
Abstract
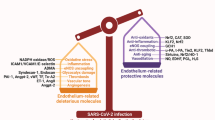
This review assesses markers of endothelial dysfunction (ED) associated with the maternal syndrome of preeclampsia (PE). We evaluate the role of antiretroviral therapy (ART) in human immunodeficiency virus (HIV)-infected preeclamptic women. Furthermore, we briefly discuss the potential of lopinavir/ritonavir (LPV/r), dolutegravir (DTG) and remdesivir (RDV) in drug repurposing and their safety in pregnancy complicated by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. In HIV infection, the trans-activator of transcription protein, which has homology with vascular endothelial growth factor, impairs angiogenesis, leading to endothelial injury and possible PE development despite neutralization of their opposing immune states. Markers of ED show strong evidence supporting the adverse role of ART in PE development and mortality compared to treatment-naïve pregnancies. Coronavirus disease 2019 (COVID-19), caused by SARS-CoV-2 infection, exploits angiotensin-converting enzyme 2 (ACE 2) to induce ED and hypertension, thereby mimicking angiotensin II-mediated PE in severe cases of infection. Upregulated ACE 2 in pregnancy is a possible risk factor for SARS-CoV-2 infection and subsequent PE development. The potential effectiveness of LPV/r against COVID-19 is inconclusive; however, defective decidualization, along with elevated markers of ED, was observed. Therefore, the safety of these drugs in HIV-positive pregnancies complicated by COVID-19 requires attention. Despite the observed endothelial protective properties of DTG, there is a lack of evidence of its effects on pregnancy and COVID-19 therapeutics. Understanding RDV-ART interactions and the inclusion of pregnant women in antiviral drug repurposing trials is essential. This review provides a platform for further research on PE in the HIV-COVID-19 syndemic.
Similar content being viewed by others


Introduction
Maternal mortality is a major concern worldwide, with its prevalence being particularly high in low- and middle-income countries (LMICs) [1, 2]. Sub-Saharan Africa bears the brunt of maternal deaths, namely, 66% of the global estimate [3]. The leading direct cause of maternal mortality in South Africa (SA) is preeclampsia (PE) [3].
Hypertensive disorders of pregnancy (HDP) are classified as follows: chronic hypertension (high blood pressure predating pregnancy or present at/or before 20 weeks of gestation); gestational hypertension, which is persistent de novo hypertension that develops at/or after 20 weeks of gestation without evidence of other organ involvement; PE without severe features; and PE with severe features [4]. PE is defined as new-onset hypertension presenting after 20 weeks of gestation in conjunction with one or more characteristic features, such as proteinuria and/or acute kidney injury, persistent headache, visual disturbances, epigastric pain, HELLP syndrome (hemolysis, elevated liver enzymes, low platelets), eclampsia (hypertension-associated seizures in pregnancy), and uteroplacental dysfunction, including fetal growth restriction [4]. Maternal mortality is present in all categories of HDP with eclampsia and PE, with severe features being the most common diagnosis before death [5]. It has been reported that PE accounts for >70,000 maternal deaths and 500,000 fetal deaths worldwide every year [4]. Globally, PE complicates 5–7% of pregnancies, and this incidence often increases to >10% in LMICs [6].
Although the exact etiology of PE remains elusive, endothelial dysfunction (ED) initiates the maternal syndrome of PE as a result of placental hypoxia, a reduction in uterine natural killer (uNK) cells, oxidative stress (OS), angiogenic imbalance and an exaggerated inflammatory response [7]. Human immunodeficiency virus (HIV) infection and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection also impact the inflammatory response and endothelial function. It is unclear whether HIV infection increases or decreases the frequency of PE. Nonetheless, the synergistic effect of these inflammatory conditions occurring concurrently requires investigation.
Pathophysiology of preeclampsia
In a normal pregnancy, the uteroplacental vasculature undergoes a significant morphological and physiological transformation to sustain fetal development [8]. Usually, cytotrophoblast (CT) cells derived from the tips of the chorionic villi migrate into the decidua and the inner myometrium in a set-time sequence [9, 10]. Thereafter, they fuse to form the multinucleated syncytiotrophoblast (ST) layer, which encloses the floating villi of the placenta and establishes the maternal-fetal interface for efficient gaseous and nutrient exchange [9, 11]. Extravillous trophoblast cells infiltrate fibrinoid-type material that replaces the musculo-elastic media of the spiral arteries, converting them into low-resistance large flaccid sinusoidal-like arteries [11, 12]. In the decidua, uNK cells regulate the depth of placentation and spiral artery remodeling [7]. The lumen of the spiral arteries is dilated five- to tenfold, ensuring an adequate supply of blood to the developing fetus [13]. These changes are typically achieved by 20 weeks of gestation [14].
Aberrant vascular remodeling predisposes the individual to PE development. PE is considered a two-stage placental disease where stage 1, often referred to as the fetoplacental or asymptomatic stage, occurs during the first and second trimesters of pregnancy [15]. In this stage, CT cells fail to take on the invasive endothelial phenotype; hence, CT migration is deficient, and there is a lack of physiological transformation of the myometrial spiral arteries [16, 17]. The resulting small arterial lumen, surrounded by vasoactive medial cells, is unable to provide adequate blood to meet the oxygen and nutrient demands of the fetus [17, 18]. This reduction in blood flow creates a hypoxic-ischemic microenvironment that marks the second stage [7, 19]. Stage 2, also referred to as the maternal stage, prompts the release of antiangiogenic factors and other mediators that initiate systemic inflammation, OS, and endothelial cell (EC) dysfunction. These mediators pre-empt the maternal syndrome of PE (presence of hypertension, proteinuria, liver dysfunction, cerebral edema, eclampsia, etc.) [4, 20]. An imbalance in circulating angiogenic factors persists during the pathogenesis of maternal syndrome [7].
Pathogenesis of the maternal syndrome in preeclampsia
Neovascularization (new blood vessel formation) results from either vasculogenesis or angiogenesis [21]. Vasculogenesis is the de novo construction of blood vessels from precursor cells, such as angioblasts, which differentiate into ECs that shape lumens to form simple blood vessels. In contrast, angiogenesis is the formation of new capillaries from the pre-existing vasculature [21, 22]. Angiogenesis is strongly associated with female reproductive conditions such as decidualization, implantation, and embryonic development [23]. Proangiogenic factors such as vascular endothelial growth factors (VEGFs) and placental growth factor (PlGF) are released into circulation, thereby increasing vascular permeability and promoting proteolysis of the extracellular matrix via proteases, leading to EC proliferation, migration and infiltration into the lumen and subsequent endothelial maturation [24, 25]. An array of VEGF isoforms, namely, VEGF-A, VEGF-B, VEGF-C, VEGF-D, and PlGF, are present in the blood circulatory system and are responsible for various vascular processes. VEGF-A binding to VEGFR-1 does not produce significant receptor activation (in this case, the receptor acts as a decoy), whereas VEGF-B binding to VEGFR-1 promotes cell survival [26]. VEGF-A can also bind to VEGFR-2, with a lower affinity, in the presence of NRP-1, a coreceptor of VEGF, thereby promoting EC migration and proliferation (Fig. 1) [26, 27]. However, VEGFs and their receptors are significantly downregulated in preeclamptic conditions due to the overexpression of their antiangiogenic counterparts [28].

Differential functions of vascular endothelial growth factor receptors. Adapted from Pandey et al. [27]. Vascular endothelial growth factor-A, B, C, D, and E (VEGF-A, B, C, D, and E), VEGF receptor-1, 2, and 3 (VEGFR-1, 2, and 3), Neuropilin-1 and 2 (NRP-1 and 2), Placental growth factor (PlGF)
Soluble fms-like tyrosine kinase (sFlt-1), also known as sVEGFR-1, is the soluble form of endothelial-bound VEGF receptors and functions as a VEGF antagonist to maintain angiogenic homeostasis [29]. Elevated sFlt-1 prevents VEGF and PlGF binding to VEGFR-2 on ECs, thus hindering angiogenic signal transduction leading to EC injury [30]. Concentrations of sFlt-1 are markedly elevated in pregnancy and are even higher in PE [31, 32]. Studies have demonstrated that the overexpression of sFlt-1 in rats induces PE-like syndrome early in pregnancy, supporting the role of antiangiogenic factors in PE development [33]. Assessment of the imbalance in the sFlt-1/PlGF ratio is currently used in the diagnosis and management of PE; however, more accurate and effective modes of early detection are urgently needed [34].
Endoglin (Eng), a coreceptor for the transforming growth factor (TGF) group of factors, is involved in vascular remodeling and hemostatic events via the activation of the endothelial nitric oxide synthase (eNOS) pathway that facilitates angiogenesis. In contrast, soluble endoglin (sEng), an extracellular variant of Eng, is highly expressed by trophoblasts and opposes TGF-β interactions with its receptor, thereby preventing vasodilation [35]. Upregulation of sEng impedes potent production of the vasodilator nitric oxide (NO) in ECs via its binding with TGF-β [36]. Therefore, exaggerated levels of sEng observed in PE may be central to the characteristic hypertension encountered during the maternal syndrome of the disease [35].
OS and nitrosative stress (NS) that causes endothelial injury in PE emanates from an imbalance between pro-oxidants and their therapeutic antagonists (antioxidants) [37]. This stress includes an increase in reactive oxygen species (ROS) and reactive nitrogen species production and/or diminished availability of antioxidant mechanisms [38]. The release of proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1, IL-6 and IL-8 from the ischemic placenta is intensified by syncytiotrophoblast microparticle (STMB) recruitment of monocytes and neutrophils to damaged EC sites [36]. These inflammatory cytokines not only decrease the bioavailability of NO and prostaglandin I2 (PGI2) but also produce ROS, which stimulates the elevation of endothelin-1 (ET-1), a potent vasoconstrictor. Vascular smooth muscle contraction results from an imbalance of endothelial vasodilators (NO and PGI2) and vasoconstrictors [Angiotensin II (Ang II), ET-1, and thromboxane A2 (TXA2)] during EC damage [39]. Vasoconstrictors decrease calcium ion efflux from smooth muscle cells through protein kinase C and Rho-kinase activation [39]. This leads to sustained vascular resistance and the hypertensive hallmark of endothelial injury observed in PE [33], depicted in Fig. 2.

Endothelial dysfunction in Preeclampsia. Adapted from Moghaddas et al. [36]. Angiotensin II (Ang II), Endothelial nitric oxide synthase (eNOS), Endothelin-1 (ET-1), Hypoxia-inducible factor (HIF), Interleukin-1, 6, and 8 (IL-1, IL-6, and IL-8), Nitric oxide (NO), Prostaglandin (PGI2), Protein kinase C (PKC), Placental growth factor (PlGF), Reactive oxygen species (ROS), Soluble endoglin (sEng), Soluble fms-like tyrosine kinase-1 (sFlt-1), Syncytiotrophoblast microparticles (STBMs), Transforming growth factor-β (TGF-β), Tumor necrosis factor-α (TNF-α), Thromboxane A2 (TXA2), Vascular endothelial growth factor (VEGF), VEGF receptor (VEGFR)
Maternal antioxidant imbalance and oxidative/nitrosative stress in preeclampsia
The EC activation encountered during PE exacerbates systemic inflammation and increases the expression of leukocyte adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), P-selectin (SELP), and E-selectin (SELE) [40, 41]. Leukocytes such as neutrophils and macrophages express nicotinamide adenine dinucleotide-phosphate (NADPH) oxidase, which generates superoxide (O2–) with subsequent production of other free radicals leading to a respiratory burst [42]. Usually, this process is tightly regulated; however, an increase in this phenomenon greatly overwhelms reducing agents such as glutathione, glutathione peroxidase, superoxide dismutase and catalase, resulting in OS and endothelial damage [43]. Elevated proinflammatory cytokines, such as TNF-α, observed in PE not only promote NO degradation leading to O2– generation but also induce free radical production during oxidative phosphorylation, further contributing to EC injury [44, 45]. Moreover, increased levels of proinflammatory cytokines such as IL-1 and TNF-α upregulate lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), which consequently elevates the receptor for oxidized low-density lipoprotein (oxLDL), thereby facilitating O2− production via NO degradation [46, 47]. In maternal circulation, STBMs evoke ED via the activation of LOX-1 with an increase in O2− and a subsequent reduction in NO-mediated vasodilation [48]. However, contradictory studies demonstrate a significant upregulation in oxLDL with reduced concentrations of LOX-1 in PE [49]. Elevated agonist autoantibodies against angiotensin receptors (AT1-AA) due to placental ischemia enhance Ang II sensitivity via angiotensin II type I receptor (AT1) in PE [50]. Higher levels of AT1-AA have demonstrated increased placental OS [51] due to superoxide production through NADPH activation [52], which may result in vascular injury, deficient trophoblast invasion, placental hypoxia, inflammation, angiogenic imbalance, and reduced bioavailability of NO [53]. In addition, free fetal hemoglobin and circulating xanthine oxidase induce ROS production through various mechanisms [38]. These pathways can lead to eNOS uncoupling, generating O2− [54], which may prompt NO-O2− interactions and the production of the potent oxidant peroxynitrite, which inevitably predisposes cells to damage and DNA fragmentation and alteration [55]. Peroxynitrite can also hinder eNOS activity and disrupt endothelium-dependent vasodilation [56]. ROS have also been shown to downregulate the calcium-activated potassium channels KCa2.3 and KCa3.1, which are vital for electrical stimulation of vascular smooth muscle to ensure effective vasodilation [38].
Severe acute respiratory syndrome coronavirus-2 and preeclampsia
The outbreak of coronavirus disease 2019 (COVID-19) was declared a pandemic in March 2020 by the World Health Organization (WHO) [57]. At present, over 43.3 million cases of COVID-19 have been confirmed, with ~1.15 million deaths in over 218 countries and territories [58]. Genetic analysis of the novel beta-coronavirus revealed that its entry mechanism exploits the renin-angiotensin system (RAS) [59, 60]. The virus thereafter induces an array of symptoms, including vasoconstriction, elevated blood pressure and profibrotic pathway activation via coagulation [61]. An observational study conducted on COVID-19-infected pregnant women revealed that severe to critical cases of COVID-19 present with PE-like symptoms exclusive to placental maladaptation [62]. PE mimicry by COVID-19 was confirmed following the alleviation of preeclamptic symptoms without delivery of the placenta, which is currently the only known method for obtaining resolution of the clinical signs and symptoms of PE [62]. This prompted further insight into COVID-19’s role in PE.
During normal RAS activation, renin catalyzes the conversion of angiotensinogen into angiotensin I (Ang I). Ang I is further cleaved by angiotensin-converting enzyme (ACE) to form Ang II [63]. The physiological antagonist of ACE and Ang II, ACE 2, serves to cleave Ang I and Ang II into angiotensin 1-9 and angiotensin 1-7 [Ang (1-7)], respectively, bringing about vasodilatory, anti-inflammatory, and antifibrotic effects upon binding to its Mas receptor [61, 64]. RAS activation is, therefore, dependent on the balance between ACE and ACE 2. Pregnant women are partially unreceptive to circulating Ang II to maintain low vascular resistance. However, this adaptation is reversed in PE, leading to an angiogenic imbalance [65]. In SARS-CoV-2 infection, ACE 2 receptors are increased and exploited for effective viral infectivity, which decreases ACE 2 function, subsequently upregulating Ang II activity [66]. The decrease in ACE 2 function, along with an increase in the Ang II/Ang (1-7) ratio, may result in hypoxia-induced upregulation of sFlt-1 [67, 68], which further sensitizes ECs to Ang II [69]. Similar to PE, COVID-19 infection shows an increase in the sFlt-1/PlGF ratio due to the pathologic Ang II/Ang (1-7) imbalance [70]. Ang II acts through its receptors (AT1 and AT2) to induce vascular impairment, which is the initiator of the maternal syndrome of PE, thereby reinforcing Ang II-mediated ED [71], as depicted in Fig. 3.

Manipulation of RAS by SARS-CoV-2 in COVID-19. Angiotensin-converting enzyme (ACE), Angiotensin-converting enzyme 2 (ACE 2), Angiotensin (1-7), (1-9), I and, II [Ang (1-7), (1-9), I, and II], Angiotensin type 1 (AT1) receptors, Angiotensin type 2 (AT2) receptors, Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)
The elevated expression of ACE 2 in STs, CTs, and the placental vasculature is imperative for blood pressure mediation for sufficient perfusion of the developing fetus. Therefore, SARS-CoV-2 infection and its alteration of ACE 2 expression may lead to dire adverse outcomes [72, 73]. A recent review highlighted that both normal pregnancy and COVID-19 infection show upregulation of ACE 2, IL-8, and IL-10; thus, pregnancy may be a risk factor for COVID-19 morbidity [74]. They also postulated that increased expression of ACE 2 receptors in the placenta might escalate the risk of vertical transmission of SAR-CoV-2 infection [74]. This suggestion is supported by the predominant localization of SARS-CoV-2 in STs at the maternal-fetal interface of the placenta, potentiating severe comorbidity among COVID-19-complicated pregnancies [75]. Conflicting evidence has revealed no significant differences in ACE 2 expression between normotensive pregnant women and preeclamptic women in the third trimester; however, the data are inconclusive, as PE onset occurs earlier in gestation [76]. Another study showed no significances in the prevalence rates of intrauterine growth restriction (IUGR) and PE between COVID-19-negative and COVID-19-positive pregnant women. The observed ED in this study was attributed to the ‘cytokine storm’ of COVID-19, similar to the proinflammatory state of PE. This is further supported by Shanes et al., who showed altered maternal vascular perfusion following placental hypoxia, conceivably due to systemic inflammation, in sixteen placentae obtained from COVID-19-infected women [77]. In contrast to PE, acute lung injury and acute respiratory distress syndrome (ARDS) have upregulated VEGF levels, which increases vascular permeability. Moreover, the same study identified VEGF-D as the most prominent indicator related to the severity of clotting in COVID-19 [78].
Soluble angiotensin-converting enzyme 2 in the therapeutic intervention of covid-19
Unlike the ACE 2 receptor, soluble angiotensin-converting enzyme 2 (sACE 2) is unable to facilitate SARS-CoV-2 entry into cells due to its lack of cell membrane interactions along with the absence of transmembrane serine protease 2 (TMPRSS2), a corequisite for SARS-CoV-2 endocytosis [79]. sACE 2 is formed through ACE 2 receptor cleavage/shedding by a disintegrin and metalloproteinase 17 (ADAM17) and is suggested to have protective effects against SARS-CoV-2 infection [80]. sACE 2 may serve as a competitive decoy for the coronavirus, thereby reducing the binding of viral particles to membrane-bound ACE 2 and consequently reducing viral infectivity [81]. Research on therapeutic interventions using sACE 2 has revealed its higher affinity for COVID-19, thereby neutralizing the virus without altering endogenous ACE 2 homeostasis [82]. However, research on the effects and safety of sACE, TMPRSS2 and ADAM17 manipulation in pregnancy and HIV-infected individuals comorbid with COVID-19 has yet to be established.
Human immunodeficiency virus infection and preeclampsia
The trans-activator of transcription (Tat) protein is a regulatory protein of HIV-1 that improves the efficiency of viral infectivity [83]. The rich arginine and lysine arrangement seen in Tat resembles the VEGF sequence [84]. Therefore, Tat mimics the role of VEGF by promoting EC adhesion and αvβ3 and α5β1 integrin expression [84, 85], which also binds osteopontin (an angiogenic factor involved in decidualization) [86]. A study conducted on HIV-1 Tat-induced angiogenesis demonstrated that Tat protein notably reduced endothelium-dependent vasorelaxation and eNOS expression and regulation in ECs of porcine coronary arteries [37]. The latter study also implicated Tat in coronary artery disease, which is associated with the long-term effect of PE [37]. In addition, Tat protein was also shown to induce the expression of ICAM-1 and VCAM-1, suggesting a possible mechanism by which HIV-1 infection contributes to endothelial injury and accelerates atherosclerosis [87, 88]. Therefore, it is plausible that Tat’s homology with VEGF affects angiogenesis in PE.
In contrast to the exaggerated immune state of PE, there is significant immune suppression after HIV infection [89, 90]. Although infection has been shown to reduce the risk of developing PE, most studies show that pregnant women receiving highly active antiretroviral therapy (HAART) have an increased prevalence of PE development [90, 91]. This increase is believed to be due to immune restoration [92]. Recent studies show no difference in the risk of PE development between treated and untreated HIV-infected pregnant women [93], but others have reported findings that do not support the notion that HIV infection has protective qualities against HDP development [94].
Role of HIV therapy in maternal endothelial dysfunction
The WHO recommends that all individuals living with HIV infection receive HAART, regardless of their CD4+ count and disease stage (including pregnant and breastfeeding women) [95]. HAART or antiretrovirals (ARVs) not only improve life expectancy but also decrease the risk of mother-to-baby (vertical) transmission of the infection in utero during birth and breastfeeding [95]. However, ARVs may trigger severe PE development [96]. A study conducted on nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), namely, azidothymidine, tenofovir disoproxil fumarate and lamivudine, revealed dysregulation of EC proliferation and migration [97]. The study also suggested that NRTIs induce mitochondrial OS, which hinders the activation and transduction of endothelial receptor tyrosine kinase signals and VEGFR-2 pathways in vascular ECs [97]. In addition, this adverse effect on angiogenesis may predispose the individual to PE development [98]. Excessive production of ROS is associated with increased trophoblast apoptosis, which may occur in placental-mediated disorders, such as PE and/or IUGR, overpowering antioxidant defenses with deleterious effects [98]. Protease inhibitors (PIs) deter HIV aspartyl protease, causing reconstitution of the immune microenvironment, which may predispose the individual to PE development [99]. In vivo, three PIs [atazanavir, lopinavir (LPV), and ritonavir (r)] significantly lower progesterone in trophoblast cells, thus indicating its hindrance of trophoblast proliferation and migration [100]. In a recent study, Kala et al. showed that LPV-based ART dysregulated uterine decidualization and spiral artery remodeling in both human ex vivo and mouse in vivo models [101]. Lower expression of the chemokines VEGF, PlGF, angiopoietin-2, granulocyte-macrophage colony-stimulating factor, interferon-gamma and matrix metalloproteinase (MMP) 9 was observed upon LPV exposure of primary decidual cell cultures [101]. They reported uNK cell depletion and deficient trophoblast invasion as a result of decreased expression of the transcription factor STAT3, which mediates decidualization [101]. These observations highlight the events that precede widespread ED in PE and its associated adverse neonatal outcomes. HAART impairs nuclear factor kappa B (NF-κB) transcription factors that decrease MMP and VEGF expression, which inevitably dysregulate angiogenesis, promoting ED and PE development [102]. The placentae of HIV-infected women receiving zidovudine-containing ART showed evidence of mitochondrial DNA depletion, elevated OS levels, and apoptosis, implicating secondary mitochondrial failure potentiating PE development and adverse perinatal outcomes [103].
Increased immune-expression of Flt-1 and sFlt-1 was observed within trophoblast cells during PE, regardless of HIV status, implying autocrine signaling in trophoblast invasion and differentiation [104]. This is believed to promote abnormal placentation with subsequent EC dysfunction in PE [104]. Pre-HAART exposure in HIV infection showed lower PlGF levels and increased sFlt-1 in women who developed PE compared to normotensive pregnant women [105]. Multivariate analysis demonstrated that PlGF and viral load were significantly related to PE development, and no significant shifts were observed in angiogenic factors following HAART among normotensive women [105]. Increased sFlt-1 and sEng levels were linked to PE regardless of HIV infection [106]. This study also elucidated a significant downregulation in PlGF levels in HIV-negative preeclamptic women compared to normotensive women. However, HIV infection downregulates PlGF in normotensive pregnant women compared to their HIV-negative counterparts (p = 0.02), thereby predisposing the individual to PE development [106]. TGF-β1 levels remain unchanged in HIV infection regardless of the increase in its coreceptor sEng [106, 107].
In contrast, a study of HIV-associated PE women revealed that HIV/HAART is linked to significant downregulation of IL-2, TNF-α and IL-6, with significant decreases in IL-2 and TNF-α observed in preeclamptic women [108]. Saums et al. found that integrase strand transfer inhibitor-containing ARTs had a greater frequency of HDP development than protease inhibitor-containing regimens [93]. Another study concluded that HIV infection, rather than its pharmacological treatment, induces alterations in markers of endothelial function [109]. The short-term duration of treatment with HAART reduces some markers of ED, including VCAM-1, with no differences between PIs and nonnucleoside reverse transcriptase inhibitors. However, SELP remained elevated upon exposure to both treatments [109].
The repurposing of various antiviral drugs (Table 1) has gained momentum as a desperate measure to prevent the deleterious effects of COVID-19 [110].
Antiretroviral therapy in pregnancy and coronavirus disease 2019
It is plausible to assume that HIV-infected individuals receiving ARVs have a lower risk of developing complications from COVID-19 infection [111,112,113,114]. Protease inhibitor-based ARVs, such as LPV/r, have shown potential against SARS-CoV-2 infection due to their ability to bind SARS-CoV-1. Studies have shown a strong sequence homology between SARS-CoV-1 and SARS-CoV-2 [115]. However, SARS-CoV-2 binds ACE 2 with a 10-20-fold greater affinity than SARS-CoV-1, which explains the high human transmission and infectivity rates of SARS-CoV-2 [116]. LPV/r lowers the risk of patients developing ARDS and subsequently dying from SARS-CoV-2 infection [117]. In various subsequent clinical trials comparing prenatal exposure to LPV/r and prenatal exposure to efavirenz (EFV), there were no significant differences in adverse outcomes in pregnancy [118] or other control measures [119]. These drugs, therefore, have become the preferred drugs of choice for pregnancy complicated by COVID-19 in China [120].
Randomized controlled trials (RCTs) are essential for providing standard guidance on clinical management, even in an emergency setting, since RCTs offer data without bias due to confounding factors, as seen in nonrandomized studies [121]. RCTs of LPV/r in severe COVID-19 showed no benefit beyond standard care [122]. Other RCTs revealed that a combination of antiviral drugs (interferon beta-1b, ribavirin, and LPV/r) was more successful in symptom alleviation than LPV/r alone in mild to moderate COVID-19 cases [123]. A systemic review of RCTs of LPV/r in COVID-19 highlighted that ARVs may reduce mortality; however, this reduction varies across different risk groups [124].
The South African National ART guidelines employ LPV/r-based ARTs as the second-line therapy in HIV-infected adults [125]. The guidelines further recommend that women who become pregnant while receiving the LPV/r-containing regimen should continue treatment with monthly clinical observations [125]. Dolutegravir (DTG), the newly established ARV in SA, together with two NRTIs, is also recommended as a second-line ART after failing a non-NRTI-based first-line regimen since DTG is suggested to be better tolerated by HIV-infected individuals than PIs such as LPV/r [126]. SA has experienced over 716,700 confirmed cases of COVID-19 as of late October 2020 [58]. Considering the slow switch from previously approved ARVs to DTG, LPV/r-containing ARTs are still readily available in SA, and clinical trials may include women in their first trimester. Notably, LPV/r has a negative influence on decidualization and placentation; therefore, the safety of these drugs in HIV-associated PE complicated by COVID-19 infection requires urgent and intensive scrutiny.
Latest antiretroviral therapy effects on endothelial dysfunction
In 2017, SA and other low/middle-income countries agreed to launch a new high-quality ART, a single-tablet regimen containing an integrase strand transfer inhibitor (INSTI), DTG, which provides rapid viral suppression [127]. Notably, the 2019 ART guidelines in SA state that the preferred first-line ART regimen is a DTG-based drug (TLD) for patients experiencing EFV side effects or those who prefer to use DTG [125]. However, the EFV-containing regimen (TEE) is recommended for use in the first 6 weeks of gestation and in women of child-bearing age due to a high risk of neural tube defects associated with TLD [125]. In vivo studies of human coronary artery endothelial cells (HCAECs) showed that DTG reduced inflammation and IL-6, IL-8, VCAM-1, and ICAM-1 secretion via NF-κB pathway inhibition and decreased senescence by repressing apoptotic pathways [128, 129]. DTG also displayed protective properties in HCAECs, such as reduced OS, inflammation, and senescence and improved ED from an aged donor with persistently elevated levels of senescence [129]. The Stockholm pregnancy cohort showed that the PE rate was normal; however, the population size was too small to make any deductions [130]. In a study on treatment-naïve HIV-infected individuals, a significant decrease in TNF-α was observed 12 months following DTG initiation in comparison to PIs and other INSTIs, such as elvitegravir [131]. This study also revealed DTG’s capacity to significantly reduce D-dimer levels [131], implicating a possible positive interaction in COVID-19-infected individuals since elevated D-dimer, a marker of clot formation, is associated with increasing severity of the disease [132]. A case study of a 63-year-old HIV-infected woman with an undetectable viral load on a DTG-containing ART showed improvement despite presenting with COVID-19 complications during admission [133]. However, the role of DTG in the treatment of COVID-19 requires further investigation.
In light of the lack of evidence that particular ARVs are clinically active against SARS-CoV-2, HIV-infected individuals are advised to refrain from changing their ART regimen in an attempt to prevent or treat COVID-19 [134].
Remdesivir in covid-19 and pregnancy
Remdesivir (RDV), initially used in the treatment of Ebola, is among the top contenders against the new coronavirus [110]. RDV is a broad-acting nucleoside analog drug that has shown inhibitory effects on pathogenic animal and human coronaviruses such as SARS-CoV-2 in vitro and inhibits Middle East respiratory syndrome coronavirus, SARS-CoV-1, and SARS-CoV-2 replication in nonhuman primates [135]. In a randomized, double-blind, placebo-controlled trial, RDV showed no significant difference in terms of clinical benefits [136]. However, a larger study population is needed to confirm the observed reduction in clinical improvement time following RDV treatment [136]. Another RCT’s final report revealed that RDV was superior to placebo in decreasing recovery time among hospitalized adults [137]. However, neither trial included pregnant women. A case report of RDV-based treatment showed the successful management of a COVID-19-positive critically ill obstetrics patient [138]. Another case report of RDV-treated COVID-19 in the third trimester of pregnancy showed no adverse outcomes apart from elevated transaminases, which is also associated with PE development [139]. PE was ruled out as a cause of transaminitis because the patient did not present with hypertension and proteinuria [139]. This report also noted that there was no clarity on whether the transaminitis observed was due to COVID-19 or RDV intake [139]. In a recent study, 86 pregnant and postpartum women with severe COVID-19 who received compassionate use of RDV showed a high rate of recovery with a low rate of serious adverse events, such as transaminitis, hypertension, and hypoxia [140]. In Ebola clinical trials, there were no adverse outcomes among pregnant women receiving RDV [80]. On October 22, 2020, the United States Food and Drug Administration (FDA) approved the emergency use of RDV for severe cases of COVID-19 [141]. However, the safety of RDV in pregnancy has not been elucidated; therefore, the inclusion of pregnant women in clinical trials is necessary to guide risk-benefit considerations of RDV treatment in COVID-19.
LMICs such as SA have a limited capacity to accommodate the daily rise in COVID-19 infections [142]. The use of RDV may be vital for the prevention of adverse outcomes and a decrease in clinical improvement time in severe COVID-19 cases while regulating intensive care unit bed capacity [142]. Recently, Gilead Sciences Incorporated, CIPLA was granted a license to manufacture and distribute a generic form of RDV for compassionate use against COVID-19 in 127 countries, including SA; however, RDV is still not readily available to all citizens [143]. There is also no knowledge of the interactions of RDV with ARVs.
Conclusion
An imbalanced angiogenic status, inflammation and OS/NS induced by placental maladaptation facilitate pervasive multiorgan ED in PE. Adverse effects associated with HIV infection and ART promote ED predisposing PE development; however, higher prevalence and mortality rates among PE cases are still associated with ART use. Pregnancies complicated by the COVID-19 exploitation of ACE 2 have a strong correlation with PE-like symptoms such as endothelial injury, implicating COVID-19 in PE onset. Despite the inconsistent data on LPV/r against COVID-19, its availability in LMICs suggests the need for further insight into its safety in HIV-associated PE complicated by COVID-19. The observed endothelial protective properties of DTG in pregnancy and its role in COVID-19 therapeutics, along with the approved compassionate use of RDV in pregnancy, have yet to be established.
References
Lewis G. Maternal mortality in the developing world: why do mothers really die? Obstet Med. 2008;1:2–6.
Girum T, Wasie A. Correlates of maternal mortality in developing countries: an ecological study in 82 countries. Matern Health Neonatol Perinatol. 2017;3:19.
World Health Organization. Maternal mortality. https://www.who.int/news-room/fact-sheets/detail/maternal-mortality. Accessed 21 February 2020.
Brown MA, Magee LA, Kenny LC, Karumanchi SA, McCarthy FP, Saito S, et al. Hypertensive Disorders of Pregnancy: ISSHP Classification, Diagnosis, and Management Recommendations for International Practice. Hypertension (Dallas, Tex: 1979). 2018;72:24–43.
National Committee for Confidential Enquiry into Maternal Deaths. Saving Mothers Report 2017. Pretoria: South African Department of Health; 2018.
Wanderer JP, Leffert LR, Mhyre JM, Kuklina EV, Callaghan WM, Bateman BT. Epidemiology of Obstetric-Related Intensive Care Unit Admissions in Maryland: 1999–2008. Crit Care Med. 2013;41:1844.
Rana S, Lemoine E, Granger JP, Karumanchi SA. Preeclampsia: pathophysiology, challenges, and perspectives. Circulation Res. 2019;124:1094–112.
Singh M, Chaudhry P, Asselin E. Bridging endometrial receptivity and implantation: network of hormones, cytokines, and growth factors. J Endocrinol. 2011;210:5–14.
Valenzuela FJ, Perez-Sepulveda A, Torres MJ, Correa P, Repetto GM, Illanes SE. Pathogenesis of preeclampsia: the genetic component. J Pregnancy. 2012;2012:632732.
Pijnenborg R. Trophoblast invasion and placentation in the human: morphological aspects. In: Trophoblast Invasion and Endometrial Receptivity. Rochester, New York: Springer; 1990. p. 33–47.
Cartwright JE, Fraser R, Leslie K, Wallace AE, James JL. Remodelling at the maternal-fetal interface: relevance to human pregnancy disorders. Reproduction. 2010;140:803–13.
Brosens I, Robertson WB, Dixon HG. The physiological response of the vessels of the placental bed to normal pregnancy. J Pathol Bacteriol. 1967;93:569–79.
Burton GJ, Woods AW, Jauniaux E, Kingdom JCP. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta. 2009;30:473–82.
Whitley GSJ, Cartwright JE. Trophoblast-mediated spiral artery remodelling: a role for apoptosis. J Anat. 2009;215:21–26.
Roberts JM, Hubel CA. The two stage model of preeclampsia: variations on the theme. Placenta. 2009;30(Suppl A):S32–37.
Meekins JW, Pijnenborg R, Hanssens M, McFadyen IR, van Asshe A. A study of placental bed spiral arteries and trophoblast invasion in normal and severe pre-eclamptic pregnancies. Br J Obstet Gynaecol. 1994;101:669–74.
Naicker T, Khedun SM, Moodley J, Pijnenborg R. Quantitative analysis of trophoblast invasion in preeclampsia. Acta Obstet Gynecol Scand. 2003;82:722–9.
Naicker T, Dorsamy E, Ramsuran D, Burton GJ, Moodley J. The role of apoptosis on trophoblast cell invasion in the placental bed of normotensive and preeclamptic pregnancies. Hypertens Pregnancy. 2013;32:245–56.
Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Sci (N. Y, NY). 2005;308:1592–4.
Wagner LK. Diagnosis and management of preeclampsia. Am Fam Physician. 2004;70:2317–24.
Ucuzian AA, Gassman AA, East AT, Greisler HP. Molecular mediators of angiogenesis. J Burn Care Res. 2010;31:158–75.
Peak C, Cross L, Singh A, Gaharwar A. Microscale Technologies for Engineering Complex Tissue Structures. In; https://doi.org/10.1007/978-3-319-20726-1_1) 2015, 3–25.
Kubis N, Levy BI. Vasculogenesis and angiogenesis: molecular and cellular controls. Part 1: growth factors. Inter Neuroradiol. 2003;9:227–37.
Reynolds LP, Grazul-Bilska AT, Redmer DA. Angiogenesis in the female reproductive organs: pathological implications. Int J Exp Pathol. 2002;83:151–63.
Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–4.
Shibuya M. Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J Biochem Mol Biol. 2006;39:469–78.
Pandey AK, Singhi EK, Arroyo JP, Ikizler TA, Gould ER, Brown J, et al. Mechanisms of VEGF (Vascular Endothelial Growth Factor) Inhibitor-Associated Hypertension and Vascular Disease. Hypertension. 2018;71:e1–e8.
Zhou Q, Liu H, Qiao F, Wu Y, Xu J. VEGF deficit is involved in endothelium dysfunction in preeclampsia. J Huazhong Univ Sci Technol Med Sci. 2010;30:370–4.
Kendall RL, Thomas KA. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci USA. 1993;90:10705–9.
Possomato-Vieira JS, Khalil RA. Mechanisms of Endothelial Dysfunction in Hypertensive Pregnancy and Preeclampsia. Adv Pharm. 2016;77:361–431.
Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350:672–83.
Chaiworapongsa T, Romero R, Kim YM, Kim GJ, Kim MR, Espinoza J, et al. Plasma soluble vascular endothelial growth factor receptor-1 concentration is elevated prior to the clinical diagnosis of pre-eclampsia. J Matern Fetal Neonatal Med. 2005;17:3–18.
Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Investig. 2003;111:649–58.
Hernández-Pacheco JA, Rosales-Zamudio CI, Borboa-Olivares H, Espejel-Núñez A, Parra-Hernández S, Estrada-Gutiérrez G, et al. The sFlt-1/PlGF ratio as a triage tool to identify superimposed preeclampsia in women with chronic hypertension in emergency rooms. Pregnancy Hypertens. 2020;21:38–42.
Levine RJ, Lam C, Qian C, Yu KF, Maynard SE, Sachs BP, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006;355:992–1005.
Moghaddas Sani H, Zununi Vahed S, Ardalan M. Preeclampsia: a close look at renal dysfunction. Biomed Pharmacother. 2019;109:408–16.
Paladugu R, Fu W, Conklin BS, Lin PH, Lumsden AB, Yao Q, et al. Hiv Tat protein causes endothelial dysfunction in porcine coronary arteries. J Vasc Surg. 2003;38:549–55. discussion 555-546
Aouache R, Biquard L, Vaiman D, Miralles F. Oxidative Stress in Preeclampsia and Placental Diseases. Int J Mol Sci. 2018;19:1496.
Touyz RM, Alves-Lopes R, Rios FJ, Camargo LL, Anagnostopoulou A, Arner A, et al. Vascular smooth muscle contraction in hypertension. Cardiovasc Res. 2018;114:529–39.
Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126–67.
Chen X, Andresen BT, Hill M, Zhang J, Booth F, Zhang C. Role of Reactive Oxygen Species in Tumor Necrosis Factor-alpha Induced Endothelial Dysfunction. Curr Hypertens Rev. 2008;4:245–55.
Robinson JM. Reactive oxygen species in phagocytic leukocytes. Histochem Cell Biol. 2008;130:281–97.
Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5:9–19.
Yoshizumi M, Perrella MA, Burnett JC Jr., Lee ME. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ Res. 1993;73:205–9.
Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evid Involv mitochondrial Radic Gener J Biol Chem. 1992;267:5317–23.
Hofnagel O, Luechtenborg B, Stolle K, Lorkowski S, Eschert H, Plenz G, et al. Proinflammatory cytokines regulate LOX-1 expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:1789–95.
Cominacini L, Rigoni A, Pasini AF, Garbin U, Davoli A, Campagnola M, et al. The binding of oxidized low density lipoprotein (ox-LDL) to ox-LDL receptor-1 reduces the intracellular concentration of nitric oxide in endothelial cells through an increased production of superoxide. J Biol Chem. 2001;276:13750–5.
Spaans F, Kao CK, Morton JS, Quon AL, Sawamura T, Tannetta DS, et al. Syncytiotrophoblast extracellular vesicles impair rat uterine vascular function via the lectin-like oxidized LDL receptor-1. PLoS ONE. 2017;12:e0180364.
Chigusa Y, Tatsumi K, Kondoh E, Fujita K, Nishimura F, Mogami H, et al. Decreased lectin-like oxidized LDL receptor 1 (LOX-1) and low Nrf2 activation in placenta are involved in preeclampsia. J Clin Endocrinol Metab. 2012;97:E1862–1870.
Campbell N, LaMarca B, Cunningham MW Jr. The Role of Agonistic Autoantibodies to the Angiotensin II Type 1 Receptor (AT1-AA) in Pathophysiology of Preeclampsia. Curr Pharm Biotechnol. 2018;19:781–5.
Brewer J, Liu R, Lu Y, Scott J, Wallace K, Wallukat G, et al. Endothelin-1, oxidative stress, and endogenous angiotensin II: mechanisms of angiotensin II type I receptor autoantibody-enhanced renal and blood pressure response during pregnancy. Hypertension. 2013;62:886–92.
Dechend R, Viedt C, Müller DN, Ugele B, Brandes RP, Wallukat G, et al. AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation. 2003;107:1632–9.
Matsubara K, Higaki T, Matsubara Y, Nawa A. Nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. Int J Mol Sci. 2015;16:4600–14.
Daiber A, Xia N, Steven S, Oelze M, Hanf A, Kröller-Schön S, et al. New Therapeutic Implications of Endothelial Nitric Oxide Synthase (eNOS) Function/Dysfunction in Cardiovascular Disease. Int J Mol Sci. 2019;20:187.
Islam BU, Habib S, Ahmad P, Allarakha S, Moinuddin, Ali A. Pathophysiological Role of Peroxynitrite Induced DNA Damage in Human Diseases: A Special Focus on Poly(ADP-ribose) Polymerase (PARP). Indian J Clin Biochem. 2015;30:368–85.
Li Q, Youn J-Y, Cai H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J Hypertens. 2015;33:1128–36.
World Health Organization. WHO Director-General’s opening remarks at the media briefing on COVID-19 - 11 March 2020. https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020. Accessed 29 October.
World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. https://covid19.who.int/. Accessed 27 October 2020.
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–7.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–80. e278
Alifano M, Alifano P, Forgez P, Iannelli A. Renin-angiotensin system at the heart of COVID-19 pandemic. Biochimie. 2020;174:30–33.
Mendoza M, Garcia-Ruiz I, Maiz N, Rodo C, Garcia-Manau P, Serrano B, et al. Pre-eclampsia-like syndrome induced by severe COVID-19: a prospective observational study. Bjog. 2020 (e-pub ahead of print 2020/06/02; https://doi.org/10.1111/1471-0528.16339).
Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM. Classical Renin-Angiotensin system in kidney physiology. Compr Physiol. 2014;4:1201–28.
Cheng H, Wang Y, Wang GQ. Organ-protective effect of angiotensin-converting enzyme 2 and its effect on the prognosis of COVID-19. J Med Virol. 2020;92:726–30.
Gant NF, Daley GL, Chand S, Whalley PJ, MacDonald PC. A study of angiotensin II pressor response throughout primigravid pregnancy. J Clin Investig. 1973;52:2682–9.
Wu J, Deng W, Li S, Yang X. Advances in research on ACE2 as a receptor for 2019-nCoV. Cell Mol Life Sci. 2020 (e-pub ahead of print 2020/08/12; https://doi.org/10.1007/s00018-020-03611-x):1-14.
Murphy SR, Cockrell K. Regulation of soluble fms-like tyrosine kinase-1 production in response to placental ischemia/hypoxia: role of angiotensin II. Physiol Rep. 2015;3:e12310.
Khaliq OP, Konoshita T, Moodley J, Naicker T. Soluble angiotensin IV receptor levels in preeclampsia: is there a variation? J Matern Fetal Neonatal Med. 2020 (e-pub ahead of print 2020/03/27; https://doi.org/10.1080/14767058.2020.1743665):1–6.
Zhou CC, Ahmad S, Mi T, Xia L, Abbasi S, Hewett PW, et al. Angiotensin II induces soluble fms-Like tyrosine kinase-1 release via calcineurin signaling pathway in pregnancy. Circ Res. 2007;100:88–95.
Giardini V, Carrer A, Casati M, Contro E, Vergani P, Gambacorti-Passerini C. Increased sFLT-1/PlGF ratio in COVID-19: a novel link to angiotensin II-mediated endothelial dysfunction. Am J Hematol. 2020;95:E188–e191.
Dimmeler S, Rippmann V, Weiland U, Haendeler J, Zeiher AM. Angiotensin II induces apoptosis of human endothelial cells. Protective effect of nitric oxide. Circ Res. 1997;81:970–6.
Abbas AM, Ahmed OA, Shaltout AS. COVID-19 and maternal pre-eclampsia: a synopsis. Scand J Immunol. 2020;92:e12918.
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–8.
Phoswa WN, Khaliq OP. Is pregnancy a risk factor of COVID-19? Eur J Obstet Gynecol Reprod Biol. 2020;252:605–9.
Hosier H, Farhadian SF, Morotti RA, Deshmukh U, Lu-Culligan A, Campbell KH, et al. SARS-CoV-2 infection of the placenta. J Clin Investig. 2020;130:4947–53.
Anton L, Brosnihan KB. Systemic and uteroplacental renin-angiotensin system in normal and pre-eclamptic pregnancies. Ther Adv Cardiovasc Dis. 2008;2:349–62.
Shanes ED, Mithal LB, Otero S, Azad HA, Miller ES, Goldstein JA. Placental pathology in COVID-19. medRxiv. 2020 (e-pub ahead of print 2020/06/09; https://doi.org/10.1101/2020.05.08.20093229).
Kong Y, Han J, Wu X, Zeng H, Liu J, Zhang H. VEGF-D: a novel biomarker for detection of COVID-19 progression. Crit Care. 2020;24:373.
Xiao L, Sakagami H, Miwa N. ACE2: the key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel? Viruses. 2020;12:491.
Mulangu S, Dodd LE, Davey RT Jr, Tshiani Mbaya O, Proschan M, Mukadi D, et al. A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics. N Engl J Med. 2019;381:2293–303.
Inal JM. Decoy ACE2-expressing extracellular vesicles that competitively bind SARS-CoV-2 as a possible COVID-19 therapy. Clin Sci (Lond). 2020;134:1301–4.
Mostafa-Hedeab G. ACE2 as Drug Target of COVID-19 Virus Treatment, Simplified Updated Review. Rep Biochem Mol Biol. 2020;9:97–105.
Debaisieux S, Rayne F, Yezid H, Beaumelle B. The ins and outs of HIV-1 Tat. Traffic. 2012;13:355–63.
Zhou F, Xue M, Qin D, Zhu X, Wang C, Zhu J, et al. HIV-1 Tat promotes Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6-induced angiogenesis and tumorigenesis by regulating PI3K/PTEN/AKT/GSK-3beta signaling pathway. PLoS ONE. 2013;8:e53145.
Albini A, Soldi R, Giunciuglio D, Giraudo E, Benelli R, Primo L, et al. The angiogenesis induced by HIV-1 tat protein is mediated by the Flk-1/KDR receptor on vascular endothelial cells. Nat Med. 1996;2:1371–5.
Yokosaki Y, Tanaka K, Higashikawa F, Yamashita K, Eboshida A. Distinct structural requirements for binding of the integrins alphavbeta6, alphavbeta3, alphavbeta5, alpha5beta1 and alpha9beta1 to osteopontin. Matrix Biol. 2005;24:418–27.
Dhawan S, Puri RK, Kumar A, Duplan H, Masson JM, Aggarwal BB. Human immunodeficiency virus-1-tat protein induces the cell surface expression of endothelial leukocyte adhesion molecule-1, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1 in human endothelial cells. Blood. 1997;90:1535–44.
Liu K, Chi DS, Li C, Hall HK, Milhorn DM, Krishnaswamy G. HIV-1 Tat protein-induced VCAM-1 expression in human pulmonary artery endothelial cells and its signaling. Am J Physiol Lung Cell Mol Physiol. 2005;289:L252–260.
Stoner MC, Vwalika B, Smid MC, George S, Chi BH, Stringer EM, et al. A retrospective study of HIV, antiretroviral therapy, and pregnancy-associated hypertension among women in Lusaka, Zambia. Int J Gynaecol Obstet. 2016;134:299–303.
Sebitloane HM, Moodley D. The impact of highly active antiretroviral therapy on obstetric conditions: a review. Eur J Obstet Gynecol Reprod Biol. 2017;210:126–31.
Naicker T, Phoswa WN, Onyangunga OA, Gathiram P, Moodley J. Angiogenesis, Lymphangiogenesis, and the Immune Response in South African Preeclamptic Women Receiving HAART. Int J Mol Sci. 2019;20:3728.
Mattar R, Amed AM, Lindsey PC, Sass N, Daher S. Preeclampsia and HIV infection. Eur J Obstet, Gynecol, Reprod Biol. 2004;117:240–1.
Saums MK, King CC, Adams JC, Sheth AN, Badell ML, Young M, et al. Combination Antiretroviral Therapy and Hypertensive Disorders of Pregnancy. Obstet Gynecol. 2019;134:1205–14.
Frank KA, Buchmann EJ, Schackis RC. Does human immunodeficiency virus infection protect against preeclampsia-eclampsia? Obstet Gynecol. 2004;104:238–42.
World Health Organization. Antiretroviral drugs for treating pregnant women and preventing HIV infection in infants. https://www.who.int/hiv/pub/mtct/antiretroviral2010/en/. Accessed 22 February 2020.
Tooke L, Riemer L, Matjila M, Harrison M. Antiretrovirals causing severe pre-eclampsia. Pregnancy Hypertens. 2016;6:266–8.
Song L, Ding S, Ge Z, Zhu X, Qiu C, Wang Y, et al. Nucleoside/nucleotide reverse transcriptase inhibitors attenuate angiogenesis and lymphangiogenesis by impairing receptor tyrosine kinases signalling in endothelial cells. Br J Pharmacol. 2018;175:1241–59.
Hernández S, Catalán-García M, Morén C, García-Otero L, López M, Guitart-Mampel M, et al. Placental mitochondrial toxicity, oxidative stress, apoptosis, and adverse perinatal outcomes in HIV pregnancies under antiretroviral treatment containing zidovudine. JAIDS J Acquired Immune Defic Syndromes. 2017;75:e113–e119.
Autran B, Carcelaint G, Li TS, Gorochov G, Blanc C, Renaud M, et al. Restoration of the immune system with anti-retroviral therapy. Immunol Lett. 1999;66:207–11.
Powis KM, Shapiro RL. Protease inhibitors and adverse birth outcomes: is progesterone the missing piece to the puzzle? J Infect Dis. 2014;211:4–7.
Kala S, Dunk C, Acosta S, Serghides L. Periconceptional exposure to lopinavir, but not darunavir, impairs decidualization: a potential mechanism leading to poor birth outcomes in HIV-positive pregnancies. Hum Reprod. 2020;35:1781–96.
Sgadari C, Barillari G, Toschi E, Carlei D, Bacigalupo I, Baccarini S, et al. HIV protease inhibitors are potent anti-angiogenic molecules and promote regression of Kaposi sarcoma. Nat Med. 2002;8:225–32.
Hernández S, Catalán-García M, Morén C, García-Otero L, López M, Guitart-Mampel M, et al. Placental Mitochondrial Toxicity, Oxidative Stress, Apoptosis, and Adverse Perinatal Outcomes in HIV Pregnancies Under Antiretroviral Treatment Containing Zidovudine. J Acquir Immune Defic Syndr. 2017;75:e113–e119.
Govender N, Moodley J, Gathiram P, Naicker T. Soluble fms-like tyrosine kinase-1 in HIV infected pre-eclamptic South African Black women. Placenta. 2014;35:618–24.
Powis KM, McElrath TF, Hughes MD, Ogwu A, Souda S, Datwyler SA, et al. High viral load and elevated angiogenic markers associated with increased risk of preeclampsia among women initiating highly active antiretroviral therapy in pregnancy in the Mma Bana study, Botswana. J Acquir Immune Defic Syndr. 2013;62:517–24.
Govender N, Naicker T, Moodley J. Maternal imbalance between pro-angiogenic and anti-angiogenic factors in HIV-infected women with pre-eclampsia. Cardiovasc J Afr. 2013;24:174–9.
Mbhele N, Moodley J, Naicker T. Role of angiopoietin-2, endoglin, and placental growth factor in HIV-associated preeclampsia. Hypertens Pregnancy. 2017;36:240–6.
Maharaj NR, Phulukdaree A, Nagiah S, Ramkaran P, Tiloke C, Chuturgoon AA. Pro-Inflammatory Cytokine Levels in HIV Infected and Uninfected Pregnant Women with and without Preeclampsia. PLoS ONE. 2017;12:e0170063.
Francisci D, Giannini S, Baldelli F, Leone M, Belfiori B, Guglielmini G, et al. HIV type 1 infection, and not short-term HAART, induces endothelial dysfunction. Aids. 2009;23:589–96.
Rosa SGV, Santos WC. Clinical trials on drug repositioning for COVID-19 treatment. Rev Panam Salud Publica. 2020;44:e40.
Joob B, Wiwanitkit V. SARS-CoV-2 and HIV. J Med Virol. 2020 (e-pub ahead of print 2020/03/29; https://doi.org/10.1002/jmv.25782).
Mascolo S, Romanelli A, Carleo MA, Esposito V. Could HIV infection alter the clinical course of SARS-CoV-2 infection? When less is better. J Med Virol. 2020 (e-pub ahead of print 2020/04/16; https://doi.org/10.1002/jmv.25881).
Ridgway JP, Farley B, Benoit JL, Frohne C, Hazra A, Pettit N, et al. A Case Series of Five People Living with HIV Hospitalized with COVID-19 in Chicago, Illinois. AIDS Patient Care STDS. 2020;34:331–5.
Tian C, Tang L, Wu J, Li W, Ming X, Zhou H, et al. An HIV-infected patient with coronavirus disease 2019 has a favourable prognosis: a case report. Ann Palliat Med. 2020 (e-pub ahead of print 2020/08/14; https://doi.org/10.21037/apm-20-576).
Zhao X, Jiang Y, Zhao Y, Xi H, Liu C, Qu F, et al. Analysis of the susceptibility to COVID-19 in pregnancy and recommendations on potential drug screening. Eur J Clin Microbiol Infect Dis. 2020;39:1209–20.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo-EM Structure of the 2019-nCoV Spike in the Prefusion Conformation. bioRxiv. 2020 (e-pub ahead of print 2020/06/09; https://doi.org/10.1101/2020.02.11.944462).
Chu CM, Cheng VC, Hung IF, Wong MM, Chan KH, Chan KS, et al. Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax. 2004;59:252–6.
Koss CA, Natureeba P, Plenty A, Luwedde F, Mwesigwa J, Ades V, et al. Risk factors for preterm birth among HIV-infected pregnant Ugandan women randomized to lopinavir/ritonavir- or efavirenz-based antiretroviral therapy. J Acquir Immune Defic Syndr. 2014;67:128–35.
Roberts SS, Martinez M, Covington DL, Rode RA, Pasley MV, Woodward WC. Lopinavir/ritonavir in pregnancy. J Acquir Immune Defic Syndr. 2009;51:456–61.
Peng F, Tu L, Yang Y, Hu P, Wang R, Hu Q, et al. Management and Treatment of COVID-19: The Chinese Experience. Can J Cardiol. 2020;36:915–30.
Oldenburg CE, Doan T. Rigorous Randomized Controlled Trial Implementation in the Era of COVID-19. Am J Trop Med Hyg. 2020;102:1154–5.
Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N Engl J Med. 2020;382:1787–99.
Hung IF, Lung KC, Tso EY, Liu R, Chung TW, Chu MY, et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet 2020;395:1695–704.
Verdugo-Paiva F, Izcovich A, Ragusa M, Rada G. Lopinavir-ritonavir for COVID-19: A living systematic review. Medwave. 2020;20:e7967.
National Department of Health South Africa. ART clinical guidelines for the management of HIV in adults, pregnancy, adolescents, children, infants and neonates. Pretoria: National Department of Health; 2019.
National Department of Health South Africa. Essential Drugs Programme. Hospital level (Adults) Standard Treatment Guidelines and Essential Medicines List. Pretoria; 2019.
World Health Organization. New high-quality antiretroviral therapy to be launched in South Africa, Kenya and over 90 low- and middle-income countries at reduced price. https://www.who.int/hiv/mediacentre/news/high-quality-arv-reduced-price/en/. Accessed 26 August 2020.
Auclair M, Guénantin AC, Fellahi S, Garcia M, Capeau J. HIV antiretroviral drugs, dolutegravir, maraviroc and ritonavir-boosted atazanavir use different pathways to affect inflammation, senescence and insulin sensitivity in human coronary endothelial cells. PLoS ONE. 2020;15:e0226924.
Afonso P, Auclair M, Caron-Debarle M, Capeau J. Impact of CCR5, integrase and protease inhibitors on human endothelial cell function, stress, inflammation and senescence. Antivir Ther. 2017;22:645–57.
Bornhede R, Soeria-Atmadja S, Westling K, Pettersson K, Navér L. Dolutegravir in pregnancy-effects on HIV-positive women and their infants. Eur J Clin Microbiol Infect Dis. 2018;37:495–500.
Quiros-Roldan E, Castelli F, Bonito A, Vezzoli M, Calza S, Biasiotto G, et al. The impact of integrase inhibitor-based regimens on markers of inflammation among HIV naïve patients. Cytokine. 2020;126:154884.
Favaloro EJ, Thachil J. Reporting of D-dimer data in COVID-19: some confusion and potential for misinformation. Clin Chem Lab Med. 2020;58:1191–9.
Cipolat MM, Sprinz E. COVID-19 pneumonia in an HIV-positive woman on antiretroviral therapy and undetectable viral load in Porto Alegre, Brazil. Braz J Infect Dis. 2020 (e-pub ahead of print 2020/09/01; https://doi.org/10.1016/j.bjid.2020.07.009).
Saag MS, Gandhi RT, Hoy JF, Landovitz RJ, Thompson MA, Sax PE, et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2020 Recommendations of the International Antiviral Society-USA Panel. Jama. 2020;324:1651–69.
de Wit E, Feldmann F, Cronin J, Jordan R, Okumura A, Thomas T, et al. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc Natl Acad Sci USA. 2020;117:6771–6.
Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395:1569–78.
Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the Treatment of Covid-19 - Final Report. N Engl J Med. 2020 (e-pub ahead of print 2020/05/24;https://doi.org/10.1056/NEJMoa2007764).
Anderson J, Schauer J, Bryant S, Graves CR. The use of convalescent plasma therapy and remdesivir in the successful management of a critically ill obstetric patient with novel coronavirus 2019 infection: a case report. Case Rep Women’s Health. 2020;27:e00221.
Maldarelli GA, Savage M, Mazur S, Oxford-Horrey C, Salvatore M, Marks KM. Remdesivir Treatment for Severe COVID-19 in Third-Trimester Pregnancy: Case Report and Management Discussion. Open Forum Infect Dis. 2020;7:ofaa345.
Burwick RM, Yawetz S, Stephenson KE, Collier AY, Sen P, Blackburn BG, et al. Compassionate Use of Remdesivir in Pregnant Women with Severe Covid-19. Clin Infect Dis. 2020 (e-pub ahead of print 2020/10/09; https://doi.org/10.1093/cid/ciaa1466).
United States Food and Drug Administration. Veklury (Remdesivir) EUA Letter of Approval. https://www.fda.gov/media/137564/download. Accessed 27 October 2020.
Nichols BE, Jamieson L, Zhang SRC, Rao GA, Silal S, Pulliam JRC, et al. The role of remdesivir in South Africa: preventing COVID-19 deaths through increasing ICU capacity. Clin Infect Dis. 2020 (e-pub ahead of print 2020/07/07; https://doi.org/10.1093/cid/ciaa937).
SABC News. Remdesivir now available in SA for COVID-19 treatment. https://www.sabcnews.com/sabcnews/remdesivir-now-available-in-sa-for-covid-19-treatment/. Accessed 27 October 2020.
Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): a Review. Jama. 2020;323:1824–36.
Marim F, Karadogan D, Eyuboglu TS, Emiralioglu N, Gurkan CG, Toreyin ZN, et al. Lessons Learned so Far from the Pandemic: A Review on Pregnants and Neonates with COVID-19. Eurasia J Med. 2020;52:202–10.
Santini-Oliveira M, Estrela Rde C, Veloso VG, Cattani VB, Yanavich C, Velasque L, et al. Randomized clinical trial comparing the pharmacokinetics of standard- and increased-dosage lopinavir-ritonavir coformulation tablets in HIV-positive pregnant women. Antimicrob Agents Chemother. 2014;58:2884–93.
McCormack SA, Best BM. Protecting the fetus against HIV infection: a systematic review of placental transfer of antiretrovirals. Clin Pharmacokinet. 2014;53:989–1004.
Louchet M, Sibiude J, Peytavin G, Picone O, Tréluyer JM, Mandelbrot L. Placental transfer and safety in pregnancy of medications under investigation to treat coronavirus disease 2019. Am J Obstet Gynecol MFM. 2020;2:100159.
Khan RJ, Jha RK, Amera GM, Jain M, Singh E, Pathak A, et al. Targeting SARS-CoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2’-O-ribose methyltransferase. J Biomol Struct Dyn. 2020 (e-pub ahead of print 2020/04/09; https://doi.org/10.1080/07391102.2020.1753577):1–14.
Rimawi BH, Johnson E, Rajakumar A, Tao S, Jiang Y, Gillespie S, et al. Pharmacokinetics and Placental Transfer of Elvitegravir, Dolutegravir, and Other Antiretrovirals during Pregnancy. Antimicrob Agents Chemother. 2017;61:02213-16.
Acknowledgements
The authors thank the UKZN College of Health Sciences and the National Research Foundation for funding.
Funding
Funded by the UKZN College of Health Sciences and the National Research Foundation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Naidoo, N., Moodley, J. & Naicker, T. Maternal endothelial dysfunction in HIV-associated preeclampsia comorbid with COVID-19: a review. Hypertens Res 44, 386–398 (2021). https://doi.org/10.1038/s41440-020-00604-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-020-00604-y
Keyword
This article is cited by
-
Renin-angiotensin system in the placenta of women with preeclampsia
Hypertension Research (2023)
-
Differential expression of the angiotensin receptors (AT1, AT2, and AT4) in the placental bed of HIV-infected preeclamptic women of African ancestry
Hypertension Research (2023)
-
Immunoexpression of neuropilin-1 in the chorionic villi of HIV-infected preeclamptic South African women of African ancestry
Histochemistry and Cell Biology (2023)
