
Abstract
An accurate diagnosis of syndromic craniosynostosis (CS) is important for personalized treatment, surveillance, and genetic counselling. We describe detailed clinical criteria for syndromic CS and the distribution of genetic diagnoses within the cohort. The prospective registry of the Norwegian National Unit for Craniofacial Surgery was used to retrieve individuals with syndromic CS born between 1 January 2002 and 30 June 2019. All individuals were assessed by a clinical geneticist and classified using defined clinical criteria. A stepwise approach consisting of single-gene analysis, comparative genomic hybridization (aCGH), and exome-based high-throughput sequencing, first filtering for 72 genes associated with syndromic CS, followed by an extended trio-based panel of 1570 genes were offered to all syndromic CS cases. A total of 381 individuals were registered with CS, of whom 104 (27%) were clinically classified as syndromic CS. Using the single-gene analysis, aCGH, and custom-designed panel, a genetic diagnosis was confirmed in 73% of the individuals (n = 94). The diagnostic yield increased to 84% after adding the results from the extended trio-based panel. Common causes of syndromic CS were found in 53 individuals (56%), whereas 26 (28%) had other genetic syndromes, including 17 individuals with syndromes not commonly associated with CS. Only 15 individuals (16%) had negative genetic analyses. Using the defined combination of clinical criteria, we detected among the highest numbers of syndromic CS cases reported, confirmed by a high genetic diagnostic yield of 84%. The observed genetic heterogeneity encourages a broad genetic approach in diagnosing syndromic CS.
Similar content being viewed by others

Introduction
Craniosynostosis (CS) is one of the most common inborn anomalies in children, affecting 1/1600–1/1800 live births [1, 2]. CS is classified into syndromic and nonsyndromic CS, where syndromic CS is reported to constitute 12–31% of all cases [3,4,5]. Individuals with syndromic CS have an increased risk of additional complications and repeat craniofacial surgery [6, 7], and need to be identified. Hence, an accurate molecular diagnosis is important for personalized treatment and surveillance, in addition to genetic counselling, family planning, social care, and support from patient organizations.
Previously, syndromic CS was defined by the occurrence of one of the frequent and well-known syndromes: Apert, Muenke, Saethre–Chotzen, Pfeiffer, or Crouzon, caused by genetic variants in the FGFR2, FGFR3, TWIST1, FGFR1/2, and FGFR2 genes, respectively [1, 8]. High-throughput sequencing (HTS) has improved and changed the diagnostics of syndromic CS over the last two decades, and genetic variants in at least 80 genes are known to cause syndromic CS [9, 10].
There is no clear consensus regarding the definition of syndromic CS. Some studies limit their cohort to a defined selection of verified genetic diagnoses [11, 12], whereas others limit their cohort to affected sutures only, as complex or coronal synostoses are more commonly associated with syndromic CS [12, 13], or use a combination of clinical criteria [5, 14]. A recent population-based epidemiological study from our group demonstrated a high proportion of syndromic cases of 27% defined by clinical criteria and a genetic detection rate of 75% after testing with array comparative genomic hybridization (aCGH) and exome-based HTS, filtering for 72 genes associated with syndromic CS [2]. We detected many midline synostoses in individuals with syndromic CS, in particular in individuals with rare genetic syndromes [2], suggesting that an affected suture alone does not provide sufficient evidence to determine whether an individual has syndromic or nonsyndromic CS. We hypothesized that a broader approach to genetic testing would further increase the diagnostic yield.
In this study, all individuals with syndromic CS born between 1 January 2002 and 30 June 2019, selected by clinical criteria, and registered in the registry of the Norwegian National Unit for Craniofacial Surgery were included. Supplemental genetic diagnostics of HTS filtering for a panel of 1570 genes informed by the Deciphering Developmental Delay study (DDG2P) were offered for negative cases. We present a large variety of genetic syndromes and aim to propose a strategy for clinical classification and genetic testing of individuals with syndromic CS.
Materials and methods
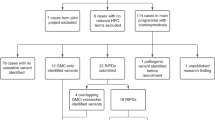
The study was approved by the Norwegian Regional Committees for Medical and Health Research Ethics (REK_2018/797) and by Oslo University Hospital (permit number P360:18/05374). Informed consent was obtained from all individuals that participated in the study. Since 2001, all individuals in Norway with suspected CS have been referred to the Norwegian National Unit for Craniofacial Surgery at Oslo University Hospital for diagnostics, treatment, and follow-up [2]. Individuals suspected of having syndromic CS are seen regularly by the unit’s multidisciplinary team, including a clinical geneticist. The unit’s registry is prospective and includes all consenting individuals diagnosed with CS (85%) [2]. Individuals with CS born between 1 January 2002 and 30 June 2019 and registered by 23 October 2019 were included in the study (n = 381). The database was updated January 2020 to include the latest genetic results. Syndromic CS was defined by a combination of clinical criteria, formulated by the authors, with one major criterion or two or more minor criteria; details are presented in Fig. 1. All individuals were classified by the same two clinical geneticists prior to inclusion (ET and KRH).The genetic analyses were offered stepwise. Individuals suspected of having one of the common and well-described CS syndromes were initially tested by single-gene analysis of FGFR2, FGFR3, TWIST1 or EFNB1. If the results came back negative, aCGH was performed. When the clinical presentation did not resemble one of the common CS syndromes, aCGH was offered initially. From 2016, exome-based HTS filtering for a custom-designed panel of 72 genes associated with syndromic CS (Supplemental Table 1) was performed if the result of the aCGH came back negative. If this did not result in a genetic diagnosis the extended trio-based HTS panel of 1570 genes was offered. A few individuals (n = 6) were diagnosed prior to assessment by the unit’s team. Their findings are presented in the results section under the diagnostic tool in which they would have been found in the stepwise approach (Tables 2–4). Ten individuals were excluded from the calculations of diagnostic yield, because they did not want genetic testing (n = 4), and were analysed with aCGH only (n = 5) or with single gene and aCGH only (n = 1). Individuals analysed with aCGH and HTS filtering for the custom-designed panel only (n = 3) were included in the calculations. All individuals with nonsyndromic CS of the coronal suture(s), or with an affected first-degree relative, were offered the custom-designed HTS panel due to the risk of having a monogenetic cause (e.g., TCF12). As genetic causes of nonsyndromic CS is not the scope of this study, these results are not included. Blood samples were obtained from all patients, followed by DNA extraction with QiaSymphony DSP DNA Mini Kit (Qiagen, Cologne, Germany). For Sanger sequencing of FGFR2, FGFR3, TWIST1, and EFNB1, primers were designed using primer3 software, sequencing was done on an ABI 3730 sequencer (Applied Biosystems, Life Technologies, CA, USA), and sequence data were analysed using SeqScape v2.7 (Life Technologies, CA, USA). For MLPA of TWIST1, the Salsa MLPA Probemix P054 (MRC Holland) was used. Array CGH was performed using Agilent 180 K SurePrint G3 Human CGH (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s recommendations. Data were processed with Feature Extraction and DNA Analytics (Agilent Technologies). Exome-based HTS was performed by using Agilent SureSelectXT Target Enrichment 50 Mb Kit (Agilent Technologies, Santa Clara, CA, USA) for library preparation and Illumina HiSeq 2500 in high‐output run mode. Bioinformatic handling of the sequencing data followed the practice from Genome Analysis Tool Kit for exome sequencing [15]. Raw reads were mapped to the reference sequence (GRCh37/hg19). Joint variant calling was performed within each trio. Variant annotation was done by Annovar [16]. Downstream filtering and analysis were done with Filtus [17] on the variants within coding regions and intron/exon boundaries of the custom-designed panel or the extended trio-based panel of 1570 genes. The extended trio-based panel was informed by the Deciphering Developmental Disorders study (DDG2P) [18] and was the largest panel available at our laboratory. We selected variants with allele frequency of less than 0.5% (for genes inherited as autosomal dominant) or less than 1% (for other inheritance patterns), as reported in gnomAD [19]. Variants were classified according to the guidelines by the American College of Medical Genetics and Genomics [20], and only class 4 (likely pathogenic) and class 5 (pathogenic) variants were included in the results. All variants were submitted to ClinVar (SCV001437545–SCV001437592).

Minor criteria are presented in the dark blue panel and major criteria in the red panels. Syndromic CS is defined by the addition of two or more minor criteria or one major criterion.
Results
In total, 381 individuals were registered with CS, of which 104 (27%) were clinically classified as syndromic based on the criteria presented in Fig. 1. A total of 94 individuals with syndromic CS (90%) accepted the stepwise genetic testing presented in the method section. By single-gene analysis, aCGH and the custom-designed panel, a genetic diagnosis was confirmed in 69 individuals (73%; Figs. 1 and 2). When including the results of the extended trio-based HTS panel, the number of genetically confirmed diagnoses increased to 79 (84%; Figs. 1 and 2, Supplemental Table 2). When excluding the CS syndromes caused by variants in the FGFR2, FGFR3, TWIST1, or EFNB1 gene, a genetic cause was confirmed in 26 individuals (28%), partitioned into 23 different genetic or chromosomal causes, 16 of these not commonly associated with CS (Tables 1–4). Fifteen individuals (16%) had negative genetic test results (Fig. 2).

The distribution of confirmed diagnoses is given in absolute numbers.
Fifty-three individuals (56%) had variants in one of the genes frequently associated with CS syndromes (FGFR2, FGFR3, TWIST1, and EFNB1). Of these, 47 individuals (89%) had a clinical phenotype in concordance with the genetic diagnosis and were diagnosed by single-gene analysis (Table 1).
Ten individuals (11%) had a de novo copy number variation associated with a known microdeletion or duplication syndrome (Fig. 2); seven of these are not commonly associated with CS (Table 2). In addition, one case of Saethre–Chotzen syndrome, caused by a deletion including the TWIST1 gene, was detected by aCGH (Table 2).
Of the 11 individuals diagnosed by the custom-designed HTS panel, seven had clinical phenotype in concordance with their genetic diagnosis, while four had unexpected clinical presentation (Table 3). A girl with an EFNB1 variant had a complex heart malformation not associated with craniofrontonasal syndrome. A boy with classic features of craniofrontonasal syndrome was not analysed by single-gene analysis due to his gender. However, HTS revealed that he was mosaic for a variant in the EFNB1 gene (Table 3) and karyotyping confirmed XY, male. In a boy with Crouzon-like appearance, with negative result of a FGFR2 analysis, HTS detected a variant in TWIST1 consistent with Saethre–Chotzen syndrome. A homozygous variant in IL11RA consistent with CS and dental anomalies syndrome was detected in a girl with late-occurring pansynostosis (4 years old) and no dental anomalies (Table 3). In addition, we detected two cases of parental mosaicism for variants in FGFR2 and ZIC1, respectively, both associated with autosomal dominant inheritance (Table 3). The individuals with the FGFR2 variant were siblings and not analysed by single-gene analysis due to the suspicion of autosomal recessive inheritance.
We performed the analysis using the extended trio-based HTS panel on 22 individuals and revealed a diagnosis in 10; these were partitioned into 9 genetic syndromes, none of them commonly reported to include CS (Table 4). We detected two individuals with variants in the AHDC1 gene, consistent with Xia–Gibbs syndrome. We further confirmed the following diagnoses: coloboma, congenital heart defects, choanal atresia, retardation of growth, developmental delay, genital abnormalities, ear abnormalities and deafness (CHARGE) syndrome, Bainbridge–Ropers syndrome (BRPS), CHDFIDD (Congenital heart defects, dysmorphic facial features, and intellectual developmental disorder, previously published [21]), Kleefstra syndrome, Genitopatellar syndrome, Floating–Harbor syndrome, Alpha-Mannosidosis (previously published [22]), and Malan syndrome (Table 4).
Discussion
In our 18-year population-based cohort of children with CS, 27% fulfilled the presented clinical criteria and were diagnosed with syndromic CS. This is the highest number of syndromic cases reported from a population-based cohort and we believe the high genetic diagnostic yield of 84% supports the clinical criteria. We found a high level of genetic heterogeneity, with variants in common and well-known genes associated with CS accounting for 67% of the solved cases; the remaining cases were distributed across a diverse range of genetic syndromes, many of which are not commonly associated with CS.
We detected mosaicism in four families: one index individual and three healthy parents (Tables 2 and 3). A variant in the EFNB1 gene was detected in a male with classic features of craniofrontonasal syndrome by HTS analysis. The variant presented as heterozygous in the analysis, suggesting mosaicism. The EFNB1 gene is located on the X chromosome and loss-of-function variants in the EFNB1 gene are assumed to cause craniofrontonasal syndrome through a paradoxical gender reversal in severity, where females usually develop typical features of craniofrontonasal syndrome and males usually have hypertelorism as the only feature. Random X-inactivation is assumed to be the cause of the severe phenotype in females, causing cellular interference as the cells have different expressions of EPHRIN-B1, generating abnormal tissue boundaries [23]. It has previously been proposed that males, being mosaic for variants in the EFNB1 gene, will present with a severe phenotype, similar to females, due to the different expression of EPHRIN-B1, which is not tolerated [23]. Our results support this. We further detected low-grade mosaicism for a variant in the FGFR2 gene in a healthy parent of two children with Crouzon syndrome and for a deletion (including the TWIST1 gene) in a healthy father of a child with Saethre–Chotzen syndrome. Parental mosaicism for FGFR2 and TWIST1 variants is previously described [24, 25]. Crouzon and Saethre–Chotzen syndrome are inherited in an autosomal dominant manner, and this finding is important for genetic guidance, as it will impact the recurrence risk. Parental mosaicism for a ZIC1 variant led to the variant initially being missed in the Trio-HTS analysis (filtering for de novo variants) in a boy with CRS6 and was only detected after manual re-evaluation of the gene due to his classical phenotype (Table 3). These cases demonstrate the need for a thorough evaluation of a well-described clinical phenotype, as diagnosis may be missed on trio analysis due to parental mosaicism.
We diagnosed syndromes not commonly associated with CS in 17 individuals, of whom 10 were detected by the extended trio-based panel and 7 by aCGH. We demonstrate an expansion of the clinical phenotype beyond CS in some cases (Table 4). Interestingly, all cases of rare syndromes detected by the extended trio-based panel, with two exceptions, had synostosis of a single midline suture only (Table 4). Likewise, seven out of ten microdeletion syndromes (Table 2) had midline synostosis only. This contrasts with the pattern typically seen in individuals with syndromic CS, where multiple suture synostosis is the most common finding [5, 26], and also with our finding in individuals with the more common CS syndromes (Tables 1 and 3). The most common reported CS syndromes have a high frequency of CS and are caused by genes acting in signalling pathways important for the development of the cranial sutures, mostly associated with osteogenic differentiation of stem cells (FGF/FGFR, Eph/Ephrin, TGFbeta/BMP, WNT) [27, 28]. The difference in affected sutures between the common CS syndromes and the rare or ultra-rare syndromes, with a low frequency of CS caused by genes acting in other pathways, might indicate that the synostoses in these two groups have different molecular mechanisms. Individuals with rare genetic syndromes which includes macrocephaly (e.g., Malan syndrome) might also be at higher risk of developing CS due to foetal head constraints that are associated with CS, especially regarding coronal premature fusion [27, 29].
Notably, in our cohort we detected several Mendelian disorders of chromatin modification (chromatinopathies), including (with the associated gene in parentheses): CHARGE (CHD7), Kleefstra (EHMT1), Floating–Harbor syndrome (SRCAP), KAT6B-related disorders (KAT6B), and 2q37 deletion syndrome (caused by haploinsufficiency of the HDAC4 gene [30]). These genes influence the epigenetic machinery by targeting the DNA or the DNA-associated histone proteins, and variants that affect function are expected to have widespread epigenetic consequences [31, 32]. Approximately 44 chromatinopathies have been described to date. The most common mechanism is presumed to be haploinsufficiency, as a majority of the individuals have a loss-of-function variant [32]; this concords with our results (Tables 2 and 4). A few of the chromatinopathies have previously been associated with CS: Kabuki syndrome, Bohring–Opitz syndrome (BOS), and two cases of KAT6B-related disorders [31,32,33,34,35,36]. To our knowledge, only one case of CS in CHARGE syndrome [37], one case in Floating–Harbor syndrome [38], one case in 2q37 deletion syndrome [30], and none in Kleefstra syndrome have been reported. This study confirms CS as a feature of CHARGE syndrome, Floating–Harbor syndrome, KAT6B-related disorders, and suggests CS as a feature in Kleefstra syndrome and 2q37 deletion syndrome. We cannot be certain that haploinsufficiency of the HDAC4 gene is the cause of CS in this case, as the individual also had a duplication on 11p15 in concordance with Silver–Russell syndrome. However, Silver–Russell syndrome is not associated with CS but rather delayed fontanelle closure. The presence of CS in several chromatinopathies at a low frequency adds to reports of other low-frequent malformations in these disorders. Their presence may be dependent on the molecular characteristics of the targeted genes, in addition to a general disruption of the epigenetic machinery; these are both suggested mechanisms for this phenotypic variability [31, 32, 39, 40]. Clinically, these findings suggest that individuals with chromatinopathies should be monitored for CS, in addition to other organ anomalies.
BRPS has phenotypic overlap with BOS. The former is caused by loss-of-function variants in the ASXL3 gene and the latter by variants in the ASXL1 gene. However, metopic synostosis, often seen in BOS, is not commonly reported in BRPS [41, 42]. Our case confirms that metopic synostosis is a rare feature in BRPS. CS has been reported in a very few individuals with CHDFIDD, Xia–Gibbs, Alpha-mannosidosis, and Malan syndrome [10, 43,44,45]. Individuals with Diamond–Blackfan anaemia have not been reported with CS.
Syndromic CS may be subdivided into syndromes with high risk of developing CS and a multitude of diverse syndromes usually defined by extracranial features with a low risk of developing CS. Due to the rarity of many syndromes, it is to be expected that the list defining the latter group is incomplete. Our results may point to a greater risk in subgroups of syndromes, such as the chromatinopathies.
Supported by our high diagnostic yield, we argue for the use of the presented clinical criteria, to ensure that all individuals with syndromic CS are identified, and thereby offered a broad genetic approach and assessment in a multidisciplinary team. For research purposes, a common clinical definition of syndromic CS is important to make reliable comparisons across cohorts. For some individuals, the features, indicating syndromic CS will not be present when the CS is evident. This argues for clinical follow-up after surgery for all individuals with CS. We recommend assessment of all individuals with syndromic CS in a multidisciplinary team to identify additional anomalies and progressive disturbances in facial growth, which may require repeat craniofacial surgeries [6, 7]. A high number of the syndromic cases in our cohort had a rare or ultra-rare genetic cause, mostly due to variants in different genes, emphasizing that syndromic CS is highly heterogeneous. This argues for a broad genetic approach. We suggest stepwise testing initiated by a custom-based HTS panel and aCGH, as the majority of the confirmed diagnoses were detected by these two analyses. In addition, our study showed that a number of variants were inherited from parents (including mosaics), all likely to be missed on the extended trio-based HTS panel. We then recommend trio-analyses, applying an extended panel of genes associated with development delay/anomalies in general, for negative cases. If the clinical presentation is highly suspicious of one of the frequent CS syndromes, one might consider testing the FGFR2, FGFR3, TWIST1, or EFNB1 genes first; however, as this and other studies [46] have shown, a number of individuals have atypical presentations.
The main strength of the study is that the data are population-based and prospectively collected. Norway has an equal-access healthcare system that ensures a high inclusion rate. The unit is organized as a centralized multidisciplinary team, including a clinical geneticist. The clinical geneticist reassesses individuals initially diagnosed with nonsyndromic CS when new findings or difficulties present. A limitation of the study is that individuals diagnosed with CS over the last two or three years may not yet have presented with additional findings; thus, some syndromic cases may have been missed and the true number might be slightly higher. In syndromes not previously associated with CS, we cannot exclude the possibility of an additional genetic diagnosis associated with CS not detected by today’s methods (e.g., deep intronic variants). Newly associated genes, such as SMAD6, recently documented to be an important cause of CS [47], were not included in the panels. In addition, MLPA of EFNB1 and TCF12 were not available at our laboratory. According to this some diagnoses may have been missed. In addition, a few individuals included in the calculations were not analysed with the extended trio-based HTS panel (n = 3). This could mean that the genetic detection rate should be even higher.
Conclusion
Using the presented clinical criteria, we identified one of the highest numbers of syndromic CS cases reported, strongly supported by a high genetic detection rate of 84%. The observed genetic heterogeneity and atypical presentations encourage a broad genetic approach in diagnosing syndromic CS. Surveillance for CS is recommended in a variety of genetic syndromes, including syndromes rarely associated with CS, such as the chromatinopathies, for the purpose of early diagnosis and treatment.
References
Kweldam CF, van der Vlugt JJ, van der Meulen JJ. The incidence of craniosynostosis in the Netherlands, 1997-2007. J Plast Reconstr Aes. 2011;64:583–8.
Tønne E, Due-Tonnessen BJ, Wiig U, Stadheim BF, Meling TR, Helseth E, et al. Epidemiology of craniosynostosis in Norway. J Neurosurg Pediatr. 2020. https://doi.org/10.3171/2020.1.PEDS2051.
Neusel C, Class D, Eckert AW, Firsching R, Gobel P, Gotz D, et al. Multicentre approach to epidemiological aspects of craniosynostosis in Germany. Br J Oral Max Surg. 2018;56:881–6.
Wilkie AOM, Johnson D, Wall SA. Clinical genetics of craniosynostosis. Curr Opin Pediatr. 2017;29:622–8.
Lattanzi W, Barba M, Di Pietro L, Boyadjiev SA. Genetic advances in craniosynostosis. Am J Med Genet A 2017;173:1406–29.
Wilkie AO, Byren JC, Hurst JA, Jayamohan J, Johnson D, Knight SJ, et al. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics 2010;126:e391–400.
Meling TR, Due-Tonnessen BJ, Hogevold HE, Skjelbred P, Arctander K. Monobloc distraction osteogenesis in pediatric patients with severe syndromal craniosynostosis. J Craniofac Surg. 2004;15:990–1000.
Heuze Y, Holmes G, Peter I, Richtsmeier JT, Jabs EW. Closing the gap: Genetic and genomic continuum from syndromic to nonsyndromic craniosynostoses. Curr Genet Med Rep. 2014;2:135–45.
Goos JAC, Mathijssen IMJ. Genetic causes of craniosynostosis: an update. Mol Syndromol. 2019;10:6–23.
Gumus E. Extending the phenotype of Xia-Gibbs syndrome in a two-year-old patient with craniosynostosis with a novel de novo AHDC1 missense mutation. Eur J Med Genet. 2020;63:103637.
Lee HQ, Hutson JM, Wray AC, Lo PA, Chong DK, Holmes AD, et al. Changing epidemiology of nonsyndromic craniosynostosis and revisiting the risk factors. J Craniofac Surg. 2012;23:1245–51.
Cornelissen M, Ottelander B, Rizopoulos D, van der Hulst R, Mink van der Molen A, van der Horst C, et al. Increase of prevalence of craniosynostosis. J Craniomaxillofac Surg. 2016;44:1273–9.
Topa A, Rohlin A, Andersson MK, Fehr A, Lovmar L, Stenman G, et al. NGS targeted screening of 100 Scandinavian patients with coronal synostosis. Am J Med Genet A 2020;182:348–56.
Johnson D, Wilkie AO. Craniosynostosis. Eur J Hum Genet. 2011;19:369–76.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Vigeland MD, Gjotterud KS, Selmer KK. FILTUS: a desktop GUI for fast and efficient detection of disease-causing variants, including a novel autozygosity detector. Bioinformatics 2016;32:1592–4.
Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–8.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–43.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Hamilton MJ, Caswell RC, Canham N, Cole T, Firth HV, Foulds N, et al. Heterozygous mutations affecting the protein kinase domain of CDK13 cause a syndromic form of developmental delay and intellectual disability. J Med Genet. 2018;55:28–38.
Riise Stensland HM, Klenow HB, Van Nguyen L, Hansen GM, Malm D, Nilssen O. Identification of 83 novel alpha-mannosidosis-associated sequence variants: functional analysis of MAN2B1 missense mutations. Hum Mutat. 2012;33:511–20.
Twigg SR, Babbs C, van den Elzen ME, Goriely A, Taylor S, McGowan SJ, et al. Cellular interference in craniofrontonasal syndrome: males mosaic for mutations in the X-linked EFNB1 gene are more severely affected than true hemizygotes. Hum Mol Genet. 2013;22:1654–62.
Goriely A, Lord H, Lim J, Johnson D, Lester T, Firth HV, et al. Germline and somatic mosaicism for FGFR2 mutation in the mother of a child with Crouzon syndrome: Implications for genetic testing in “paternal age-effect” syndromes. Am J Med Genet A 2010;152a:2067–73.
Apostolopoulou D, Kaxira OS, Hatzaki A, Panagopoulos KP, Alexandrou K, Stratoudakis A, et al. Genetic analysis of syndromic and nonsyndromic patients with craniosynostosis identifies novel mutations in the TWIST1 and EFNB1 Genes. Cleft Palate Craniofac J. 2018;55:1092–102.
Di Rocco F, Arnaud E, Meyer P, Sainte-Rose C, Renier D. Focus session on the changing “epidemiology” of craniosynostosis (comparing two quinquennia: 1985-1989 and 2003-2007) and its impact on the daily clinical practice: a review from Necker Enfants Malades. Childs Nerv Syst. 2009;25:807–11.
Wu X, Gu Y. Signaling mechanisms underlying genetic pathophysiology of craniosynostosis. Int J Biol Sci. 2019;15:298–311.
Twigg SR, Wilkie AO. A genetic-pathophysiological framework for craniosynostosis. Am J Hum Genet. 2015;97:359–77.
Sanchez-Lara PA, Carmichael SL, Graham JM Jr., Lammer EJ, Shaw GM, Ma C, et al. Fetal constraint as a potential risk factor for craniosynostosis. Am J Med Genet A 2010;152a:394–400.
Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, McLeod DR, et al. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am J Hum Genet. 2010;87:219–28.
Zollino M, Lattante S, Orteschi D, Frangella S, Doronzio PN, Contaldo I, et al. Syndromic Craniosynostosis can define new candidate genes for suture development or result from the non-specifc effects of pleiotropic genes: rasopathies and chromatinopathies as examples. Front Neurosci. 2017;11:587.
Bjornsson HT. The Mendelian disorders of the epigenetic machinery. Genome Res. 2015;25:1473–81.
Topa A, Samuelsson L, Lovmar L, Stenman G, Kolby L. On the significance of craniosynostosis in a case of Kabuki syndrome with a concomitant KMT2D mutation and 3.2 Mbp de novo 10q22.3q23.1 deletion. Am J Med Genet A 2017;173:2219–25.
Dong C, Umar M, Bartoletti G, Gahankari A, Fidelak L, He F. Expression pattern of Kmt2d in murine craniofacial tissues. Gene Expr Patterns. 2019;34:119060.
Dangiolo SB, Wilson A, Jobanputra V, Anyane-Yeboa K. Bohring-Opitz syndrome (BOS) with a new ASXL1 pathogenic variant: Review of the most prevalent molecular and phenotypic features of the syndrome. Am J Med Genet A 2015;167a:3161–6.
Bashir RA, Dixit A, Goedhart C, Parboosingh JS, Innes AM, Ferreira P, et al. Lin-Gettig syndrome: craniosynostosis expands the spectrum of the KAT6B related disorders. Am J Med Genet A 2017;173:2596–604.
Siakallis L, Tan AP, Chorbachi R, Mankad K. A unique case of CHARGE syndrome with craniosynostosis. Childs Nerv Syst. 2019;35:11–3.
Hersh JH, Groom KR, Yen FF, Verdi GD. Changing phenotype in Floating-Harbor syndrome. Am J Med Genet. 1998;76:58–61.
Poot M. Structural genome variations related to craniosynostosis. Mol Syndromol. 2019;10:24–39.
Law MJ, Lower KM, Voon HP, Hughes JR, Garrick D, Viprakasit V, et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 2010;143:367–78.
Bacrot S, Mechler C, Talhi N, Martin-Coignard D, Roth P, Michot C, et al. Whole exome sequencing diagnoses the first fetal case of Bainbridge-Ropers syndrome presenting as pontocerebellar hypoplasia type 1. Birth Defects Res. 2018;110:538–42.
Chinen Y, Nakamura S, Ganaha A, Hayashi S, Inazawa J, Yanagi K, et al. Mild prominence of the Sylvian fissure in a Bainbridge-Ropers syndrome patient with a novel frameshift variant in ASXL3. Clin Case Rep. 2018;6:330–6.
Bostwick BL, McLean S, Posey JE, Streff HE, Gripp KW, Blesson A, et al. Phenotypic and molecular characterisation of CDK13-related congenital heart defects, dysmorphic facial features and intellectual developmental disorders. Genome Med. 2017;9:73.
Klaassens M, Morrogh D, Rosser EM, Jaffer F, Vreeburg M, Bok LA, et al. Malan syndrome: Sotos-like overgrowth with de novo NFIX sequence variants and deletions in six new patients and a review of the literature. Eur J Hum Genet. 2015;23:610–5.
Grabb PA, Albright AL, Zitelli BJ. Multiple suture synostosis, macrocephaly, and intracranial hypertension in a child with alpha-D-mannosidase deficiency. Case Rep J Neurosurg. 1995;82:647–9.
Miller KA, Twigg SR, McGowan SJ, Phipps JM, Fenwick AL, Johnson D, et al. Diagnostic value of exome and whole genome sequencing in craniosynostosis. J Med Genet. 2017;54:260–8.
Calpena E, Cuellar A, Bala K, Swagemakers SMA, Koelling N, McGowan SJ, et al. SMAD6 variants in craniosynostosis: genotype and phenotype evaluation. Genet Med. 2020;22:1498–506.
Acknowledgements
We thank the Department of Medical Genetics at Haukeland University Hospital and the Department of Medical Genetics at the University Hospital of North Norway for diagnosing some individuals in our cohort. We thank Grete Furseth and Elisabeth Elgesem for their excellent secretarial work. We greatly appreciate the contribution and goodwill from included individuals and families.
Funding
This work is supported by the Norwegian National Advisory Unit on Rare Disorders with a grant covering salary for one year to Elin Tønne.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tønne, E., Due-Tønnessen, B.J., Mero, IL. et al. Benefits of clinical criteria and high-throughput sequencing for diagnosing children with syndromic craniosynostosis. Eur J Hum Genet 29, 920–929 (2021). https://doi.org/10.1038/s41431-020-00788-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-00788-4
This article is cited by
-
De novo mutations in the BMP signaling pathway in lambdoid craniosynostosis
Human Genetics (2023)
-
Clinical genomics—but faster
European Journal of Human Genetics (2021)
-
Missense and truncating variants in CHD5 in a dominant neurodevelopmental disorder with intellectual disability, behavioral disturbances, and epilepsy
Human Genetics (2021)
