
Abstract
The genetic structure of 13 populations of the amphiatlantic sea urchin Arbacia lixula, as well as temporal genetic changes in three of these localities, were assessed using ten hypervariable microsatellite loci. This thermophilous sea urchin is an important engineer species triggering the formation of barren grounds through its grazing activity. Its abundance seems to be increasing in most parts of the Mediterranean, probably favoured by warming conditions. Significant genetic differentiation was found both spatially and temporally. The main break corresponded to the separation of western Atlantic populations from those in eastern Atlantic and the Mediterranean Sea. A less marked, but significant differentiation was also found between Macaronesia (eastern Atlantic) and the Mediterranean. In the latter area, a signal of differentiation between the transitional area (Alboran Sea) and the rest of the Mediterranean was detected. However, no genetic structure is found within the Mediterranean (excluding Alboran) across the Siculo-Tunisian Strait, resulting from either enough gene flow to homogenize distance areas or/and a recent evolutionary history marked by demographic expansion in this basin. Genetic temporal variation at the Alboran Sea is as important as spatial variation, suggesting that temporal changes in hydrological features can affect the genetic composition of the populations. A picture of genetic homogeneity in the Mediterranean emerges, implying that the potential expansion of this keystone species will not be limited by intraspecific genetic features and/or potential impact of postulated barriers to gene flow in the region.
Similar content being viewed by others
Introduction
Arbacia lixula (Linnaeus 1758) is a warm-temperate water species occurring from the western Atlantic in Brazil (Tommasi 1964) to the other side of the Atlantic where it is present in the Macaronesian archipelagos (Mortensen 1935; Lessios et al. 2012), African Atlantic coast from Gibraltar to Angola, and the Mediterranean Sea (Tortonese 1965). Marine species with amphiatlantic distributions (i.e., those inhabiting both eastern and western Atlantic shorelines) provide interesting tests of the permeability of the mid-Atlantic dispersal barrier. Barring cases of cryptic speciation (e.g. Carmona et al. 2011), historical, hydrological and developmental features are usually called for to explain trans-Atlantic dispersal. In this sense, Arbacia is an interesting genus with fossil record dating from the Palaeocene (Kroh and Smith 2010). Its five extant species occur in the eastern Pacific and both sides of the Atlantic (Lessios et al. 2012). The two Atlantic species, A. punctulata (western Atlantic) and A. lixula (amphiatlantic) diverged some 1.5–3.3 mya at both sides of the mid-Atlantic barrier (Lessios et al. 2012), likely by a range expansion event from western to eastern Atlantic of the lineage that would become A. lixula, which nevertheless crossed again the mid-Atlantic barrier to establish the present-day Brazilian populations (Lessios et al. 2012; Wangensteen et al. 2012).
Arbacia lixula is an ecosystem engineer species (i.e., those that change availability of resources to other species; Jones et al. 1994, 1997), capable of transforming littoral communities into barren grounds due to its grazing activity (Bulleri et al. 1999; Gianguzza et al. 2011; Bonaviri et al. 2011). Mitochondrial genetic data (Wangensteen et al. 2012) and the absence of fossil records (Stefanini 1911; Mortensen 1935; Madeira et al. 2011) support the idea of a relatively recent colonization of this sea urchin in the Mediterranean Sea, likely during the last interglacial period (Wangensteen et al. 2012). The Mediterranean is a semi-enclosed sea subject to important anthropogenic impacts (e.g. Lejeusne et al. 2010; Coll et al. 2010). In turn, these threats interact in complex ways with the ongoing climate change that favours the progressive tropicalization of this sea (Francour et al. 1994). Among the key drivers of structure and function in littoral Mediterranean communities is the grazing activity of sea urchins, which induce regime shifts between macroalgal beds and sea urchin barrens (Bonaviri et al. 2011). Human-derived impacts can exacerbate the risk and irreversibility of such dramatic changes (Ling et al. 2014).
The thermophilous nature of A. lixula has long been recognized (Kempf 1962; Tortonese 1965), and this species is listed among those being currently favoured by the warming of the Mediterranean (Wangensteen 2013). Its abundance has been increasing in several areas of this sea in the past (Petit et al. 1950; Boudouresque et al. 1989; Francour et al. 1994). Its reproduction is enhanced by high temperatures (Gianguzza et al. 2011, Wangensteen et al. 2013b) and larval development features indicate that warming, modulated by other factors such as pH and food availability, may favour A. lixula development (Privitera et al. 2011; Wangensteen et al. 2013a; Gianguzza et al. 2014; Visconti et al. 2017). Although recent results showed a regression of marine invertebrate populations at the coast of Israel (eastern Mediterranean) due to the whole ecosystem collapsing (Yeruham et al. 2015; Rilov 2016), the general scenario is a progressive increase of abundance of A. lixula in most areas of the Mediterranean (Privitera et al. 2011; Wangensteen et al. 2013a; Visconti et al. 2017), which will result in significant changes in ecosystem functioning.
Under this scenario, it is of utmost importance to ascertain the genetic structure of A. lixula. In a previous study, Wangensteen et al. (2012) identified phylogeographic patterns in A. lixula using sequences of the mitochondrial gene cytochrome oxidase I (COI). That study identified three haplogroups in worldwide populations, one of them shared between eastern and western Atlantic populations. The mitochondrial structure of the species appeared to be shaped by Pleistocene demographic expansions, isolation between the eastern Atlantic, western Atlantic and Mediterranean Sea, and genetic homogeneity across the Mediterranean. Nevertheless, the lack of genetic differentiation across the Mediterranean basin (Wangensteen et al. 2012; Deli et al. 2017) needs to be compared with nuclear markers to confirm the information on gene-flow patterns and genetic signals in this species. Mitochondrial DNA only retains half of the species’ evolutionary history (Avise 2000), and due to the potential differential selection (Silva et al. 2014; Consuegra et al. 2015) and stochasticity of the coalescence processes between nuclear and mitochondrial DNA, these two types of markers can show different evolutionary signatures (e.g. Glynn et al. 2015; Garcia-Cisneros et al. 2016; Pérez-Portela et al. 2017). Therefore, combining both mitochondrial and nuclear information should provide complementary information to unravel both recent and historical processes shaping the genetic structure of A. lixula.
Population analyses should additionally include information about temporal changes in genetic make-up to understand whether the structure observed is stable over contemporary time periods. Currently, there is still a scarce number of temporal genetic studies in marine species, despite being a fundamental information for interpreting their long-term genetic distribution (e.g. Pérez-Portela et al. 2012; Pineda et al. 2016; Pascual et al. 2016). It is known that the stochasticity of reproduction, recruitment and survival of larvae and juveniles can potentially change the genetic composition of populations over the generations (e.g. Calderón et al. 2012; Aglieri et al. 2014; Couvray and Coupé 2018). Additionally, temporal variation across oceanographic discontinuities can also promote variation of gene-flow patterns over time (Olivar et al. 2003; Calderón et al. 2012). An outstanding example of interannual oceanographic variation is that across the Atlantic-Mediterranean transition, associated with shifts in Atlantic and Mediterranean water contributions across the Alboran Sea (Renault et al. 2012; Oguz et al. 2014). These marine circulation variations determine different levels of genetic mixing between Atlantic and Mediterranean genetic stocks over the years (Pascual et al. 2016). Therefore, spatio-temporal structuring patterns can provide valuable information about the future evolution of the populations, identifying connectivity patterns over time, and reservoirs of genetic diversity, among other important features.
In the present work, we use hypervariable nuclear microsatellite loci to investigate in detail the genetic structure of A. lixula across most of its distribution range using the same samples analysed by Wangensteen et al. (2012), but also extending these analyses to a temporal perspective. With new nuclear markers and samples, we specifically tested: (a) the disruptive effect of major oceanographic breaks, including the mid-Atlantic barrier, as well as migration patterns across them, which were used to determine the coherence of genetic divergence patterns between the nuclear and mitochondrial data, and (b) the relevance of the genetic change over time in two sites at the Alboran Sea (Atlantic-Mediterranean transition) and in another non-transitional Mediterranean site, which were sampled at two time points. We were particularly interested in inferring spatio-temporal population structure at the Atlanto-Mediterranean transition where other marine invertebrates have shown significant interannual variation in genetic structure (Pascual et al. 2016). The data generated in this study can be useful to infer present-day and future processes in the ongoing expansion of this keystone engineer species.
Materials and methods
Sample collection and microsatellite genotyping
Spatial genetic structure
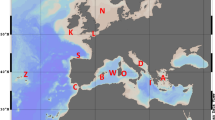
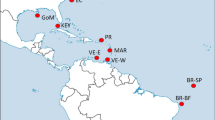
The collection sites included 13 different localities across most of the distribution range of the species: two localities on the western Atlantic (Brazil), three sites on the eastern Atlantic: Cape Verde, Canary Islands and Azores (Macaronesian Islands), five at the western Mediterranean (including two populations from the transitional zone at the Alboran Sea), and three in the eastern Mediterranean (see details in Fig. 1 and Table 1). Samples analysed correspond to a subset of 278 out of 604 individuals previously sequenced (mitochondrial COI gene) by Wangensteen et al. (2012) between 2009 and 2011, with an additional location from Sicily collected by SCUBA diving for the present study at the end of 2011. This sampling scheme included several major oceanographic breaks and/or transitions with observed disruptive effect in populations of other echinoderms (e.g. Calderón et al. 2008; Pérez‐Portela et al. 2013, 2017; Taboada and Pérez-Portela 2016; Garcia-Cisneros et al. 2016, 2017): the mid-Atlantic barrier that divides the eastern and western Atlantic; the Gibraltar Strait that marks the geographical partition between the Atlantic Ocean and Mediterranean Sea; the Almeria-Oran front, described as the biogeographical break between the Atlantic and Mediterranean basins in most marine species; and the Siculo-Tunisian Strait between the eastern and western Mediterranean sub-basins.

Collection sites and general genetic structure in Arbacia lixula. a Sampling sites and pie charts of the percentage of each cluster (K = 3) per site obtained from STRUCTURE as represented below, and b STRUCTURE barplot with the posterior probabilities of individual assignment of the most probable number of clusters for the whole data set (K = 3). Clusters are represented by different colours (yellow, red and blue), and each bar represents a different individual. Numbers and red lines represent the four different marine barriers considered: Mid-Atlantic barrier (1), Gibraltar Strait (2), Almeria-Oran front (3) and Siculo-Tunisian Strait (4)
Temporal genetic trends
For testing potential changes in genetic structure and diversity over time, three of the Mediterranean populations sampled in 2009 were re-sampled for this study in 2014: Colera at the northwestern Mediterranean, and La Herradura and Torremuelle at the Alboran Sea−Atlantic−Mediterranean transition. These sites were selected because we were specifically interested in exploring the potential effect of interannual oceanographic variation on populations’ divergence at the Atlantic−Mediterranean transition, an area where A. lixula populations displayed significant mitochondrial differences (Wangensteen et al. 2012) despite the short geographical distances separating them to other Atlantic and Mediterranean sites. We analysed the two Alboran sites for which samples from 2009 were available (Wangensteen et al. 2012) and one northwestern Mediterranean site far away from this Atlantic−Mediterranean transition for comparison with the first two sites.
Tissue samples were collected and fixed as described in Wangensteen et al. 2012. Total DNA was extracted from 302 individuals for the “spatial” study, plus 77 individuals of the 2014 sampling used for the “temporal” study. The REDExtract-N-Amp Tissue PCR kit (from Sigma-Aldrich, www.sigmaaldrich.com/) was used, following the protocol described by the manufacturer. All individuals were genotyped at ten microsatellite loci (ALM2, ALM4, ALM5, ALM7, ALM8, ALM9, ALM11, ALM14, ALM15 and ALM17) described in Garcia-Cisneros et al. (2013).
Amplification of fragments containing microsatellites was performed by polymerase chain reaction (PCR) in a final volume of 10 μl, containing 5 μl of ReadyMix Taq PCR Reaction Mix (Sigma-Aldrich), 2–8 ng of DNA, 0.4 μl (10 μM) of each primer (forward and reverse) and 3.2 μl of ultrapure water. Samples were amplified in a thermocycler (Bio-Rad MyCycler, http://www.bio-rad.com) with an initial 2 min denaturation step at 94 °C, and 35 amplification cycles: 45 s at 94 °C, 50 s at the locus-specific annealing temperature (51–58 °C; see Garcia-Cisneros et al. 2013) and 40 s at 72 °C, followed by 4 min of final extension at 72 °C.
Successful amplifications were genotyped in an automated sequencer (Applied Biosystems, www.thermofisher.com) in the Science and Technology Centres of the University of Barcelona (CCiTUB). Allele length was estimated relative to the internal size standard 70–500 ROX (Bioventures) using the software Peak-Scanner v 1.0 (Applied Biosystems).
Data analyses
The number of alleles per population, observed heterozygosity (Ho), expected heterozygosity (He), inbreeding coefficients (FIS), and number of private alleles per geographical area were calculated using GenAlex v 6.41 (Peakall and Smouse 2006) and Genepop v 4.2 webserver (Raymond and Rousset 1995). The exact test for departure from Hardy−Weinberg equilibrium (HWE) was performed in Arlequin v 3.5.1.2 (Excoffier et al. 2005). The potential correlation between the FIS and number of missing data per population was explored to understand the impact of missing data on this statistic.
Spatial genetic structure
We used different approaches based on Bayesian clustering, genetic distances and discriminant analyses of principal components. Whereas methods based on genetic distances (e.g FST) are affected by the populations’ Hardy−Weinberg disequilibrium, and assume absence of linkage disequilibrium among all loci within populations, other multivariate methods are free from these two assumptions. Therefore, we compared here different methods to minimize potential bias of using only one approach.
The software STRUCTURE v 2.3.4 (Pritchard et al. 2000) was used to infer an optimal number of homogeneous genetic units (K) based on Bayesian clustering analyses. It was run with the whole data set, with a K number from 1 to 16, and 200,000 Markov chain Monte Carlo (MCMC) steps were performed following 80,000 burn-in iterations in ten independent replicates under the “admixture model” and the “correlated allele frequencies mode” implemented by the software. The same strategy was separately applied to selected subsets of the populations in order to obtain a finer-scale analysis within major marine areas: (a) the eastern Atlantic and Mediterranean populations to better explore genetic partition across the Atlantic−Mediterranean arch and, (b) only Mediterranean sites to investigate potential divergence within this basin and across the Almeria-Oran Front and the Siculo-Tunisian Strait. The most likely value of “real” clusters was identified comparing the rate of change in the likelihood of K. The optimal K values were determined using the ad hoc statistic ΔK (Evanno et al. 2005). Ten independent replicates per run were averaged using the clumpak server (Kopelman et al. 2015), and results were graphically represented with the same software.
Genetic clusters were also delineated using “find.clusters” of the adegenet package for R software (Jombart 2008; Team R Core 2013) using a K-means clustering algorithm. A range of cluster numbers was chosen and the optimal number was selected using a Bayesian Information Criterion (BIC). Group assignment probabilities were then displayed with the “compoplot” function of adegenet. As before, further analyses were performed with “find.clusters” considering only eastern Atlantic and Mediterranean populations and, finally, only Mediterranean populations. Additionally, we ran a discriminant analysis of principal components (DAPC, Jombart et al. 2010) using populations as groups with the adegenet package. This method allows the visual identification of genetic clusters of individuals and can outperform Bayesian clustering approaches in detecting genetic substructure (Jombart et al. 2010). The optimal number of principal components (PC) from the PCA step passed onto the discriminant analysis was determined by the cross-validation method, and by comparison of a-scores for a set of increasing numbers of PCs and a spline interpolation using the “a-score” function of adegenet. DAPCs were performed separately for the whole data set, for the eastern Atlantic plus Mediterranean populations, and for the Mediterranean populations alone.
The software Arlequin was used to estimate population distances with the FST statistic between pairs of populations based on an allele infinite model. The Jost’s Dest estimator (Jost 2008) was also obtained with the package DEMEtics in R (Gerlach et al. 2010). A false discovery rate correction was applied for the p values (Benjamini−Yekutieli method, Narum 2006) to account for multiple tests. The genetic dissimilarity matrices generated with both estimators were represented with cluster analyses and heatmaps obtained with the gplots package for R (Warnes et al. 2016).
To test the concordance between nuclear and mitochondrial genetic distances, we performed correlation analyses for FST and Dest matrixes obtained from microsatellite loci (this study) and COI sequences (COI distance matrixes obtained from Wangensteen et al. 2012).
Null allele frequencies were estimated following the Expectation Maximization (EM) algorithm implemented in FreeNA (Chapuis and Estoup 2007). Using this information, the corrected estimations of FST values were calculated applying the ENA and INA methods with the same software.
Analyses of molecular variance (AMOVA) were computed using an allele infinite model, and their significance tested with 20,000 permutations in Arlequin. For the AMOVAs we grouped populations in different sets according to the FST results, geographical origin and known oceanographic barriers. We initially tested differences among western Atlantic, eastern Atlantic and Mediterranean Sea, considering two major marine breaks: the mid-Atlantic barrier and the Gibraltar Strait. In a second analysis we removed populations from western Atlantic and compared east Atlantic populations with Mediterranean populations. We then compared the populations from the Alboran Sea with the rest of the Mediterranean to test differentiation across the Almeria-Oran front. Finally, we analysed only Mediterranean populations excluding Alboran Sea, comparing the eastern and the western sub-basins to explore the potential disruptive effect of the Siculo-Tunisian Strait.
The potential effect of genetic isolation of populations by geographical distance, independently of oceanographic barriers, was assessed for the whole data set, and separately for different population subsets (eastern Atlantic and Mediterranean Sea, and only the Mediterranean Sea), using the correlation of linearized genetic distances (FST / 1 – FST) with geographical distances (as measured in Wangensteen et al. 2012) between localities. The significance of the correlations was tested by a Mantel test, as implemented in Arlequin with 20,000 permutations per analysis.
To estimate gene flow among marine areas, we calculated mutation-scaled effective migration rates (M) based on Bayesian inference using the software MIGRATE v 3.6.11 (Beerli 2006; Beerli and Felsenstein 2001). We estimated asymmetric M among the three major geographical areas: the western Atlantic (Brazilian sites), eastern Atlantic (Macaronesian islands) and the Mediterranean Sea. Migration estimates per generation can be expressed as 4Nm for nuclear markers, in which N is the effective population size and m the immigration rate. Three preliminary runs were performed to infer initial parameters and check convergence before performing a final run. For the latter, we used a Brownian motion mutation model with constant mutation rate for all loci, three different replicates with one long chain, 3,000,000 iterations (9,000,000 final sampled parameters) with the first 30,000 iterations discarded, and an adaptive heating scheme of four different temperature chains.
Temporal genetic trends
For the three populations sampled in 2009 and again in 2014 (Colera, Torremuelle and La Herradura), we computed a DAPC representation using populations from each sampling year as groups (with the adegenet package in R) and pairwise tests using FST (calculated with Arlequin) and Dest (calculated with DEMEtics) as described above.
We also estimated effective population sizes (Ne) for these three populations (Colera, Torremuelle and La Herradura) using the temporal method, based in shifts in allele frequencies between samples taken a number of generations apart (Jorde and Ryman 2007). We used NeEstimator v.2.01 (Do et al. 2014) to calculate Ne based on allele frequency changes between the two sampling years using three different estimators that differ in precision and bias (Do et al. 2014): those of Nei and Tajima (1981), Pollak (1983), and Jorde and Ryman (2007). We considered a generation per year (Wangensteen et al. 2013b) and removed alleles below a frequency threshold of 0.05 to reduce random error (likely at the cost of a slight downward bias in the estimates, Do et al. 2014). Arbacia lixula has overlapping generations, which adds complexity to the computation of Ne estimates originally devised for discrete generations. Ideally, a correction should be made on measures of temporal change in allele frequency that incorporates the different contributions of the co-existing cohorts (Jorde and Ryman 1995). Calculating this correction requires precise biological knowledge of the cohort structure, age-specific survival rates, and age-specific reproduction rates (e.g., Calderón et al. 2009), parameters that were not available for A. lixula. We nevertheless applied temporal methods without correction as, first, we sampled the sea urchins randomly with respect to age and, second, we sampled at a wide interval of generations (five generations apart, from 2009 to 2014). Jorde and Ryman (1995) showed how sampling over long time intervals greatly reduces the bias in temporal methods for overlapping generations. In any case, our estimates should still be useful for comparative purposes among populations, as biological parameters are unlikely to be very different between populations and, therefore, any remaining bias should be similar.
Results
The ten microsatellite loci were highly polymorphic, with a total number of alleles ranging between 16 (locus ALM11) and 38 (locus ALM4). Details of genetic descriptors for each locus and population are presented as supplementary material (Table S1). Populations of A. lixula were in general characterized by high genetic diversity and a large number of alleles (mean number per locus ranged from 9.3 to 14.3 alleles, Table 1). Allele richness, used to compare allelic diversity among marine areas with large differences in sample size, showed that the eastern Atlantic retained the highest richness, followed by the Mediterranean and the western Atlantic areas. Regarding private alleles, the eastern Atlantic showed the lowest value, with only 6.77% (13 alleles) of private alleles, whereas the Mediterranean and western Atlantic had 14.2% (31 alleles) and 10.7% (14 alleles) of private alleles, respectively (Supplementary Fig. S1).
In all populations observed heterozygosity was lower than expected, as demonstrated by the significant values of the FIS, with significant deviation from the HWE in all populations (p < 0.001) (see Table 1). All microsatellite loci considered individually had overall positive values of FIS, significant in all cases (FIS values > 0.11) except in the locus ALM2 (FIS = 0.021, p = 0.157). A low overall percentage of missing data (2.25%), distributed across all microsatellites but mostly concentrated in the Brazilian populations, makes unlikely that null alleles underlie this general deficit of heterozygotes. Interestingly, the two populations showing the highest percentage of missing data also displayed the lowest FIS values, also suggesting that missing data are not related to positive and significant FIS (Supplementary Fig. S2).
The Bayesian analyses detected an optimal K value of 3 based on the ΔK plot (Supplementary Fig. S3). The composition of the different populations in terms of these three genetic groups (sum of individual membership probabilities to each group) is represented in form of pie charts in Fig. 1a. One of the three genetic clusters detected sharply separated the populations from the western Atlantic (yellow group in Fig. 1), while the rest of populations were mainly composed of the other two genetic clusters. In most individuals, however, the most probable group had a membership probability above 75%, with few admixed individuals (Fig. 1b).
The situation is similar when genetic groups are delineated using the “find.clusters” function in adegenet. The number of clusters (BIC criterion) that better explains our data is 6 (Fig. S4), but the plot of membership probabilities shows clear differences between western Atlantic and all other Atlantic and Mediterranean samples, and some differentiation between the eastern Atlantic (Macaronesia) and Mediterranean based on group membership (Fig. S4). Hence, both the Bayesian clustering analysis and “find.clusters” function detected a strong disruptive effect of the mid-Atlantic barrier and a smaller effect of the eastern Atlantic (Macaronesia)−Mediterranean transition. Analyses performed separately for the different marine areas, the whole data set, eastern Atlantic and Mediterranean and only Mediterranean Sea, did not provide additional information (results not shown).
Results from FreeNA showed that, in most cases, the correction of FST values was minimal and the significance of the FST statistic did not change in any case. Therefore, we consider that null alleles do not have a large effect on genetic distance estimations in this study, and that uncorrected values can be used for further analyses. The values of population differentiation using FST and Dest estimators are shown in Table S2 and graphically depicted as dendrograms and heatmaps in Fig. 2. Both estimators provide basically the same information, and are highly correlated (r = 0.979, p < 0.001). Moreover, they are highly correlated with previous genetic distance results from mitochondrial DNA obtained from Wangensteen et al. (2012) (r = 0.832 and r = 0.816, p < 0.01 for FST and Dest values, respectively), showing congruent results between microsatellites and COI. Pairwise comparisons using microsatellite loci showed significant differentiation in all comparisons involving Brazilian populations (western Atlantic) with the rest, indicating a strong disruptive effect of the mid-Atlantic barrier. In addition, 17 comparisons (out of 24) between eastern Atlantic (Macaronesian) and Mediterranean populations were significant with both estimators, and 6 comparisons (out of 12) of the Alboran Sea populations (La Herradura and Torremuelle) with the rest of the Mediterranean were also significant for both indices, suggesting limited gene flow across two additional marine barriers: the Gibraltar Strait and the Almeria-Oran front. Furthermore, the two sites from the Alboran Sea, the transition area between the eastern Atlantic and Mediterranean Sea, were significantly different from each other for both indices. Only one significant pairwise difference within Macaronesia was found between Los Gigantes (Gig—Canary Islands) and Boavista (Cav—Cape Verde Islands) with Jost’s estimator (Dest). No significant divergence was found in any comparison within the western Atlantic. Within the Mediterranean Sea, no significant divergence was detected between sites, discarding the Siculo-Tunisian Strait as a genetic barrier in this species.

Genetic differentiation between sites in Arbacia lixula. a Heatmap and dendrogram based on pairwise FST values, and b heatmap and dendrogram based on pairwise Dest values. Values of FST and Dest and their associated p values are included as Supplementary Table S2. *Highlights the different clustering of Torremuelle (Tor) with other marine areas between heatmaps
The heatmaps and dendrograms show clearly the distinction between western Atlantic populations and the remaining ones. Among the latter, the Macaronesian populations (eastern Atlantic- Faials, Los Gigantes and Boavista) formed a cluster, while Mediterranean populations appeared well mixed, with no inter-basin structure, although Alboran Sea populations (Torremuelle and La Herradura) were in general slightly more differentiated. In particular, the Torremuelle population was somewhat more divergent and was separated from the rest of Mediterranean populations (Dest) or even external to the eastern Atlantic plus Mediterranean clusters with the FST estimator (Fig. 2).
The spatial representation of the DAPC considering all populations (Fig. 3a, 51 PCs retained) showed again this pattern of separation between western Atlantic and eastern Atlantic plus Mediterranean in the first axis, while along the second axis the populations of the Macaronesian archipelagos are separated, albeit with some overlap, from the Mediterranean populations.

DAPCs of Arbacia lixula. Graphs represent DAPC results from three different analyses: a the whole data set, b eastern Atlantic and Mediterranean sites, and c only Mediterranean sites (including the Alboran Sea). On the graph, points represent different individuals, and point patterns different sampling sites. Blue pattern = Mediterranean sites; red-orange pattern = eastern Atlantic; and yellow pattern = western Atlantic
A DAPC graph excluding the Brazilian populations (Fig. 3b, 28 PCs retained) also showed a separation of the Macaronesian populations along the first axis, with overlap of the inertia ellipses. Torremuelle appeared also partially separated from the rest on the second axis. Finally, a DAPC considering only the Mediterranean populations (Fig. 3c, 26 PCs retained) showed less differentiation than the previous graphs. The two populations from the Alboran Sea appeared somewhat offset from the others, Torremuelle at one extreme of the first axis, La Herradura at one extreme along the second axis. No differentiation was apparent among populations of eastern and western Mediterranean, which showed interspersed centroids and widely overlapping inertia ellipses.
The results of the AMOVA analyses are coherent with the results from clustering and ordination methods (Table 2). An AMOVA considering as groups the Brazilian (western Atlantic), Macaronesian (eastern Atlantic), and Mediterranean populations (thus including the whole data set) showed low but highly significant percentage of variation between groups and among populations within groups. The same outcome was found when excluding western Atlantic populations and considering the Macaronesian (eastern Atlantic) and the Mediterranean populations as different groups. However, in an analysis comparing the Alboran Sea with the rest of the Mediterranean populations the “among group” component explained only 0.54% of the variance and was not significant, while the among populations within groups component was still significant (p = 0.002). Finally, if we restrict the analysis to the Mediterranean populations excluding the Alboran Sea and compare western with eastern Mediterranean populations, the “among group” and the “among populations within groups” components were not significant (p = 0.393 and p = 0.472, respectively), pointing to a lack of gene-flow restriction across the Siculo-Tunisian Strait. In all cases, most of the variation was contained within populations (29.32–32.58%) and, particularly, within individuals (FIT) (66.58–67.51%) (Table 2).
Assessing the hypothesis of isolation by distance through the Mantel test revealed significant correlation between genetic and geographic distances (r = 0.859, p < 0.001) when considering all populations. The correlation was weaker, but still significant, when removing the Brazilian populations (r = 0.384, p = 0.025), and no correlation was found when considering just the Mediterranean Sea (r = 0.189, p = 0.179) (see correlation graphs in Supplementary Fig. S5).
The results of migration patterns between western Atlantic, eastern Atlantic and Mediterranean Sea are presented in Table 3. Migration outputs showed a general overlapping of the 95% confidential intervals around the M estimates between areas. Only M estimations from the Mediterranean to eastern Atlantic, and from the Mediterranean to the western Atlantic, which were also the highest values of M (mean 24.182 and 18.336, respectively), did not include zero within the confidence interval. These results may suggest a potential pattern of asymmetric and long distance migration that mainly occurs westwards. All the other estimations presented lower values of the M mean, ranging from 7.145 to 14.919, and wide confidence intervals that always included zero (Table 3).
Temporal genetic trends
For the three populations that were re-sampled in 2014 (Torremuelle and La Herradura at the Alboran Sea, and Colera at the northwestern Mediterranean), the discerned genetic diversity was higher than that recorded in 2009, in terms of observed heterozygosity and mean allele number (except Colera for the latter parameter). Likewise, FIS values were lower, likely indicating less inbreeding (Table 1). Both FST and Dest estimators showed significant genetic differentiation between Torremuelle and the other two populations in 2009 (p < 0.015), whereas La Herradura and Colera did not show significant differences between them in 2009. In 2014, the three populations displayed no significant differences in genetic structure (Supplementary Table S3). Genetic distances also revealed that the northwestern Mediterranean population of Colera did not significantly change in genetic structure between 2009 and 2014, whereas both populations at the Alboran Sea, Torremuelle and La Herradura, demonstrated significant differences in genetic structure between 2009 and 2014. Therefore, Alboran Sea populations significantly changed their genetic structure over time (Supplementary Table S3 for FST and Dest, and Fig. 4). Mean differentiation values between years in the three populations (FST: 0.040 ± 0.015, Dest: 0.103 ± 0.029, mean ± SE) were higher, but of the same order, than mean genetic divergence detected in the spatial study among the Mediterranean populations (FST: 0.015 ± 0.002, Dest: 0.087 ± 0.007, mean ± SE).

Genetic differences over time in Arbacia lixula. a Heatmaps based on FST (left) and Dest (right) values between years and three sites, and b DAPC analyses for the three populations at two time points (2009 and 2014)
A heatmap representation of the FST and Dest values (Fig. 4a) highlighted this pattern of marked interannual differences, but showed also that the three populations were more divergent among them in 2009 than in 2014. A DAPC representation (Fig. 4b, 20 PCs retained) revealed this same pattern: the three populations were more separated in 2009 (particularly Tor), but clustered tightly in 2014.
Considering one generation per year, the different estimators of effective population size (Table 4) revealed low values in all populations (approximate range 30–400 individuals). There were consistently higher sizes in the northern population of Colera (177.3–387.9 individuals, according to the different methods) than in the Alboran sea populations of La Herradura (33.9–38.3) and Torremuelle (34.2–38.8). The three estimators yielded remarkably similar estimates (and confidence intervals) in the southern populations, but varied by a factor of ca. 2 for the Colera population, for which defined confidence intervals could be obtained only with the unbiased Jorde/Ryman’s estimator.
Discussion
The amphiatlantic sea urchin, Arbacia lixula displayed significant nuclear divergence among the western Atlantic, eastern Atlantic and Mediterranean Sea. Additionally, variable structure across the transitional area of the Alboran Sea was also detected, which can be attributed to the interannual variation in the oceanographic circulation across this area.
Populations of A. lixula showed a high degree of genetic diversity. There was, however, a strong deficit of heterozygotes in all populations, with significant departure from HWE. This is unexpected for species with long pelagic larval duration. However, Addison and Hart (2005), reviewing data for 124 marine invertebrates, showed a prevalence of positive FIS values even in species with planktonic larvae. It can be explained by several factors, such as null alleles, mating among relatives, or unrecognized spatial and temporal structure within samples (Wahlund effects). The scarcity of null alleles indicates that our result is not an artefact of the markers. A potential explanation in our case is that assortative mating occurs linked to different gamete recognition proteins. Bindin, the sperm protein implicated in the fertilization of the egg, is well known in sea urchins (Metz et al. 1998; Zigler and Lessios 2003; Zigler et al. 2005; Lessios et al. 2012). Calderón and Turon (2010) showed that assortative mating linked to selected positions in the bindin gene of Paracentrotus explained inter-cohort differentiation. Such non-random mating structures, as well as the presence of spatial breeding groups, linked to stochasticity in reproductive success, patchiness in gamete distribution and the collective dispersal of genetically related larvae in the plankton (e.g. Broquet et al. 2013; Couvray and Coupé 2018), can explain the lack of HWE detected in Arbacia. In A. lixula, as in many other species, most genetic diversity was retained within populations and individuals (e.g. Calderón et al. 2008; Garcia-Cisneros et al. 2016).
Our nuclear results showed a sharp divergence between the western and eastern Atlantic areas, likely due to the combined effect of isolation by distance and the strong disruptive effect of the deep mid-Atlantic barrier. This sharp genetic divergence is similar to the one observed in other amphiatlantic echinoderms with large dispersal potential (e.g. Lessios et al. 2001; Garcia-Cisneros et al. 2017). The nuclear divergence in A. lixula was also largely congruent with COI mitochondrial data, but historical migration patterns and allele frequencies highlighted interesting insights in its phylogeography. Lessios et al. (2012) and Wangensteen et al. (2012) hypothesized the colonization of the western Atlantic coast, across the mid-Atlantic barrier, from eastern Atlantic stocks. However, neither migration nor allele distribution patterns from our new nuclear results fully supported this hypothesis and suggested instead the Mediterranean as a potential source of colonizers. Migration patterns estimated from microsatellites showed asymmetric gene flow among areas, with the most important historical migration likely flowing westward from the Mediterranean to the eastern and western Atlantic. Our results discard large historical connectivity between eastern and western Atlantic regions, which showed a low value of M. In addition, the Mediterranean origin of the western Atlantic populations can be also supported by 14 alleles shared (out of 250) between these two areas, whereas only two alleles were found in common between the eastern and western Atlantic stocks that can be indicative of long-term isolation between populations at both sides of the Atlantic. Interestingly, a detailed re-evaluation of the COI network also points out the potential origin of the Brazilian haplotype cluster from some of the most frequent Mediterranean haplotypes. Therefore all current genetic evidences suggest divergence of the western Atlantic populations of A. lixula from the Mediterranean area, which likely happened after the Pleistocene colonization and demographic expansion in the Mediterranean Sea (93.8–205.2 kya) (Wangensteen et al. 2012). Nonetheless, further investigations are necessary to discard other unexplored genetic stocks and to confirm the Mediterranean origin of the western Atlantic lineages.
Additionally, subtler structure is also found in the Atlantic−Mediterranean area, with significant differentiation between the Macaronesian islands and the Mediterranean. The biogeographic break between Atlantic and Mediterranean leaves a strong signature in the genetic structure of many species of fish and invertebrates with different biological characteristics (Patarnello et al. 2007; Pascual et al. 2017), including sea urchins, sea stars, brittle-stars and sea cucumbers (Borrero‐Pérez et al. 2011; Pérez-Portela et al. 2010; Calderón et al. 2012; Taboada and Pérez-Portela 2016; Garcia-Cisneros et al. 2016, 2017). However, the Mediterranean Sea also has a number of internal oceanographic barriers that can restrict species dispersal. Among the better identified oceanographic barriers within the Mediterranean are: the Gibraltar Strait and the Almeria-Oran Front between the Atlantic and Mediterranean basins, the Ibiza Channel and Balearic Front dividing the north- and southwestern Mediterranean sub-basins, the Siculo-Tunisian Front between the western and eastern Mediterranean, and the Otranto Strait and Aegean Front delimiting the Adriatic and Aegean seas, respectively (e.g., Villamor et al. 2014; Riesgo et al. 2016; Garcia-Cisneros et al. 2016; and a review in Pascual et al. 2017).
Nevertheless, these oceanographic fronts do not have equal effect on all marine species. Pascual et al. (2017), reviewing published information for 70 species, found that the reduction of gene flow linked to the presence of the abovementioned oceanographic fronts is more important in species with long planktonic durations. This unexpected pattern is likely because these larvae move off-shore, along the continental shelf and slope, and are thus more affected by major oceanographic circulation and marine fronts than larvae that remain close to the coastline (Pascual et al. 2017). In our case, we detected genetic divergence between both sides of the Almeria-Oran front, as observed in other echinoderms (Calderón et al. 2012; Garcia-Cisneros et al. 2016, 2017), although the divergence detected in Alboran populations of A. lixula may actually be a transient process, as discussed below for the temporal analyses, rather than a permanent one. Nevertheless, we could not find any evidence of genetic divergence between the western and eastern Mediterranean sub-basins, nor was there any significant isolation by distance effect in the Mediterranean, a pattern that contrasts with other echinoderms with large dispersal potential across the same geographical area (e.g. Garcia-Cisneros et al. 2016, 2017). This may suggest that A. lixula is not largely affected by discontinuities between the Mediterranean populations, representing a well-mixed genetic pool within this sea, and/or it reflects the recent evolutionary history within this basin, marked by a demographic expansion (Wangensteen et al. 2012), with not enough time to diverge within the Mediterranean basins.
The temporal genetic patterns among the two populations from the Atlantic−Mediterranean transition and the one from the north-western Mediterranean indicate that populations were more divergent, particularly Torremuelle (Alboran Sea), in 2009 than in 2014. Interannual variations in the hydrological features along the Iberian Mediterranean shores are well known (Pascual et al. 2002; Pinot et al. 2002; Bouffard et al. 2010; Balbín et al. 2014), and have been held responsible for temporal patterns of genetic variation in organisms such as the fish Sardina pilchardus (Olivar et al. 2003), the sea urchin Paracentrotus lividus (Calderón et al. 2012), or the crab Liocarcinus depurator (Pascual et al. 2016). In particular, in the Alboran area, there is a complex structure with two main anticyclonic gyres and a central cyclonic gyre (Sanchez-Vidal et al. 2004; Sánchez-Garrido et al. 2013). The relative intensity of these gyres changes over time, and it determines a temporally variable system of hydrological fronts in the area (Renault et al. 2012; Oguz et al. 2014). These features affect the interplay between Atlantic and Mediterranean waters, leading to variable patterns of distribution of water masses in the Alboran Sea. This can explain our finding of significant temporal genetic differences in Torremuelle and La Herradura located in the Alboran Sea, while the northern population of Colera, outside of this transitional area, remained more stable. Such temporal changes in genetic composition of southern Iberian populations relative to more northern populations were also detected for Paracentrotus lividus (Calderón et al. 2012). Torremuelle, in particular, lies in western Alboran Sea, in a relatively isolated spot just outside the frontal system generated by the western anticyclonic gyre (Sánchez-Garrido et al. 2013; Oguz et al. 2014). Thus, arrival of larvae to this locality is subject to stochastic and oceanographic changes among years, which may explain its higher genetic distance compared to other Mediterranean populations.
The effective population sizes (Ne) detected examining temporal variation in genetic composition were small (from tens to a few hundred individuals), and similar to Ne estimates for P. lividus (Calderón et al. 2009). In this study, we did not specifically measure A. lixula abundances but information obtained from other studies showed densities that vary across space and time from low-density populations (0.2–0.3 individuals/m2) to densely populated sites (over 1.0 individuals/m2) (Palacín et al. 1998; Hereu et al. 2012). It is common for invertebrates and fish to have effective population sizes 2–6 orders of magnitude smaller than census sizes (Turner et al. 2002; Hauser and Carvalho 2008; Plough 2016), which is often explained by large variance of reproductive success, whereby only few adults are able to produce successful progeny (sweepstake reproduction, Hedgecock 1994). Statistic methods to calculate effective population size based on genetic data at two time points are appropriate to estimate contemporary Ne that reflects the effective number of parental specimens from which the collected sample comes from (e.g. Casilagan et al. 2013). Thus, the stochastic events that can take place during the reproduction together with the long planktonic period and the settlement and recruitment phases of A. lixula can likely explain the small effective population sizes detected. It is noteworthy that the hydrologically more stable northern population of Colera had ca. 6−10 times larger effective population sizes than the two southern populations.
From the last few years, there is increasing evidence of the importance of A. lixula in the formation and maintenance of bare habitats (Bulleri et al. 1999; Gianguzza et al. 2011; Bonaviri et al. 2011). Arbacia lixula is a thermophilous species likely to be enhanced by warming temperatures (Francour et al. 1994; Gianguzza et al. 2011; Wangensteen et al. 2013a) and a generalist species with a catholic diet that qualifies it as omnivore (Wangensteen et al. 2011; Agnetta et al. 2013) not affected by a commercial fishing industry. Thus, although some populations of A. lixula at the Levant basin are in decline due to the ecosystem collapsing (Rilov 2016), under the current scenario of the ongoing tropicalization of the Mediterranean, A. lixula can be favoured, leading to important changes in ecosystem structure and functioning.
This study shows a main genetic break in A. lixula between both sides of the Atlantic, and smaller differentiation signals associated with the Atlanto-Mediterranean transition. However, no genetic structure was found within the Mediterranean populations, suggesting that either the species’ dispersal abilities suffice to break the hydrological barrier separating the two Mediterranean sub-basins and/or the genetic homogeneity is the result of the recent evolutionary history of the species, although both hypotheses are not mutually exclusive. A picture of genetic homogeneity across the Mediterranean implies that the species may safely overcome occasional adverse local conditions and quickly replenish populations from neighbouring and distant locations. Future research, including whole-genome scans and the inclusion of populations from other areas (such as the Adriatic sea, Levant basin and/or the Atlantic African shores) will likely show a more nuanced picture of the underlying genetic structure associated with adaptation (e.g. Carreras et al. 2017). Overall, however, the patterns found suggest that the spread potential of A. lixula in the Mediterranean is large and the ongoing expansion of this thermophilous species will not be restricted by the potential impact of postulated barriers to gene flow.
Data archiving
Data sets are available from Mendeley at: https://doi.org/10.17632/kzrgwmpzzt.1 and https://doi.org/10.17632/ksgcv7dd4y.1
References
Avise JC (2000) Phylogeography: the history and formation of species. Harvard University Press, Cambridge, Massachusetts.
Addison JA, Hart MW (2005) Spawning, copulation and inbreeding coefficients in marine invertebrates. Biol Lett 1:450–3
Aglieri G, Papetti C, Zane L, Milisenda G, Boero F, Piraino S (2014) First evidence of inbreeding, relatedness and chaotic genetic patchiness in the holoplanktonic jellyfish Pelagia noctiluca (Scyphozoa, Cnidaria). PLoS ONE 9:e99647
Agnetta D, Bonaviri C, Badalamenti F, Scianna C, Vizzini S, Gianguzza P (2013) Functional traits of two co-occurring sea urchins across a barren/forest patch system. J Sea Res 76:170–177
Balbín R, López-Jurado JL, Flexas MM, Reglero P, Vélez-Velchí P, González-Pola C et al. (2014) Interannual variability of the early summer circulation around the Balearic Islands: Driving factors and potential effects on the marine ecosystem. J Mar Syst 138:70–81
Beerli P,Felsenstein J,(2001) Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach Proc Natl Acad Sci USA 98:4563–4568
Beerli P (2006) Comparison of Bayesian and maximum likelihood inference of population genetic parameters. Bioinformatics 22:341–345
Bonaviri C, Vega Fernández T, Fanelli G, Badalamenti F, Gianguzza P (2011) Leading role of the sea urchin Arbacia lixula in maintaining the barren state in southwestern Mediterranean. Mar Biol 158:2505–2513
Borrero‐Pérez GH, González‐Wangüemert M, Marcos C, Pérez‐Ruzafa A (2011) Phylogeography of the Atlanto‐Mediterranean sea cucumber Holothuria (Holothuria) mammata: the combined effects of historical processes and current oceanographical pattern. Mol Ecol 20:1964–1975
Boudouresque CF, Verlaque M, Azzolina JF, Meinesz A, Nédélec H, Rico V (1989) Evolution des populations de Paracentrotus lividus et d’Arbacia lixula (Echinoidea) le long d’un transect permanent à Galeria (Corse). rav Sci Parc Nat Rég Rés Nat Corse 22:65–82
Bouffard J, Pascual A, Ruiz S, Faugère Y, Tintoré J (2010) Coastal and mesoscale dynamics characterization using altimetry and gliders: a case study in the Balearic Sea. J Geophys Res 115:C10029
Broquet T, Viard F, Yearsley JM (2013) Genetic drift and collective dispersal can result in chaotic genetic patchiness. Evolution 67:1660–1675
Bulleri F, Benedetti-Cecchi L, Cinelli F (1999) Grazing by the sea urchins Arbacia lixula L. and Paracentrotus lividus Lam. in the Northwest Mediterranean. J Exp Mar Bio Ecol 241:81–95
Calderón I, Turon X (2010) Temporal genetic variability in the Mediterranean common sea urchin Paracentrotus lividus. Mar Ecol Prog Ser 408:149–159
Calderón I, Giribet G, Turon X (2008) Two markers and one history: phylogeography of the edible common sea urchin Paracentrotus lividus in the Lusitanian region. Mar Biol 154:137–151
Calderón I, Palacín C, Turon X (2009) Microsatellite markers reveal shallow genetic differentiation between cohorts of the common sea urchin Paracentrotus lividus (Lamarck) in northwest Mediterranean. Mol Ecol 18:3036–3049
Calderón I, Pita L, Brusciotti S, Palacín C, Turon X (2012) Time and space: genetic structure of the cohorts of the common sea urchin Paracentrotus lividus in Western Mediterranean. Mar Biol 159:187–197
Carmona L, Malaquias MAE, Gosliner TM, Pola M, Cervera JL (2011) Amphi-Atlantic distributions and cryptic species in Sacoglossan sea slugs. J Mollusca Stud 77:401–412
Carreras C, Ordóñez V, Zane L, Kruschel C, Nasto I, Macpherson E, Pascual M (2017) Population genomics of an endemic Mediterranean fish: differentiation by fine scale dispersal and adaptation. Sci Rep 7:43417
Casilagan ILN, Juinio-Meñez MA, Crandall ED (2013) Genetic diversity, population structure, and demographic history of exploited sea urchin populations (Tripneustes gratilla) in the Philippines. J Exp Mar Bio Ecol 449:284–293
Chapuis M-P, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Coll M, Piroddi C, Steenbeek J, Kaschner K, Ben Rais Lasram F, Aguzzi J et al. (2010) The biodiversity of the Mediterranean Sea: estimates, patterns, and threats. PLoS One 5:e11842
Consuegra S, John E, Verspoor E, De Leaniz CG (2015) Patterns of natural selection acting on the mitochondrial genome of a locally adapted fish species. Genet Sel Evol 47:58
Couvray S, Coupé S (2018) Three-year monitoring of genetic diversity reveals a micro-connectivity pattern and local recruitment in the broadcast marine species Paracentrotus lividus. Heredity 120:110
Deli T, Mohamed AB, Attia MHB, Zitari-Chatti R, Said K, Chatti N (2017) High genetic connectivity among morphologically differentiated populations of the black sea urchin Arbacia lixula (Echinoidea: Arbacioida) across the central African Mediterranean coast. Mar Biodivers. https://doi.org/10.1007/s12526-017-0832-y.
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator V2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Francour P, Boudouresque CF, Harmelin JG, Harmelin-Vivien ML, Quignard JP (1994) Are the Mediterranean waters becoming warmer? Information from biological indicators. Mar Pollut Bull 28:523–526
Garcia-Cisneros A, Valero-Jiménez C, Palacín C, Pérez-Portela R (2013) Characterization of thirty two microsatellite loci for three Atlanto-Mediterranean echinoderm species. Conserv Genet Resour 5:749–753
Garcia-Cisneros A, Palacín C, Khadra YB, Pérez-Portela R (2016) Low genetic diversity and recent demographic expansion in the red starfish Echinaster sepositus (Retzius 1816). Sci Rep 6:33269
Garcia-Cisneros A, Palacín C, Ventura CRR, Feital B, Paiva PC, Pérez-Portela R (2017) Intraspecific genetic structure, divergence and high rates of clonality in an amphi-Atlantic starfish. Mol Ecol. https://doi.org/10.1111/mec.14454.
Gerlach G, Jueterbock A, Kraemer P, Deppermann J, Harmand P (2010) Calculations of population differentiation based on G(ST) and D: forget G(ST) but not all of statistics! Mol Ecol 19:3845–3852
Gianguzza P, Agnetta D, Bonaviri C, Di Trapani F, Visconti G, Gianguzza F et al. (2011) The rise of thermophilic sea urchins and the expansion of barren grounds in the Mediterranean Sea. Chem Ecol 27:129–134
Gianguzza P, Visconti G, Gianguzza F, Vizzini S, Sarà G, Dupont S (2014) Temperature modulates the response of the thermophilous sea urchin Arbacia lixula early life stages to CO2-driven acidification. Mar Environ Res 93:70–77
Glynn F, Houghton JD, Provan J (2015) Population genetic analyses reveal distinct geographical blooms of the jellyfish Rhizostoma octopus (Scyphozoa). Biol J Linn Soc 116:582–592
Hauser L, Carvalho GR (2008) Paradigm shifts in marine fisheries genetics: ugly hypotheses slain by beautiful facts. Fish Fish 9:333–362
Hedgecock D (1994) Does variance in reproductive success limit effective population size of marine organisms? In: Beaumont A (ed) Genetics and evolution of aquatic organisms. Chapman and Hall, London, p 122–134
Hereu B, Linares C, Sala E, Garrabou J, Garcia-Rubies A, Diaz D, Zabala M (2012) Multiple processes regulate long-term population dynamics of sea urchins on Mediterranean rocky reefs. PLoS ONE 7:e36901
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Jones CG, Lawton JH, Shachak M (1994) Organisms as ecosystem engineers. In: Ecosystem management. Springer New York, NY, p 130–147
Jones CG, Lawton JH, Shachak M (1997) Positive and negative effects of organisms as physical ecosystem engineers. Ecology 78:1946–1957
Jorde E, Ryman N (1995) Temporal allele frequency change and estimation of effective size in populations with overlapping generations. Genetics 139:1077–1090
Jorde PE, Ryman N (2007) Unbiased estimator for genetic drift and effective population size. Genetics 177:927–935
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Kempf M (1962) Recherches d’écologie comparée sur Paracentrotus lividus (Lmk.) et Arbacia lixula (L.). Rec Trav Stat Mar Endoume 25:47–116
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191
Kroh A, Smith AB (2010) The phylogeny and classification of post-Palaeozoic echinoids. J Syst Palaeontol 8:147–212
Lejeusne C, Chevaldonné P, Pergent-Martini C, Boudouresque CF, Pérez T (2010) Climate change effects on a miniature ocean: the highly diverse, highly impacted Mediterranean Sea. Trends Ecol Evol 25:250–260
Lessios HA, Kessing BD, Pearse JS (2001) Population structure and speciation in tropical seas: global phylogeography of the sea urchin Diadema. Evolution 55:955–975
Lessios HA, Lockhart S, Collin R, Sotil G, Sánchez-Jerez P, Zigler KS et al. (2012) Phylogeography and bindin evolution in Arbacia, a sea urchin genus with an unusual distribution. Mol Ecol 21:130–144
Ling SD, Scheibling RE, Rassweiler A, Johnson CR, Shears N, Connell SD, et al. (2014) Global regime shift dynamics of catastrophic sea urchin overgrazing. Philos Trans R Soc B Biol Sci 370:20130269
Madeira P, Kroh A, Cordeiro R, Meireles R, Avila SP (2011) The fossil echinoids of Santa Maria Island, Azores (Northern Atlantic Ocean). Acta Geol Pol 61:243–264
Metz EC, Gomez-Gutierrez G, Vacquier VD (1998) Mitochondrial DNA and bindin gene sequence evolution among allopatric species of the sea urchin genus Arbacia. Mol Biol Evol 15:185–195
Mortensen T (1935) A monograph of the echinoidea II: Bothriocidaroida, Melonechinoida, Lepidocentroida and Stirodonta. C.A. Reitzel, Copenhagen
Narum SR (2006) Beyond Bonferroni: less conservative analyses for conservation genetics. Conserv Genet 7:783–787
Nei M, Tajima F (1981) Genetic drift and estimation of effective population size. Genetics 98:625–40
Oguz T, Macias D, Garcia-Lafuente J, Pascual A, Tintore J (2014) Fueling plankton production by a meandering frontal jet: a case study for the Alboran Sea (Western Mediterranean). PLoS ONE 9:e111482
Olivar MP, Catalán IA, Emelianov M, Fernández de Puelles ML (2003) Early stages of Sardina pilchardus and environmental anomalies in the Northwestern Mediterranean. Est Coast Shelf Sci 56:609–19
Palacín C, Turon X, Ballesteros M, Giribet G, López S (1998) Stock evaluation of three littoral echinoid species on the Catalan Coast North‐Western Mediterranean. Mar Ecol 19:163–177
Pascual A, Buongiorno Nardelli B, Larnicol G, Emelianov M, Gomis D (2002) A case of an intense anticyclonic eddy in the Balearic Sea (western Mediterranean). J Geophys Res 107:3183
Pascual M, Palero F, García-Merchán VH, Macpherson E, Robainas-Barcia A, Mestres F et al. (2016) Temporal and spatial genetic differentiation in the crab Liocarcinus depurator across the Atlantic-Mediterranean transition. Sci Rep 6:29892
Pascual M, Rives B, Scgunter C, Macpherson E (2017) Impact of life history traits on gene flow: a multispecies systematic review across oceanographic barriers in the Mediterranean Sea. PLoS ONE 12:e0176419
Peakall R, Smouse PE (2006) GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Patarnello T, Volckaert FA, Castilho R (2007) Pillars of Hercules: is the Atlantic-Mediterranean transition aphylogeographical break? Mol Ecol 16:4426–4444
Pérez-Portela R, Villamor A, Almada V (2010) Phylogeography of the sea star Marthasterias glacialis (Asteroidea, Echinodermata): deep genetic divergence between mitochondrial lineages in the north-western Mediterranean. Mar Biol 157:2015–2028
Pérez-Portela R, Turon X, Bishop JDD (2012) Bottlenecks and loss of genetic diversity: spatio-temporal patterns of genetic structure in an ascidian recently introduced in Europe. Mar Ecol Prog Ser 451:93–105
Pérez‐Portela R, Almada V, Turon X (2013) Cryptic speciation and genetic structure of widely distributed brittle stars (Ophiuroidea) in Europe. Zool Scr 42:151–169
Pérez-Portela R, Rius M, Villamor A (2017) Lineage splitting, secondary contacts and genetic admixture of a widely distributed marine invertebrate. J Biogeogr 44:446–460
Petit G, Delamare-Deboutteville C, Bougis P (1950) Le fichier faunistique du laboratoire Arago. Vie Milieu Série A Biol Mar 1:356–360
Pineda MC, Turon X, Pérez-Portela R, López-Legentil S (2016) Stable populations in unstable habitats: temporal genetic structure of the introduced ascidian Styela plicata in North Carolina. Mar Biol 163:59
Pinot JM, López-Jurado J, Riera M (2002) The CANALES experiment (1996-1998). Interannual, seasonal, and mesoscale variability of the circulation in the Balearic Channels. Prog Oceanogr 55:335–370
Plough LV (2016) Genetic load in marine animals: a review. Curr Zool 62:567–579
Pollak E (1983) A new method for estimating the effective population size from allele frequency changes. Genetics 104:531–48
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Privitera D, Noli M, Falugi C, Chiantore M (2011) Benthic assemblages and temperature effects on Paracentrotus lividus and Arbacia lixula larvae and settlement. J Exp Mar Bio Ecol 407:6–11
Raymond M, Rousset F (1995) GENEPOP: a population genetics software for exact tests and ecumenism. Vers. 1.2. J Hered 86:248–249
Renault L, Oguz T, Pascual A, Vizoso G, Tintore J (2012) Surface circulation in the Alborán Sea (western Mediterranean) inferred from remotely sensed data. J Geophys Res Ocean 117:n/a–n/a
Riesgo A, Pérez-Portela R, Pita L, Blasco G, Erwin PM, López-Legentil S (2016) Population structure and connectivity in the Mediterranean sponge Ircinia fasciculata are affected by mass mortalities and hybridization. Heredity 117:427
Rilov G (2016) Multi-species collapses at the warm edge of the warming sea. Sci Rep 6:36897
Sánchez-Garrido JC, García Lafuente J, Álvarez Fanjul E, Sotillo MG, de los Santos FJ (2013) What does cause the collapse of the Western Alboran Gyre? Results of an operational ocean model. Prog Oceanogr 116:142–153
Sanchez-Vidal A, Calafat A, Canals M, Fabres J (2004) Particle fluxes in the Almeria-Oran Front: control by coastal upwelling and sea surface circulation. J Mar Syst 52:89–106
Silva G, Lima FP, Martel P, Castilho R (2014) Thermal adaptation and clinal mitochondrial DNA variation of European anchovy. P Roy Soc Lond B Biol 281:20141093
Stefanini G (1911) Di alcune Arbacia fossili. Riv Ital di Paleontol 17:51–52
Taboada S, Pérez-Portela R (2016) Contrasted phylogeographic patterns on mitochondrial DNA of shallow and deep brittle stars across the Atlantic-Mediterranean area. Sci Rep 6:32425
Team R Core (2013) A language and environment for statistical computing.
Tommasi LR (1964) Observações sôbre Equinóides do Brasil. Rev Bras Biol 24:83–93
Tortonese E (1965) Fauna d’Italia: Vol. VI, Echinodermata. Edizioni Calderini, Bologna.
Turner TF, Wares JP, Gold JR (2002) Genetic effective size is three orders of magnitude smaller than adult census size in an abundant, Estuarine-dependent marine fish (Sciaenops ocellatus). Genetics 162:1329–39
Villamor A, Costantini F, Abbiati M (2014) Genetic structuring across marine biogeographic boundaries in rocky shore invertebrates. PLoS ONE 9:e101135
Visconti G, Gianguzza F, Butera E, Costa V, Vizzini S, Byrne M et al. (2017) Morphological response of the larvae of Arbacia lixula to near-future ocean warming and acidification. ICES J Mar Sci 74:1180–1190
Wangensteen OS, Turon X, García-Cisneros A, Recasens M, Romero J, Palacín C (2011) A wolf in sheep’s clothing: carnivory in dominant sea urchins in the Mediterranean. Mar Ecol Prog Ser 441:117–128
Wangensteen OS, Turon X, Pérez-Portela R, Palacín C (2012) Natural or Naturalized? Phylogeography suggests that the abundant sea urchin Arbacia lixula is a recent colonizer of the Mediterranean. PLoS ONE 7:e45067
Wangensteen OS (2013). Biology and phylogeography of the black sea urchin Arbacia lixula (Echinoidea: Arbacioida). PhD thesis, University of Barcelona.
Wangensteen OS, Dupont S, Casties I, Turon X, Palacín C (2013a) Some like it hot: Temperature and pH modulate larval development and settlement of the sea urchin Arbacia lixula. J Exp Mar Bio Ecol 449:304–311
Wangensteen OS, Turon X, Casso M, Palacín C (2013b) The reproductive cycle of the sea urchin Arbacia lixula in northwest Mediterranean: potential influence of temperature and photoperiod. Mar Biol 160:3157–3168
Warnes GR et al. (2016). gplots: various R programming tools for plotting data. R package version 3.0.1. The Comprehensive R Archive Network
Yeruham E, Rilov G, Shpigel M, Abelson A (2015) Collapse of the echinoid Paracentrotus lividus populations in the Eastern Mediterranean result of climate change? Sci Rep 5:13479
Zigler KS, Lessios HA (2003) 250 million years of bindin evolution. Biol Bull 205:8–15
Zigler KS, McCartney MA, Levitan DR, Lessios HA (2005) Sea urchin binding divergence predicts gamete compatibility. Evolution 59:2399
Acknowledgements
This research was financially supported by the Spanish Government projects CTM2013-48163 and CTM2017-88080 (AEI, FEDER-UE) and by a “Juan de la Cierva” contract from the Spanish Government to RPP. We are indebted to Carlos Renato Rezende Ventura for supplying us with the Brazilian samples, and to Jacob González-Solís for providing the samples from Cape Verde.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Pérez-Portela, R., Wangensteen, O.S., Garcia-Cisneros, A. et al. Spatio-temporal patterns of genetic variation in Arbacia lixula, a thermophilous sea urchin in expansion in the Mediterranean. Heredity 122, 244–259 (2019). https://doi.org/10.1038/s41437-018-0098-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0098-6
This article is cited by
-
Shedding light on variation in reproductive success through studies of population genetic structure in a Southeast Pacific Coast mussel
Heredity (2023)
-
Exceptional population genomic homogeneity in the black brittle star Ophiocomina nigra (Ophiuroidea, Echinodermata) along the Atlantic-Mediterranean coast
Scientific Reports (2023)
-
Spatial coalescent connectivity through multi-generation dispersal modelling predicts gene flow across marine phyla
Nature Communications (2022)
-
Genetic homogeneity, lack of larvae recruitment, and clonality in absence of females across western Mediterranean populations of the starfish Coscinasterias tenuispina
Scientific Reports (2021)
-
Individual-based population genomics reveal different drivers of adaptation in sympatric fish
Scientific Reports (2020)
