a phenomenological theory of phase transformations in multi-component, crystalline solids. npj Comput. Mater. 2, 16012 (2016).
Thomas, J. C. & der Ven, A. V. The exploration of nonlinear elasticity and its efficient parameterization for crystalline materials. J. Mech. Phys. Solids 107, 76–95 (2017).
Natarajan, A. R., Thomas, J. C., Puchala, B. & der Ven, A. V. Symmetry-adapted order parameters and free energies for solids undergoing order-disorder phase transitions. Phys. Rev. B 96, 134204 (2017).
Grach, G. & Fried, E. An order-parameter-based theory as a regularization of a sharp-interface theory for solid-solid phase transitions. Arch. Ration. Mech. Anal. 138, 355–404 (1997).
Elder, K. R., Grant, M., Provatas, N. & Kosterlitz, J. M. Sharp interface limits of phase-field models. Phys. Rev. E 64, 021604 (2001).
Braides, A. Gamma Convergence for Beginners (Oxford Univ. Press, 2002).
Hildebrand, F. & Miehe, C. A phase field model for the formation and evolution of martensitic laminate microstructure at finite strains. Philos. Mag. 92, 4250–4290 (2012).
Yang, Y., Ragnvaldsen, O., Bai, Y., Yi, M. & Xu, B.-X. 3D non-isothermal phase-field simulation of microstructure evolution during selective laser sintering. npj Comput. Mater. 5, 81 (2019).
Yang, Y., Kühn, P., Yi, M., Egger, H. & Xu, B.-X. Non-isothermal phase-field modeling of heat-melt-microstructure-coupled processes during powder bed fusion. J. Mater. 72, 1719–1733 (2020).
Penrose, O. & Fife, P. C. Thermodynamically consistent models of phase-field type for the kinetics of phase transitions. Phys. D. 43, 44–62 (1990).
Penrose, O. & Fife, P. C. On the relation between the standard phase-field model and a “thermodynamically consistent" phase-field model. Phys. D. 69, 107–113 (1993).
Wang, S.-L. et al. Thermodynamically-consistent phase-field models for solidification. Phys. D Nonlinear Phenom. 69, 189–200 (1993).
Gladkov, S., Kochmann, J., Hütter, M., Reese, S. & Svendsen, B. Thermodynamic model formulations for inhomogeneous solids with application to non-isothermal phase field modeling. J. Non-Equilb. Thermody. 41, 131–139 (2016).
Svendsen, B. Phase-field extension of crystal plasticity with application to hardening modeling. In Continuum Scale Simulation of Engineering Materials 501–511 (John Wiley & Sons, Ltd, 2004). https://doi.org/10.1002/3527603786.ch24.
Yalcinkaya, T., Brekelmans, W. A. M. & Geers, M. G. D. Deformation patterning driven by rate dependent non-convex strain gradient plasticity. J. Mech. Phys. Solids 59, 1–17 (2011).
Miehe, C. A multi-field incremental variational framework for gradient-extended standard dissipative solids. J. Mech. Phys. Solids 59, 898–923 (2011).
Klusemann, B. & Yalcinkaya, T. Plastic deformation induced microstructure evolution through gradient enhanced crystal plasticity based on a non-convex Helmholtz energy. Int. J. Plast. 48, 168–188 (2013).
Shanthraj, P., Eisenlohr, P., Diehl, M. & Roters, F. Numerically robust spectral methods for crystal plasticity simulations of heterogeneous materials. Int. J. Plast. 66, 31–45 (2015).
Shanthraj, P., Sharma, L., Svendsen, B., Roters, F. & Raabe, D. A phase field model for damage in elasto-viscoplastic materials. Comput. Methods Appl. Mech. Eng. 312, 167–185 (2016).
García, R. E., Bishop, C. M. & Carter, W. C. Thermodynamically consistent variational principles with applications to electrically and magnetically active systems. Acta Mater. 52, 11–21 (2004).
Bucci, G., Chiang, Y.-M. & Carter, W. C. Formulation of the coupled electrochemical-mechanical boundary-value problem with applications to transport of multiple charged species. Acta Mater. 104, 33–51 (2016).
Svendsen, B., Shanthraj, P. & Raabe, D. Finite-deformation phase-field chemomechanics for multiphase, multicomponent solids. J. Mech. Phys. Solids 112, 619–636 (2018).
Liu, I.-S. Method of lagrange multipliers for exploitation of the entropy principle. Arch. Ration. Mech. Anal. 46, 131–148 (1972).
Abinandanan, T. A. & Haider, F. An extended Cahn-Hilliard model for interfaces with cubic anisotropy. Philos. Mag. 81, 2457–2479 (2001).
Roy, A., Nani, E. S., Lahiri, A. & Gururajan, M. P. Interfacial free energy anisotropy driven faceting of precipitates. Philos. Mag. 97, 2705–2735 (2017).
Nani, E. S. & Gururajan, M. P. On the incorporation of cubic and hexagonal interfacial energy anisotropy in phase field models using higher order tensor terms. Philos. Mag. 94, 3331–3352 (2014).
Maugin, G. On internal variables and dissipative structures. J. Non-equil. Thermody. 15, 173–192 (1990).
Steinbach, I. et al. A phase field concept for multiphase systems. Phys. D Nonlinear Phenom. 94, 135–147 (1996).
Moelans, N. A quantitative and thermodynamically consistent phase-field interpolation function for multi-phase systems. Acta Mater. 59, 1077–1086 (2011).
Hütter, M. & Svendsen, B. Quasi-linear versus potential-based formulations of force-flux relations and the GENERIC for irreversible processes: comparisons and examples. Contin. Mech. Thermodyn. 25, 803–816 (2013).
Svendsen, B. On the thermodynamic- and variational-based formulation of models for inelastic continua with internal lengthscales. Comput. Methods Appl. Mech. Eng. 48, 5429–5452 (2004).
Svendsen, B. Continuum Thermodynamic and Rate Variational Formulation of Models forExtended Continua. In Proc. Advances in Extended and Multifield Theories for Continua 1–18 (Springer Berlin Heidelberg, Berlin, Heidelberg 2011).
Gladkov, S. & Svendsen, B. Thermodynamic and rate variational formulation of models for inhomogeneous gradient materials with microstructure and application to phase field modeling. Acta Mech. Sin. 31, 162–172 (2015).
Cocks, A. C. F., Gill, S. P. A. & Pan, J. Modelling microstructure evolution in engineering materials. Adv. Appl. Mech. 36, 81–162 (1999).
Hackl, K. & Fischer, F. D. On the relation between the principle of maximum dissipation and inelastic evolution given by dissipation potentials. Proc. R. Soc. Lond. A Math. Phys. Sci. 464, 117–132 (2008).
Fischer, F. D., Svoboda, J. & Petryk, H. Thermodynamic extremal principles for irreversible processes in materials science. Acta Mater. 67, 1–20 (2014).
Hackl, K., Fischer, F. D., Zickler, G. A. & Svoboda, J. Are Onsager’s reciprocal relations necessary to apply thermodynamic extremal principles? J. Mech. Phys. Solids 135, 103780 (2020).
Clayton, J. D. & Knap, J. A phase field model of deformation twinning: Nonlinear theory and numerical simulations. Phys. D Nonlinear Phenom. 240, 841–858 (2011).
Steinbach, I. & Apel, M. Multi phase field model for solid state transformation with elastic strain. Phys. D Nonlinear Phenom. 217, 153–160 (2006).
Mosler, J., Shchyglo, O. & Hojjat, H. M. A novel homogenization method for phase field approaches based on partial rank-one relaxation. J. Mech. Phys. Solids 68, 251–266 (2014).
Schneider, D. et al. Phase-field elasticity model based on mechanical jump conditions. Comput. Mech. 55, 887–901 (2015).
Bartels, A. & Mosler, J. Efficient variational constitutive updates for allen-cahn-type phase field theory coupled to continuum mechanics. Comput. Methods Appl. Mech. Eng. 317, 55–83 (2017).
Schneider, D. et al. On the stress calculation within phase-field approaches: a model for finite deformations. Comput. Mech. 60, 203–217 (2017).
Hakim, V. & Karma, A. Laws of crack motion and phase-field models of fracture. J. Mech. Phys. Solids 57, 342–368 (2009).
Miehe, C., Welschinger, F. & Hofacker, M. Thermodynamically consistent phase-field models of fracture: variational principles and multi-field fe implementations. Int. J. Numer. Methods Eng. 83, 1273–1311 (2010).
Shanthraj, P., Svendsen, B., Sharma, L., Roters, F. & Raabe, D. Elasto-viscoplastic phase field modelling of anisotropic cleavage fracture. J. Mech. Phys. Solids 99, 19–34 (2017).
Durga, A., Wollants, P. & Moelans, N. A quantitative phase-field model for two-phase elastically inhomogeneous systems. Comput. Mater. Sci. 99, 81–95 (2015).
Zhang, L. & Steinbach, I. Phase-field model with finite interface dissipation: Extension to multi-component multi-phase alloys. Acta Mater. 60, 2702–2710 (2012).
Ma, N., Shen, C., Dregia, S. A. & Wang, Y. Segregation and wetting transition at dislocations. Metall. Mater. Trans. A 37A, 1773–1783 (2006).
Mianroodi, J. R. et al. Atomistic phase field chemomechanical modeling of dislocation-solute-precipitate interaction in Ni–Al–Co. Acta Mater. 175, 250–261 (2019).
Cahn, J. W. On spinodal decomposition. Acta metall. 9, 795–801 (1961).
Mianroodi, J. R., Shanthraj, P., Svendsen, B. & Raabe, D. Phase-field modeling of chemoelastic binodal/spinodal relations and solute segregation to defects in binary alloys. Materials 14, 1787 (2021).
Titus, M. S. et al. Solute segregation and deviation from bulk thermodynamics at nanoscale crystalline defects. Sci. Adv. 2, e1601796 (2016).
Choudhury, S., Li, Y. L., Krill III, C. E. & Chen, L.-Q. Phase-field simulation of polarization switching and domain evolution in ferroelectric polycrystals. Acta Mater. 53, 1415–1426 (2005).
Miehe, C. & Rosato, D. A rate-dependent incremental variational formulation of ferroelasticity. Int. J. Eng. Sci. 49, 466–496 (2011).
Li, J. Y., Lei, C. H., Li, L. J., Shu, Y. C. & Liu, Y. Y. Unconventional phase field simulations of transforming materials with evolving microstructures. Acta Mech. Sin. 28, 915–927 (2012).
Tsou, N. T., Huber, J. E. & Cocks, A. C. F. Evolution of compatible laminate domain structures in ferroelectric single crystals. Acta Mater. 61, 670–682 (2013).
Chen, L. et al. A phase-field model coupled with large elasto-plastic deformation: application to lithiated silicon electrodes. J. Electrochem. Soc. 161, F3164–F3172 (2014).
Leo, C. V. D., Rejovitzky, E. & Anand, L. A Cahn-Hilliard-type phase-field theory for species diffusion coupled with large elastic deformations: application to phase-separating Li-ion electrode materials. J. Mech. Phys. Solids 70, 1–29 (2014).
Miehe, C., Dal, H., Schänzel, L.-M. & Raina, A. A phase-field model for chemo-mechanical induced fracture in lithium-ion battery electrode particles. Int. J. Numer. Methods Eng. 106, 683–711 (2016).
Zhao, Y., Xu, B.-X., Stein, P. & Gross, D. Phase-field study of electrochemical reactions at exterior and interior interfaces in Li-ion battery electrode particles. Comput. Methods Appl. Mech. Eng. 312, 428–446 (2016).
OpenPhase. http://www.icams.de/content/software-development/openphase. Accessed: 2021-09-06.
Vakili, S., Steinbach, I. & Varnik, F. Multi-phase-field simulation of microstructure evolution in metallic foams. Sci. Rep. 10, 19987 (2020).
Darvishi Kamachali, R. & Steinbach, I. 3-D phase-field simulation of grain growth: topological analysis versus mean-field approximations. Acta Mater. 60, 2719–2728 (2012).
Zhang, L. & Steinbach, I. Phase-field model with finite interface dissipation: extension to multi-component multi-phase alloys. Acta Mater. 60, 2702–2710 (2012).
Vakili, S., Steinbach, I. & Varnik, F. Controlling bubble coalescence in metallic foams: a simple phase field-based approach. Comput. Mater. Sci. 173, 109437 (2020).
Schiedung, R., Kamachali, R. D., Steinbach, I. & Varnik, F. Multi-phase-field model for surface and phase-boundary diffusion. Phys. Rev. E 96, 012801 (2017).
Ali, M. A. et al. Effect of \({\gamma }^{\prime}\) precipitate size on hardness and creep properties of Ni-base single crystal superalloys: experiment and simulation. Materialia 12, 100692 (2020).
Salama, H. et al. Role of inclination dependence of grain boundary energy on the microstructure evolution during grain growth. Acta Mater. 188, 641–651 (2020).
Borukhovich, E. et al. Microstructure design of tempered martensite by atomistically informed full-field simulation: from quenching to fracture. Materials 9, 673 (2016).
Roters, F. et al. Damask – the Düsseldorf advanced material simulation kit for modeling multi-physics crystal plasticity, thermal, and damage phenomena from the single crystal up to the component scale. Comput. Mater. Sci. 158, 420–478 (2019).
PETSc users manual. https://www.mcs.anl.gov/petsc. Accessed: 2021-09-06.
DREAM3D. http://dream3d.bluequartz.net. Accessed: 2021-09-06.
MTEX Toolbox. https://mtex-toolbox.github.io. Accessed: 2021-09-06.
Neper: polycrystal generation and meshing. https://neper.info. Accessed: 2021-09-06.
Gmsh: a three-dimensional finite element mesh generator with built-in pre- and post-processing facilities. https://gmsh.info. Accessed: 2021-09-06.
ParaView. https://www.paraview.org. Accessed: 2021-09-06.
Permann, C. J. et al. MOOSE: enabling massively parallel multiphysics simulation. SoftwareX 11, 100430 (2020).
Bai, Y., Zhao, Y., Liu, W. & Xu, B.-X. Two-level modeling of lithium-ion batteries. J. Power Sources 422, 92–103 (2019).
Bai, Y. et al. A chemo-mechanical damage model at large deformation: numerical and experimental studies on polycrystalline energy materials. Int. J. Solids Struct. 228, 111099 (2021).
Amitex general documentation. http://www.maisondelasimulation.fr/projects/amitex/general/_build/html/index.html. Accessed: 2021-09-15.
Prisms-plasticity. https://github.com/prisms-center/plasticity. Accessed: 2021-09-15.
PRISMS-PF. https://github.com/prisms-center/phaseField. Accessed: 2021-09-15.
FiPy: A finite volume pde solver using python. https://www.ctcms.nist.gov/fipy. Accessed: 2021-09-15.
FEniCS project. https://fenicsproject.org. Accessed: 2021-09-15.
MicroSim. https://github.com/ICME-India/MicroSim. Accessed: 2022-02-13.
COMSOL - software for multiphysics simulation. https://www.comsol.com. Accessed: 2021-09-15.
Ansys engineering simulation software. https://www.ansys.com. Accessed: 2021-09-15.
Abaqus multiphysics technology. https://www.3ds.com/products-services/simulia/products/abaqus/multiphysics. Accessed: 2021-09-15.
Wheeler, A. A., Boettinger, W. J. & McFadden, G. B. Phase-field model for isothermal phase transitions in binary alloys. Phys. Rev. A 45, 7424–7439 (1992).
Kim, S. G., Kim, W. T. & Suzuki, T. Phase-field model for binary alloys. Phys. Rev. E 60, 7186–7197 (1999).
Alber, H.-D. & Zhu, P. Comparison of a rapidely converging phase field model for interfaces in solids with the Allen-Cahn model. J. Elast. 111, 153–221 (2013).
Finel, A. et al. Sharp phase field method. Phys. Rev. Lett. 121, 025501 (2018).
NIST CALPHAD assessments. https://materialsdata.nist.gov/handle/11256/8. Accessed: 2021-09-06.
Crystallography open database. http://www.crystallography.net/cod. Accessed: 2021-09-06.
Inorganic crystal structure database. https://icsd.products.fiz-karlsruhe.de/en. Accessed: 2021-09-06.
The materials data facility. https://materialsdatafacility.org. Accessed: 2021-09-06.
Interatomic potentials repository. https://www.ctcms.nist.gov/potentials/resources.html. Accessed: 2021-09-06.
Sedighiani, K. et al. An efficient and robust approach to determine material parameters of crystal plasticity constitutive laws from macro-scale stress–strain curves. Int. J. Plast. 134, 102779 (2020).
Kamachali, R. D., Borukhovich, E., Shchyglo, O. & Steinbach, I. Solutal gradients in strained equilibrium. Philos. Mag. Lett. 93, 680–687 (2013).
Wilkinson, M. D. et al. The FAIR guiding principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016).
Khachaturyan, A. G., Semenovskaya, S. & Tsakalakos, T. Elastic strain energy of inhomogeneous solids. Phys. Rev. B 52, 15909–15919 (1995).
Cahn, J. W. Nucleation on dislocations. Acta metall. 5, 169–172 (1957).
Larché, F. & Cahn, J. Overview no. 41 the interactions of composition and stress in crystalline solids. Acta metall. 33, 331–357 (1985).
Ubachs, R., Schreurs, P. & Geers, M. Phase field dependent viscoplastic behaviour of solder alloys. Int. J. Solids Struct. 42, 2533–2558 (2005).
Di Leo, C. V., Rejovitzky, E. & Anand, L. Diffusion–deformation theory for amorphous silicon anodes: the role of plastic deformation on electrochemical performance. Int. J. Solids Struct. 67-68, 283–296 (2015).
Anand, L. A Cahn–Hilliard-type theory for species diffusion coupled with large elastic–plastic deformations. J. Mech. Phys. Solids 60, 1983–2002 (2012).
Anand, L. A thermo-mechanically-coupled theory accounting for hydrogen diffusion and large elastic–viscoplastic deformations of metals. Int. J. Solids Struct. 48, 962–971 (2011).
Liu, C. et al. An integrated crystal plasticity–phase field model for spatially resolved twin nucleation, propagation, and growth in hexagonal materials. Int. J. Plast. 106, 203–227 (2018).
Liu, C. et al. On the interaction of precipitates and tensile twins in magnesium alloys. Acta Mater. 178, 146–162 (2019).
Cottura, M. et al. A phase field model incorporating strain gradient viscoplasticity: application to rafting in Ni-base superalloys. J. Mech. Phys. Solids 60, 1243–1256 (2012).
Cottura, M., Appolaire, B., Finel, A. & Le Bouar, Y. Coupling the phase field method for diffusive transformations with dislocation density-based crystal plasticity: application to Ni-based superalloys. J. Mech. Phys. Solids 94, 473–489 (2016).
Cottura, M., Appolaire, B., Finel, A. & Le Bouar, Y. Microstructure evolution under [110] creep in Ni-base superalloys. Acta Mater. 212, 116851 (2021).
Wang, C., Ali, M. A., Gao, S., Goerler, J. V. & Steinbach, I. Combined phase-field crystal plasticity simulation of P- and N-type rafting in Co-based superalloys. Acta Mater. 175, 21–34 (2019).
Ali, M. A., Amin, W., Shchyglo, O. & Steinbach, I. 45-degree rafting in Ni-based superalloys: a combined phase-field and strain gradient crystal plasticity study. Int. J. Plast. 128, 102659 (2020).
Di Leo, C. V., Rejovitzky, E. & Anand, L. A Cahn–Hilliard-type phase-field theory for species diffusion coupled with large elastic deformations: application to phase-separating Li-ion electrode materials. J. Mech. Phys. Solids 70, 1–29 (2014).
Anand, L., Mao, Y. & Talamini, B. On modeling fracture of ferritic steels due to hydrogen embrittlement. J. Mech. Phys. Solids 122, 280–314 (2019).
Wang, Y., Jin, Y., Cuitiño, A. & Khachaturyan, A. Nanoscale phase field microelasticity theory of dislocations: model and 3D simulations. Acta Mater. 49, 1847–1857 (2001).
Martínez-Pañeda, E., Golahmar, A. & Niordson, C. F. A phase field formulation for hydrogen assisted cracking. Comput. Methods Appl. Mech. Eng. 342, 742–761 (2018).
Kristensen, P. K., Niordson, C. F. & Martínez-Pañeda, E. A phase field model for elastic-gradient-plastic solids undergoing hydrogen embrittlement. J. Mech. Phys. Solids 143, 104093 (2020).
Kristensen, P. K., Niordson, C. F. & Martínez-Pañeda, E. Applications of phase field fracture in modelling hydrogen assisted failures. Theor. Appl. Fract. Mech. 110, 102837 (2020).
Stainier, L., Leygue, A. & Ortiz, M. Model-free data-driven methods in mechanics: material data identification and solvers. Comput. Mech. 64, 381–393 (2019).
Bessa, M. et al. A framework for data-driven analysis of materials under uncertainty: countering the curse of dimensionality. Comput. Methods Appl. Mech. Eng. 320, 633–667 (2017).
Mishin, Y. Machine-learning interatomic potentials for materials science. Acta Mater. 214, 116980 (2021).
Fernández, M., Rezaei, S., Mianroodi, J. R., Fritzen, F. & Reese, S. Application of artificial neural networks for the prediction of interface mechanics: a study on grain boundary constitutive behavior. Adv. Model. Simul. Eng. Sci. 7, 1 (2020).
Mianroodi, J. R., Rezaei, S., Siboni, N. H., Xu, B.-X. & Raabe, D. Lossless multi-scale constitutive elastic relations with artificial intelligence. npj Comput. Mater. 8, 67 (2022).
Wang, H., Planas, R., Chandramowlishwaran, A. & Bostanabad, R. Mosaic flows: a transferable deep learning framework for solving pdes on unseen domains. Comput. Methods Appl. Mech. Eng. 389, 114424 (2022).
Mianroodi, J. R., H. Siboni, N. & Raabe, D. Teaching solid mechanics to artificial intelligence—a fast solver for heterogeneous materials. npj Comput. Mater. 7, 1–10 (2021).
Zhu, Q., Liu, Z. & Yan, J. Machine learning for metal additive manufacturing: predicting temperature and melt pool fluid dynamics using physics-informed neural networks. Comput. Mech. 67, 619–635 (2021).
Liu, X., Réthoré, J., Baietto, M.-C., Sainsot, P. & Lubrecht, A. A. An efficient strategy for large scale 3d simulation of heterogeneous materials to predict effective thermal conductivity. Comput. Mater. Sci. 166, 265–275 (2019).
Liu, X., Réthoré, J., Baietto, M.-C., Sainsot, P. & Lubrecht, A. A. An efficient finite element based multigrid method for simulations of the mechanical behavior of heterogeneous materials using CT images. Comput. Mech. 66, 1427–1441 (2020).
Franceschini, A., Paludetto Magri, V. A., Mazzucco, G., Spiezia, N. & Janna, C. A robust adaptive algebraic multigrid linear solver for structural mechanics. Comput. Methods Appl. Mech. Eng. 352, 389–416 (2019).
Paludetto Magri, V. A., Franceschini, A. & Janna, C. A novel algebraic multigrid approach based on adaptive smoothing and prolongation for ill-conditioned systems. SIAM J. Sci. Comput. 41, A190–A219 (2019).
Isotton, G., Janna, C. & Bernaschi, M. A gpu-accelerated adaptive FSAI preconditioner for massively parallel simulations. Int. J. High Perform. Comput. Appl. 36, 10943420211017188 (2021).
Lee, B. Algebraic multigrid for systems of elliptic boundary-value problems. Numer. Linear Algebra Appl. 28, e2303 (2021).
Duerinckx, M., Gloria, A. & Otto, F. The structure of fluctuations in stochastic homogenization. Commun. Math. Phys. 377, 259–306 (2020).
Chiumenti, M., Cervera, M., Moreira, C. & Barbat, G. Stress, strain and dissipation accurate 3-field formulation for inelastic isochoric deformation. Finite Elem. Anal. Des. 192, 103534 (2021).
Rosenbrock, C. W. et al. Machine-learned interatomic potentials for alloys and alloy phase diagrams. npj Comput. Mater. 7, 24 (2021).
van der Giessen, E. et al. Roadmap on multiscale materials modeling. Model. Simul. Mater. Sci. Eng. 28, 043001 (2020).
Bai, Y., Zhao, K., Liu, Y., Stein, P. & Xu, B.-X. A chemo-mechanical grain boundary model and its application to understand the damage of li-ion battery materials. Scr. Mater. 183, 45–49 (2020).
Wong, S. L., Madivala, M., Prahl, U., Roters, F. & Raabe, D. A crystal plasticity model for twinning- and transformation-induced plasticity. Acta Mater. 118, 140–151 (2016).
Acknowledgements
Financial support of the modeling and simulation work reported on here in Subproject M5 and M3 (RA 659/23) of the Priority Program 1713 “Strong Coupling of Thermochemical and Thermo-mechanical States in Applied Materials” of the Deutsche Forschungsgemeinschaft (DFG) is gratefully acknowledged. The thermo-mechanical simulations in DAMASK in this research were carried out under project number T17019e in the framework of the Research Program of the Materials innovation institute (M2i) (www.m2i.nl) supported by the Dutch government.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
J.R.M., D.R., M.D., and F.R. conceived the idea and developed the framework of the review. B.S., J.R.M., and M.D. contributed to the theory review part. S.V. and Y.B. contributed to OpenPhase and MOOSE review sections, respectively. S.R. and C.L. provided the DAMASK review and examples. J.R.M., N.H.S., and N.P. contributed to the section on emerging avenues. All authors provided critical feedback and contributed to the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mianroodi, J.R., Shanthraj, P., Liu, C. et al. Modeling and simulation of microstructure in metallic systems based on multi-physics approaches. npj Comput Mater 8, 93 (2022). https://doi.org/10.1038/s41524-022-00764-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-022-00764-0
