
Abstract
α-Tertiary aliphatic amides are key elements in organic molecules, which are abundantly present in natural products, pharmaceuticals, agrochemicals, and functional organic materials. Enantioconvergent alkyl-alkyl bond-forming process is one of the most straightforward and efficient, yet highly challenging ways to build such stereogenic carbon centers. Herein, we report an enantioselective alkyl-alkyl cross-coupling between two different alkyl electrophiles to access α-tertiary aliphatic amides. With a newly-developed chiral tridentate ligand, two distinct alkyl halides were successfully cross-coupled together to forge an alkyl-alkyl bond enantioselectively under reductive conditions. Mechanistic investigations reveal that one alkyl halides exclusively undergo oxidative addition with nickel versus in-situ formation of alkyl zinc reagents from the other alkyl halides, rendering formal reductive alkyl-alkyl cross-coupling from easily available alkyl electrophiles without preformation of organometallic reagents.
Similar content being viewed by others


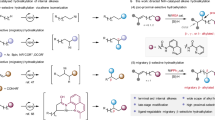
Introduction
α-Tertiary aliphatic amides with a α-saturated stereogenic carbon center are key structural units in chemistry, functional materials and many related areas1,2,3,4,5. Thus, the development of versatile and straightforward methods to access saturated stereogenic centers in a highly enantioenriched manner has been attracting long-term interests from chemistry community6. Early efforts have been paid to the employing of chiral auxiliaries to control the desired stereochemistry, resulting in the use of stoichiometric amount of chiral auxiliaries as well as additional steps for their installation and removal from the target molecules7. Over the past decades, studies have been increasingly focused on catalytic approaches to access such stereogenic centers8,9, including Ni-catalysed enantioconvergent cross-coupling between an alkyl electrophile and an alkyl nucleophile (Fig. 1a)10,11. Over the past years, significant progress has been achieved in nickel-catalysed enantioselective cross-coupling of racemic secondary alkyl electrophiles with organometallic reagents12,13,14,15,16,17,18,19,20. This reaction mode has been well-developed and evolved into an inevitable tool for constructing saturated stereogenic carbon centers. Although the significant advances, this reaction mode requires stoichiometric, reactive, and often sensitive organometallic reagents, which usually require time-consuming preformation. To this end, one alternative is to use alkenes as masked alkyl nucleophiles in the presence of metal hydride to undergo enantioselective alkyl-alkyl cross-coupling21,22,23,24. Hydrometallation of alkenes through metal hydride insertion generates alkyl metallic intermediates in situ as alkyl nucleophiles. In 2019, Fu group reported a seminal work on Ni-H catalysed enantioselective alkyl-alkyl cross-couplings of 1-substituted alkenes as a surrogate of carbon nucleophile to couple with secondary alkyl bromides adjacent to amides and esters (Fig. 1b)25,26. More recently, secondary alkyl bromides next to phosphates and ethers were successfully involved27,28,29,30,31,32,33. Accordingly, this strategy has evolved into an efficient cross-coupling of diverse alkenes with alkyl electrophiles to build saturated stereogenic carbon centers in the presence of metal hydrides34,35.

a Enantioselective Csp3-Csp3 bond-forming from alkyl-M and alkyl electrophiles. b Enantioselective Csp3-Csp3 bond-forming from alkenes and alkyl electrophiles. c Enantioselective Csp3-Csp3 bond-forming from two alkyl electrophiles.
However, direct reductive cross-coupling between two distinct electrophiles is still one of the most straightforward, cost-effective, thus ideal alternatives to construct saturated stereogenic carbon centers36,37,38. Ni-catalysed cross-coupling reactions between organ-electrophiles under reductive conditions have been extensively investigated for Csp2-Csp3 bond formation39,40,41,42,43,44. To date, no example of non-enzyme-catalysed enantioselective Csp3-Csp3 bond formation was reported45. Herein, we report a Ni-catalysed intermolecular cross-coupling between two different alkyl electrophiles under reductive conditions (Fig. 1c). The use of newly-developed chiral ligand enables construction of Csp3-Csp3 bond by selective coupling of two distinct alkyl electrophiles, without the preformation of organometallic reagents.
Results
Optimization of the reaction conditions
To prove the concept, we commenced the investigation using 1a and 2a as the prototype substrates using nickel catalysis to evaluate the reaction parameters. After extensive preliminary evaluation (See Supplementary Tables 1–5), we found the use of pyridine-BOX type ligands gave better results compared to other types of ligands in the presence of zinc (3.0 equiv) as sacrificing reductant, ferrous chloride (25 mol%), and cesium iodide (3.0 equiv) as additives. Among the tested known ligands, L1 gave the best result, delivering the desired cross-electrophile coupling product 3a in 67% yield with 70% ee (Table 1, entry 1). Modifying the substituents at α-position to oxygen on the oxazolidine ring of iPr-PyBOX significantly altered the efficiency of the ligand for this reaction (Table 1, entries 2–7). Linear substituents at α-position to oxygen of iPr-PyBOX (L2-L4) substantially diminished the yield and enantioselectivity (Table 1, entries 2–4). Cyclic substituents slightly increased the enantioselectivity of 3a from 70% to 76% and 71%, respectively (Table 1, entries 5 and 6). Introducing two phenyl groups onto α-position to oxygen (L7) led to trace amount of 3a (Table 1, entry 7). Next, Bn-PyBOX derived ligands (L8-L12) were applied to this asymmetric cross-electrophile coupling reaction (Table 1, entries 8-12). Bn-PyBOX delivered the desired product 3a in 29% yield with 60% ee (Table 1, entry 8). Increasing the steric hindrance at the α-position to oxygen improved the enantiomeric excess of 3a to 84% (Table 1, entry 10). Ligands derived from iBu-PyBOX (L13-L15) gave inferior yields and enantioselectivity (Table 1, entries 13-15). When Et-PyBOX based ligand L16 was used, 3a was obtained in 29% yield with 90% ee (Table 1, entry 16). Then, Me-PyBOX derived ligands (L17-L23) were tested (Table 1, entries 17-23). The use of propyl Me-PyBOX (L20) furnished 3a in 21% yield with 94% ee (Table 1, entry 20). Further evaluation of additive and solvent effect revealed that the addition of 15-crown-5 (10 mol%) in a mixture of DMA and diglyme (1:3) afforded 3a in 85% yield with 94% ee (See Supplementary Tables, 7-11). The use of ferrous chloride may facilitate the cross-coupling of 1a with 2a. In addition, the addition of 15-crown-5 may serve as an additive to enhance the solubility of inorganic salts in organic phase.
Scope of the reaction
With the optimized conditions in hand, we turned to test the scope of this reaction (Figs. 2 and 3). First, we examined the viability of α-bromoamides 1 (Fig. 2). Various substituted aniline derived α-bromoamides were good substrates for this enantioselective cross-electrophile alkyl-alkyl coupling reaction (3b-3r). Electron-donating group substituted aniline based amides delivered the desired enantioenriched α-alkylated amides in 58%–67% yields with 88–94% ee (3b-3e). Electron-withdrawing groups were also tolerated in the reaction, delivering the corresponding α-tertiary amides in 51–73% yields with 93–94% ee (3f-3k). Ketones and esters were compatible under the reaction conditions, giving the ketone and ester containing α-tertiary amides in 60% and 71% yields with 93% ee (3g-3h). Halides, such as fluorine, chlorine, and bromine, were also well-tolerated in this nickel-catalysed reductive process (3i-3k), leaving halides as a chemical handle for further elaboration. Notably, free phenol was tolerated in the catalytic process, furnishing desired enantioenriched amide 3l in 51% yield with 91% ee. Moreover, meta- and ortho-substituted as well as 2-naphthylamine derived α-bromoamides could be converted to corresponding α-alkylated amides in 58–71% yields with 92–94% ee (3m-3q). Thiophene amine was tolerated in the reaction, giving the desired product 3r in 67% yield with 94% ee. Aliphatic amines, including the linear, branched, and benzylic amine based α-bromoamides were all good substrates, giving the desired products in synthetic useful yields with 82–88% ee (3s-3v). α-Bromoamide from chiral amine was converted to α-alkylated amide 3w in 63% yield with 9:1 dr. Impressively, unprotected α-bromoamides, which are challenging for enantioselective coupling reactions, could be tolerated to deliver corresponding reductive cross alkyl-alkyl coupling product 3x in 49% yield with 87% ee. In addition, α-bromo-N,N-disubstituted amide is applicable in this reaction, affording the cross-coupling product (3y) in 52% yield with 92% ee. Unfortunately, α-bromo ester failed to deliver the desired cross-coupling product (3z) under the reaction conditions. Next, we embarked to test the scope of α-substituent of amides. Diverse alkyl substituents with different chain length were good substrates, delivering corresponding α-alkylated amides (4a and 4b) in 69% and 65% yields with 94% ee, respectively. Notably, α-chloroamides successfully underwent asymmetric alkyl-alkyl cross-coupling with 2a to give 4a in 61% yield with 92% ee. More steric demanding substituents, such as isopropyl, cyclopentylmethyl were also compatible in the reaction, giving 4c and 4d in 50% and 63% yields with 89% and 90% ee. Benzyl, phenylethyl, chloroethyl, and allyl could be tolerated in the reaction, giving the desired products 4e-4h in 58%-67% yields with 91%-92% ee. Notably, the enantioenriched amides with similar steric hindrance at α-position could be achieved with excellent enantioselectivity (4f). The absolute configuration of the product was further confirmed by the X-ray diffraction analysis of 4f.

The reaction was performed on a 0.2 mmol scale under the conditions in Table 1, entry 24. 15C5 = 15-crown-5. DME dimethoxyethane, DMA dimethylacetamide. Note: aα-Chloroamide was used instead of α-bromoamide. b3-Phenyl-1-iodopropane was used instead of 2a.

The reaction was performed on 0.2 mmol scale under the conditions in Table 1, entry 24. 15C5 = 15-crown-5. DME dimethoxyethane, DMA dimethylacetamide.
Next, the scope of the other alkyl electrophile was evaluated (Fig. 3). Different alkyl bromides were good substrates for this enantioselective cross-electrophile coupling reaction, giving corresponding alkyl-alkyl products (5a-5c) in 47–66% yields with 89–92% ee. Many functional groups, such as chlorine, nitrile, amide, alkene, alkyne, acetal, ester, ether containing alkyl bromides could be coupled to deliver desired products in moderate to good yields with 90–94% ee (5d-5m). Heterocycles, such as thiophene and furan substituted alkyl halides were transformed into corresponding products (5n and 5o) in 65% and 62% yields with 90% and 93% ee, respectively. Unfortunately, secondary unactivated alkyl halides remain unsuccessful for the reaction. In addition, both isomers of 6 were obtained under identical reaction conditions with the same chiral ligand. In the presence of (S,S)-L20, the reaction of 1a with 1-iodopropane gave R-6 in 70% yield with 86% ee, while the reaction of 1b with ethyl bromide furnished the other isomer S-6 in 59% yield with 88% ee.
Mechanistic study
In order to gain insight into the mechanism of the reaction, we set up a series of reactions to shed light on the reaction pathways (Fig. 4). First, the reaction of 1a with 2a in the presence of a radical scavenger TEMPO under otherwise identical to standard conditions was conducted (Fig. 4a). The desired intermolecular cross-coupling product 3a was not formed. Instead, the adduct 7 of TEMPO with 1a was obtained in 85% yield, indicating α-bromoamides underwent a single electron transfer process in this transformation. Next, the reactions of alkyl bromides with preformed alkyl zinc reagents under the standard conditions were tested (Fig. 4b). When alkyl zinc reagent 8 was used instead of 1a to couple with 2a under standard conditions, no desired product 3a was detected, and only protonated product 8’ was formed quantitatively, indicating alkyl zinc reagent 8 could not mediate the reaction under the standard reaction conditions. In contrast, the reaction of 1a with alkyl zinc reagent 9 under standard conditions delivered the desired cross-coupling product 3a in 87% yield with 89% ee. Further conducting the reaction with slow addition of alkyl zinc reagent 9 led to the formation of 3a in 83% yield with 94% ee, which is identical to the standard reaction conditions. These results suggest slow formation of alkyl zinc intermediate 9 in-situ to serve as intermediate for the reaction. To further prove the formation of alkyl zinc intermediates during the reaction, a real-time reaction course was conducted (Fig. 4c). The monitor the reaction process of 1a with 2a under standard conditions showed that no formation of 3a in the first 30 min, although the consumption of 2a was observed, indicating the induction time to form significant amount of alkyl intermediate to initiate the coupling reaction to generate 3a.

a Quench of radical intermediates. TEMPO tetramethylpiperidine oxide. b Reactions with alkyl zinc reagents. c Time course for 2a and 3a under standard conditions.
On the basis of experimental results and literature precedence46,47,48, a plausible mechanism is depicted in Fig. 5. First, Ni(II) was reduced by zinc to generate the ligated nickel (I) species (Int-A) in the presence of chiral ligand (L), which could undergo single electron transfer to 1 to give alkyl radical intermediate Int-B and Ni (II) intermediate Int-C. In the meantime, alkyl zinc reagent Int-D could be formed from 2 and zinc in the assistance with iodide, which could undergo transmetalation with Int-C to generate alkyl Ni(II) species Int-E. The rebound of intermediates Int-B and Int-E could form dialkyl Ni (III) intermediate Int-F, which would facilitate reductive elimination to furnish the final product 3 and regenerate Ni (I) species.

A plausible reaction mechanism for the intermolecular alkyl-alkyl cross-coupling between distinct alkyl halides based on all experimental results and previous literature evidence.
Discussion
In summary, an intermolecular enantioselective alkyl-alkyl cross-coupling between two alkyl electrophiles has been developed enabled by the efficient and selective cross-coupling reaction between two distinct alkyl halides under reductive conditions, representing an alternative for the construction of chiral Csp3-Csp3 bonds. One alkyl halides in-situ formed alkyl nucleophiles with reducing metal to cross-couple with the other alkyl halides in a chemo- and enantioselective manner, circumventing the tedious and time-consuming preformation of alkyl metal species. We anticipate this will inspire enantioselective in-situ cross-coupling between alkyl electrophiles under reductive conditions to be evolved into one of the major strategies to build saturated carbon centers via enantioselective Csp3-Csp3 bond-formation.
Methods
General procedure for Ni-catalysed enantioconvergent intermolecular alkyl-alkyl cross-coupling
In a nitrogen-filled glovebox, NiCl2·glyme (0.016 mmol, 8 mol%), chiral ligand L20 (0.016 mmol, 8 mol%) and diglyme (1.0 mL) were added to a 10-mL vial equipped with a stir bar. The mixture was allowed to stir for 1 h, after which it was an orange solution. Then, FeCl2 (0.05 mmol, 25 mol%), 15C5 (0.02 mmol, 10 mol%), CsI (0.6 mmol, 300 mol%), Zn (0.4 mmol, 200 mol%), 1 (0.2 mmol), 2 (0.6 mmol, 300 mol%), DMA (0.5 mL) and diglyme (0.5 mL) were added. The reaction mixture was transferred out of the glovebox and stirred (~1400 rpm) at room temperature for 24 h. Next, ethyl acetate (20.0 mL) was added, and the mixture was washed with water (10.0 mL) and brine (10.0 mL), dried over Na2SO4, filtered, and concentrated under vacuum. The residue was purified by flash chromatography on silica gel to afford the enantioselective alkyl-alkyl cross-coupling product.
Data availability
The X-ray crystallographic coordinates for structures that support the findings of this study have been deposited at the Cambridge Crystallographic Data Center (CCDC) with the accession code CCDC 2089117 (4f) and CCDC 2239323 (7) (www.ccdc.cam.ac.uk/data_request/cif). The authors declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author upon request.
References
Carreira, E. M. & Yamamoto, H. Comprehensive chirality; Academic: Amsterdam, 2012.
Lin, G.-Q., You, Q.-D. & Cheng, J.-F. Chiral drugs: Chemistry and biological action; Wiley: New York, 2011.
Lovering, F., Bikker, J. & Humblet, C. Escape from flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756 (2009).
Tobert, J. A. Lovastatin and beyond: The history of the HMG–CoA reductase inhibitors. Nat. Rev. Drug Discov. 2, 517–526 (2003).
Ganellin, C. R., Jefferis, R. & Roberts, S. M. Introduction to biological and small molecule drug research and development; Elsevier: Amsterdam, 339-416, 2013.
Fiorito, D., Liu, Y., Besnard, C. & Mazet, C. Direct access to chiral secondary amides by copper-catalyzed borylative carboxamidation of vinylarenes with isocyanates. J. Am. Chem. Soc. 142, 623–632 (2020).
Sibi, M. P., Petrovic, G. & Zimmerman, J. Enantioselective radical addition/trapping reactions with α,β-disubstituted unsaturated imides. synthesis of anti-propionate aldols. J. Am. Chem. Soc. 127, 2390–2391 (2005).
Stoltz, B. M. et al. in Comprehensive Organic Synthesis 2nd edn, Vol. 3 (eds Knochel, P. et al.) 1-55 (Elsevier, Amsterdam, 2014).
MacMillan, D. W. C. & Watson, A. J. B. in Science of synthesis: Stereoselective synthesis. Vol. 3 (ed. Evans, P. A.) 675-745 (Thieme, New York, 2011).
Cherney, A. H., Kadunce, N. T. & Reisman, S. E. Enantioselective and enantiospecific transition-metal-catalyzed cross-coupling reactions of organometallic reagents to construct C-C bonds. Chem. Rev. 115, 9587–9652 (2015).
Fu, G. C. Transition-metal catalysis of nucleophilic substitution reactions: A radical alternative to SN1 and SN2 processes. ACS Cent. Sci. 3, 692–700 (2017).
Fischer, C. & Fu, G. C. Asymmetric nickel-catalyzed Negishi cross-couplings of secondary α-bromo amides with organozinc reagents. J. Am. Chem. Soc. 127, 4594–4595 (2005).
Arp, F. O. & Fu, G. C. Catalytic enantioselective Negishi reactions of racemic secondary benzylic halides. J. Am. Chem. Soc. 127, 10482–10483 (2005).
Son, S. & Fu, G. C. Nickel-catalyzed asymmetric Negishi cross-couplings of secondary allylic chlorides with alkylzincs. J. Am. Chem. Soc. 130, 2756–2757 (2008).
Owston, N. A. & Fu, G. C. Asymmetric alkyl-alkyl cross-couplings of unactivated secondary alkyl electrophiles: Stereoconvergent Suzuki reactions of racemic acylated halohydrins. J. Am. Chem. Soc. 132, 11908–11909 (2010).
Zultanski, S. L. & Fu, G. C. Catalytic asymmetric γ-alkylation of carbonyl compounds via stereoconvergent Suzuki cross-couplings. J. Am. Chem. Soc. 133, 15362–15364 (2011).
Wilsily, A., Tramutola, F., Owston, N. A. & Fu, G. C. New directing groups for metal-catalyzed asymmetric carbon-carbon bond-forming processes: Stereoconvergent alkyl-alkyl Suzuki cross-couplings of unactivated electrophiles. J. Am. Chem. Soc. 134, 5794–5797 (2012).
Cordier, C. J., Lundgren, R. J. & Fu, G. C. Enantioconvergent cross-couplings of racemic alkylmetal reagents with unactivated secondary alkyl electrophiles: Catalytic asymmetric Negishi α‑alkylations of N‑Boc-pyrrolidine. J. Am. Chem. Soc. 135, 10946–10949 (2013).
Mu, X., Shibata, Y., Makida, Y. & Fu, G. C. Control of vicinal stereocenters through nickel-catalyzed alkyl-alkyl cross-coupling. Angew. Chem. Int. Ed. 56, 5821–5824 (2017).
Tong, X., Schneck, F. & Fu, G. C. Catalytic enantioselective α‑alkylation of amides by unactivated alkyl electrophiles. J. Am. Chem. Soc. 144, 14856–14863 (2022).
Lu, X. et al. Practical carbon-carbon bond formation from olefins through nickel-catalyzed reductive olefin hydrocarbonation. Nat. Commun. 7, 11129 (2016).
Sun, S.-Z., Börjesson, M., Martin-Montero, R. & Martin, R. Site-selective Ni-catalyzed reductive coupling of α‑haloboranes with unactivated olefins. J. Am. Chem. Soc. 140, 12765–12769 (2018).
Zhou, F., Zhu, J., Zhang, Y. & Zhu, S. NiH-catalyzed reductive relay hydroalkylation: A strategy for the remote C(sp3)-H alkylation of alkenes. Angew. Chem. Int. Ed. 57, 4058–4062 (2018).
Bera, S. & Hu, X. Nickel-catalyzed regioselective hydroalkylation and hydroarylation of alkenyl boronic esters. Angew. Chem. Int. Ed. 58, 13854–13859 (2019).
Wang, Z., Yin, H. & Fu, G. C. Catalytic enantioconvergent coupling of secondary and tertiary electrophiles with olefins. Nature 563, 379–383 (2018).
Zhou, F., Zhang, Y., Xu, X. & Zhu, S. NiH-catalyzed remote asymmetric hydroalkylation of alkenes with racemic α-bromo amides. Angew. Chem. Int. Ed. 58, 1754–1758 (2019).
He, S.-J. et al. Nickel-catalyzed enantioconvergent reductive hydroalkylation of olefins with α‑heteroatom phosphorus or sulfur alkyl electrophiles. J. Am. Chem. Soc. 142, 214–221 (2020).
Yang, Z.-P. & Fu, G. C. Convergent catalytic asymmetric synthesis of esters of chiral dialkyl carbinols. J. Am. Chem. Soc. 142, 5870–5875 (2020).
Shi, L., Xing, L.-L., Hu, W.-B. & Shu, W. Regio- and enantioselective Ni-catalyzed formal hydroalkylation, hydrobenzylation, and hydropropargylation of acrylamides to α-tertiary amides. Angew. Chem. Int. Ed. 60, 1599–1604 (2021).
Bera, S., Mao, R. & Hu, X. Enantioselective C(sp3)-C(sp3) cross-coupling of non-activated alkyl electrophiles via nickel hydride catalysis. Nat. Chem. 13, 270–277 (2021).
Qian, D., Bera, S. & Hu, X. Enantioselective C(sp3)-C(sp3) cross-coupling of non-activated alkyl electrophiles via nickel hydride catalysis. J. Am. Chem. Soc. 143, 1959–1967 (2021).
Wang, J.-W. et al. Catalytic asymmetric reductive hydroalkylation of enamides and enecarbamates to chiral aliphatic amines. Nat. Commun. 12, 1313 (2021).
Wang, S. et al. Enantioselective access to chiral aliphatic amines and alcohols via Ni-catalyzed hydroalkylations. Nat. Commun. 12, 2771 (2021).
Wang, X.-X., Lu, X., Li, Y., Wang, J.-W. & Fu, Y. Recent advances in nickel-catalyzed reductive hydroalkylation and hydroarylation of electronically unbiased alkenes. Sci. China Chem. 63, 1586–1600 (2020).
He, Y., Chen, J., Jiang, X. & Zhu, S. Enantioselective NiH-catalyzed reductive hydrofunctionalization of alkenes. Chin. J. Chem. 40, 651–661 (2022).
Weix, D. J. Methods and mechanisms for cross-electrophile coupling of Csp2 halides with alkyl electrophiles. Acc. Chem. Res. 48, 1767–1775 (2015).
Lucas, E. L. & Jarvo, E. R. Stereospecific and stereoconvergent cross-couplings between alkyl electrophiles. Nat. Rev. Chem. 1, 0065 (2017).
Poremba, K. E., Dibrell, S. E. & Reisman, S. E. Nickel-catalyzed enantioselective reductive cross-coupling reactions. ACS Catal. 10, 8237–8246 (2020).
Wang, K., Ding, Z., Zhou, Z. & Kong, W. Ni-catalyzed enantioselective reductive diarylation of activated alkenes by domino cyclization/cross-coupling. J. Am. Chem. Soc. 140, 12364–12368 (2018).
Jin, Y. & Wang, C. Nickel-catalyzed asymmetric reductive arylalkylation of unactivated alkenes. Angew. Chem. Int. Ed. 58, 6722–6726 (2019).
Tian, Z.-X. et al. Highly enantioselective cross-electrophile aryl-alkenylation of unactivated alkenes. J. Am. Chem. Soc. 141, 7637–7643 (2019).
Fan, P., Lan, Y., Zhang, C. & Wang, C. Nickel/photo-cocatalyzed asymmetric acyl-carbamoylation of alkenes. J. Am. Chem. Soc. 142, 2180–2186 (2020).
Wu, X., Qu, J. & Chen, Y. Quinim: A new ligand scaffold enables nickel-catalyzed enantioselective synthesis of α‑alkylated γ‑lactam. J. Am. Chem. Soc. 142, 15654–15660 (2020).
Guan, H., Zhang, Q., Walsh, P. J. & Mao, J. Nickel/photoredox-catalyzed asymmetric reductive cross-coupling of racemic α-chloro esters with aryl iodides. Angew. Chem. Int. Ed. 59, 5172–5177 (2020).
Fu, H. et al. An asymmetric sp3–sp3 cross-electrophile coupling using ‘ene’-reductases. Nature 610, 302–307 (2022).
Schley, N. D. & Fu, G. C. Nickel-catalyzed Negishi arylations of propargylic bromides: A mechanistic investigation. J. Am. Chem. Soc. 136, 16588–16593 (2014).
Yin, H. & Fu, G. C. Mechanistic investigation of enantioconvergent Kumada reactions of racemic α-bromoketones catalyzed by a nickel/bis(oxazoline) complex. J. Am. Chem. Soc. 141, 15433–15440 (2019).
Yang, Z.-P., Freas, D. J. & Fu, G. C. The asymmetric synthesis of amines via nickel-catalyzed enantioconvergent substitution reactions. J. Am. Chem. Soc. 143, 2930–2937 (2021).
Acknowledgements
We sincerely acknowledge NSFC (22171127 and 21971101), Guangdong Basic and Applied Basic Research Foundation (2022A1515011806), Department of Education of Guangdong Province (2022JGXM054 and 2021KTSCX106), The Pearl River Talent Recruitment Program (2019QN01Y261), Shenzhen Science and Technology Innovation Committee (JCYJ20220519201425001), Thousand Talents Program for Young Scholars, Guangdong Provincial Key Laboratory of Catalysis (2020B121201002) for financial support. We acknowledge the assistance of SUSTech Core Research Facilities, Dr. Xiaoyong Chang for X-Ray diffraction anaylsis of 4f (CCDC 2089117) and 7 (CCDC 2239323), and Dr. Lou Shi (SUSTech) for reproducing the results of 3n, 4c, 5b and 5n.
Author information
Authors and Affiliations
Contributions
W.T.Z. discovered and developed the reaction. W.S. conceived and directed the project. W.T.Z. performed the experiments and collected the data. J.X.Z. and B.H.C. synthesized the substrate materials. All authors disccussed and analyzed the data. W.S and W.T.Z. wrote the manuscript with contribution from other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, WT., Zhang, JX., Chen, BH. et al. Ligand-enabled Ni-catalysed enantioconvergent intermolecular Alkyl-Alkyl cross-coupling between distinct Alkyl halides. Nat Commun 14, 2938 (2023). https://doi.org/10.1038/s41467-023-38702-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-38702-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
