
Abstract
Astrocytes are involved in the neuroinflammation of neurodegenerative diseases, such as Parkinson’s disease (PD). Among the numerous inflammatory cytokines, interleukin-1β (IL-1β) produced by astrocytic Nod-like receptor protein (NLRP) inflammasome is crucial in the pathogenesis of PD. β-arrestin2-mediated dopamine D2 receptor (Drd2) signal transduction has been regarded as a potential anti-inflammatory target. Our previous study revealed that astrocytic Drd2 suppresses neuroinflammation in the central nervous system. However, the role of Drd2 in astrocytic NLRP3 inflammasome activation and subsequent IL-1β production remains unclear. In the present study, we used 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced PD mouse model to investigate whether Drd2 could suppress astrocytic NLRP3 inflammasome activation. We showed that Drd2 agonist inhibited NLRP3 inflammasome activation, evidenced by decreased caspase-1 expression and reduced IL-1β release in the midbrain of wild type mice. The anti-inflammasome effect of Drd2 was abolished in β-arrestin2 knockout and β-arrestin2 small interfering RNA-injected mice, suggesting a critical role of β-arrestin2 in Drd2-regulated NLRP3 inflammasome activation. We also found that Drd2 agonists suppressed the upregulation of caspase-1 and IL-1β expression in primary cultured mouse astrocytes in response to the activation of NLRP3 inflammasome induced by lipopolysaccharide plus adenosine triphosphate. Furthermore, we demonstrated that β-arrestin2 mediated the inhibitory effect of Drd2 on NLRP3 inflammasome activation via interacting with NLRP3 and interfering the inflammasome assembly. Collectively, our study illustrates that astrocytic Drd2 inhibits NLRP3 inflammasome activation through a β-arrestin2-dependent mechanism, and provides a new strategy for treatment of PD.
Similar content being viewed by others

Introduction
Astrocytes are the dominant cells in quantity in the central nervous systems (CNS). They are derived from the neuroectoderm and are of great significance in CNS homeostasis and pathology [1, 2]. Upon the inflammatory stimulations, astrocytes are activated to induce the upregulation of glial fibrillary acidic protein (GFAP) expression [3], secretion of inflammatory mediators [4], and altered expression of astrocytic G protein-coupled receptors (GPCRs) [5]. Increasing evidence suggests that activated astrocytes play a vital role in neuroinflammation in the ageing brain and most neurodegenerative diseases (NDDs). As one of the most common NDDs, Parkinson’s disease (PD) is pathologically characterized by the progressive loss of dopaminergic neurons in the substantia nigra [6] and the intracytoplasmic Lewy body inclusions [7]. Although the exact pathogenic mechanisms of PD remain unclear [8], reactive astrogliosis and astrocyte-mediated neuroinflammation have been recognized to play critical role in the pathogenesis of PD [9, 10].
Neuroinflammation is a common feature of the ageing brain and most NDDs [11, 12]. It is mediated predominantly by activated glial cells and is accompanied by the production of inflammatory cytokines. Specifically, astrocytes can amplify microglia-derived inflammatory response and lead to feedback loop of neuroinflammation [13]. Among the numerous inflammatory cytokines, interleukin-1β (IL-1β) produced by astrocytic Nod-like receptor protein (NLRP) inflammasome exerts a central role in regulating neuroinflammation [14]. Upon stimulation by adenosine triphosphate (ATP), reactive oxygen species, lysosomal contents or other factors, NLRP3 recruits the adapter molecule apoptosis-related speck-like protein (ASC) and pro-caspase-1 to promote caspase-1 activation. This process leads to the maturation of the pro-inflammatory cytokines (IL-1β, IL-18). Secretion of IL-1β by glial cells contributes towards the destruction of dopaminergic neurons in the brain of PD patients [15] and the initiation of cell pyroptotic death [16, 17]. Hence, suppression of NLRP3 inflammasome-derived IL-1β production may be beneficial to PD patients.
Dopamine D2 receptor (Drd2) has been regarded as a potential anti-inflammatory target in the therapy of NDDs [18,19,20]. Drd2 couples to Gαi protein and expresses both in neurons and astrocytes [21]. Our previous study revealed that astrocytic Drd2 controls the expression of anti-inflammatory protein αB-crystallin and helps maintaining the immune homeostasis in the CNS [22]. However, the role of Drd2 in astrocytic NLRP3 inflammasome activation and subsequent IL-1β production has not been defined yet. There are typically two types of signal transduction pathways that Drd2 participates to trigger cellular responses: the classic G protein-dependent pathway and the β-arrestins-dependent pathway [23]. β-arrestins are recruited to GPCRs and triggers receptor desensitization and internalization [24]. Besides, β-arrestins also trigger diverse signaling pathways regulating cell proliferation and apoptosis, leading to multitudinous physiological responses [25]. Over the last decade, the anti-inflammatory roles of β-arrestins in the treatment of NDDs have become increasingly appreciated [24, 26]. A recent study showed that β-arrestin2 acts downstream of GPR120 and GPR40 to inhibit inflammasome activation in bone-marrow-derived macrophages [27]. However, the inhibitory role of β-arrestins on inflammasome in astrocytes has not been well studied. We hypothesize that β-arrestin2 may negatively regulate astrocytic NLRP3 inflammasome activation in the pathogenesis of PD.
In the present study, we used 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD mouse model to investigate whether Drd2 can suppress astrocytic NLRP3 inflammasome activation. We showed that Drd2 agonist inhibited NLRP3 inflammasome activation in vivo and in vitro, and the anti-inflammasome effect of Drd2 was abolished in β-arrestin2 deficiency mice, suggesting the critical role of β-arrestin2 in Drd2-regulated NLRP3 inflammasome activation. Furthermore, we found that β-arrestin2 interacted with NLRP3 and interfered inflammasome assembly. These findings illustrate that astrocytic Drd2 suppresses NLRP3 inflammasome activation through a β-arrestin2-dependent mechanism, and provide a new strategy for targeting the astrocyte-mediated inflammatory response in the treatment of NDDs.
Results
Drd2 activation inhibits NLRP3 inflammasome in cultured astrocytes
Over the last decade, Drd2 has been reported as an important anti-inflammatory target for the management of NDDs. In the present study, we first studied the effect of Drd2 agonists on NLRP3 inflammasome activation in lipopolysaccharide (LPS) plus ATP-primed mouse primary astrocytes. As shown in Fig. 1a, treatment of Drd2 agonist LY171555 (10, 20, 40 μM) blocked caspase-1 activation and IL-1β maturation in a concentration-dependent manner. Similarly, Quinelorane and Bromocriptine, another two Drd2 agonists, inhibited caspase-1 and IL-1β production after astrocytes were stimulated with ATP and LPS (Fig. 1b–c).

The effect of Drd2 agonists on NLRP3 inflammasome activation in cultured astrocytes. a–c LPS (100 ng/ml) primed-primary astrocytes were treated with different concentrations of LY171555 (10, 20, 40 μM) a, Quinelorane (10, 50, 100 μM) b, Bromocriptine (10, 50, 100 μM) c, and then stimulated with ATP (5 mM). IL-1β and caspase-1 from medium supernatants (SN) and pro-IL-1β and pro-caspase-1 from cell extracts (Input) were analyzed by immunoblotting. d–e Densitometric analysis of IL-1β and caspase-1. f Primary astrocytes were transfected with siRNA targeting the mRNA encoding Drd2 or empty vector. LPS (100 ng/ml) primed-primary astrocytes were treated with LY171555 and then stimulated with ATP (5 mM). IL-1β and caspase-1 from medium supernatants (SN) and pro-IL-1β and pro-caspase-1 from cell extracts (Input) were analyzed by immunoblotting. g–h Densitometric analysis of IL-1β and caspase-1. **p < 0.01 vs Con group. #p < 0.05 vs LPS + ATP group. Values are means ± SEM. Data are representative of at least three independent experiments
In order to confirm the effect of Drd2 in anti-inflammasome, small interfering RNA (siRNA) targeting Drd2 was transfected in astrocytes (Figure S1a). After 72 h, we observed that the silencing of Drd2 prevented the downregulation of caspase-1 and IL-1β levels by LY171555 treatment (Fig. 1f). This result indicates that Drd2 is responsible for LY171555-induced NLRP3 inflammasome inhibition.
Drd2 agonist inhibits multiple stimulators-induced NLRP3 inflammasome activation in cultured astrocytes
To confirm the inhibitory effect of Drd2 agonist on NLRP3 inflammasome in astrocytes, we used two other stimulators of NLRP3 inflammasome, nigericin, and monosodium urate (MSU), to induce NLRP3 inflammasome activation. Consistently, LY171555 also significantly inhibited nigericin- and MSU-induced caspase-1 cleavage and IL-1β production in astrocytes (Fig. 2a). Since the oligomerization of ASC is required for NLRP3 inflammasome activation to induce pro-caspase-1 recruitment, we further examined the effect of Drd2 agonist on the oligomerization of ASC after LPS and ATP stimulation. As shown in Fig. 2d, treatment of Drd2 agonist LY171555 (40 μM) inhibited the accumulation of ASC specks. This result confirms the inhibitory effect of Drd2 agonist on NLRP3 inflammasome activation in astrocytes.

The effect of Drd2 agonist on multiple stimulators-induced NLRP3 inflammasome activation in cultured astrocytes. a LPS (100 ng/ml) primed-primary astrocytes were treated with LY171555 (40 μM) and then stimulated with ATP (5 mM), MSU (monosodium urate) (250 μg/ml), or NIG (nigericin) (20 nM). IL-1β and caspase-1 from medium supernatants (SN) and pro-IL-1β and pro-caspase-1 from cell extracts (Input) were analyzed by immunoblotting. b, c Densitometric analysis of IL-1β and caspase-1. d LPS (100 ng/ml)-primed-primary astrocytes were treated with LY171555 (40 μM) and then stimulated with ATP (5 mM). Cells were analyzed by Immunostaining. The white arrow represents ASC specks. Scale bar represents 20 μm. *p < 0.05, **p < 0.01 vs Con group. #p < 0.05 vs LPS + ATP group. $p < 0.05, $$p < 0.01 vs LPS + MSU group. &p < 0.05 vs LPS + NIG group. Values are means ± SEM. Data are representative of at least three independent experiments
Anti-inflammasome effect of Drd2 is independent of classical G protein pathways
Activation of Drd2 stimulates classical G protein pathways alongside β-arrestins-dependent signaling, leading to diverse downstream events [28, 29]. Thus, we explored the possible involvement of both signaling pathways in the observed anti-inflammasome effect. First, we assessed the role of the classical G protein pathway. As shown in Fig. 3a, we observed that treatment of forskolin, an adenylate cyclase agonist that increases level of cyclic AMP (cAMP) (Figure S2), inhibited the expression of caspase-1 and IL-1β in a dose-dependent manner after astrocytes were incubated with LPS and ATP. Similarly, dibutyryl cyclic AMP (dbcAMP), a cyclic nucleotide derivative that mimics the action of endogenous cAMP, and pertussis toxin (PTX), a Gαi protein inhibitor, also inhibited NLRP3 activation (Fig. 3d). As coupling to Gαi protein, active Drd2 suppressed astrocyte cAMP levels both in normal and forskolin stimulated conditions (Fig. 3g). In theory, as (1) Drd2 suppresses cAMP levels and (2) cAMP inhibits NLRP3 inflammasome, it can be deduced that Drd2 agonists may accelerate NLRP3 inflammasome activation induced by LPS and ATP, which contradicts the observation showed in Figs. 1 and 2. Thus, we speculated that instead of the classical G protein pathway, it is more likely that the β-arrestins-dependent signaling pathway mediated the Drd2-inhibiting NLRP3 inflammasome activation.

The effect of cAMP on NLRP3 inflammasome activation. a LPS (100 ng/ml) primed-primary astrocytes were treated with different concentrations of Forskolin (10, 50, 100 μM) and then stimulated with ATP (5 mM). IL-1β and caspase-1 from medium supernatants (SN) and pro-IL-1β and pro-caspase-1 from cell extracts (Input) were analyzed by immunoblotting. b, c Densitometric analysis of IL-1β and caspase-1. d LPS (100 ng/ml) primed-primary astrocytes were treated with different doses of dbcAMP (10, 50, 100 μM) and then stimulated with ATP (5 mM). IL-1β and caspase-1 from medium supernatants (SN) and pro-IL-1β and pro-caspase-1 from cell extracts (Input) were analyzed by immunoblotting. e, f Densitometric analysis of IL-1β and caspase-1. g LPS (100 ng/ml) primed-primary astrocytes were treated with PTX (100 ng/ml) and then stimulated with ATP (5 mM). IL-1β and caspase-1 from medium supernatants (SN) and pro-IL-1β and pro-caspase-1 from cell extracts (Input) were analyzed by immunoblotting. h–i Densitometric analysis of IL-1β and caspase-1. **p < 0.01 vs Con group. #p < 0.05, ##p < 0.01 vs LPS + ATP group. j Primary astrocytes were pretreated with LY171555 (40 μM) for 1 h and stimulated with 100 µM forskolin. Culture medium was analyzed for intracellular cAMP levels. *p < 0.05, **p < 0.01 vs Con group, ##p < 0.01 vs Forskolin group. Values are means ± SEM. Data are representative of at least three independent experiments
Anti-inflammasome effect of Drd2 is dependent on β-arrestin2-mediated signaling
β-arrestin2 and β-arrestin1 share 78% amino-acid sequence, with most differences in the C-terminus [24]. Studies over the past decades have shown that their functions in GPCR signaling differ in diverse conditions [30, 31]. Here, we explored the roles of β-arrestin2 and β-arrestin1 in anti-inflammasome, respectively. As shown in Figure S3, we observed that neither LY171555 nor LPS and ATP treatment could change the expressions of β-arrestin2 and β-arrestin1 in primary cultured astrocytes.
We further delineated the effects of β-arrestin2 and β-arrestin1 on NLRP3 inflammasome activation by interfering them separately. We observed that knockdown of β-arrestin2 (Figure S1c) abrogated the downregulation of caspase-1 and IL-1β levels by LY171555 treatment (Fig. 4), whereas interference of β-arrestin1 (Figure S1e) directly inhibited NLRP3 inflammasome activation (Figure S4). To further explore the mechanisms of Drd2 in anti-inflammasome, we selected β-arrestin2 as our research object in the subsequent experiments as β-arrestin1 might act on upstream pathways irrelevant to Drd2.

Interference of β-arrestin2 has influence on the effect of Drd2 agonist on NLRP3 inflammasome activation. a Primary astrocytes were transfected with siRNA targeting the mRNA encoding β-arrestin2 or empty vector. LPS (100 ng/ml) primed-primary astrocytes were treated with LY171555 and then stimulated with ATP (5 mM). IL-1β and caspase-1 from medium supernatants (SN) and pro-IL-1β and pro-caspase-1 from cell extracts (Input) were analyzed by immunoblotting. b, c Densitometric analysis of IL-1β and caspase-1. *p < 0.05, **p < 0.01 vs Con group. #p < 0.05 vs Mock LPS + ATP group. Values are means ± SEM. Data are representative of at least three independent experiments
Drd2 activation induces the interaction of β-arrestin2 and NLRP3
As β-arrestin2 is a significant scaffolding protein involved in signaling pathways both GPCR dependent and independent, it can interact with various intracellular molecules such as c-Jun N-terminal kinases in mitogen-activated protein kinase family and inhibitory κBα (IκBα) in NF-κB pathway etc [24]. Thus, we tested the interaction between β-arrestin2 and NLRP3. As showed in Fig. 5a, b, after the plasmids co-transfection, NLRP3 interacted with β-arrestin2 and exhibited colocalization within HEK293T cells. Similarly, LY171555 promoted the interaction of NLRP3 and β-arrestin2, as well as suppressed the bonding between NLRP3 and ASC in mouse primary astrocytes (Fig. 5c–e). Furthermore, we observed that LY171555 induced Drd2 internalization and subsequent binding with NLRP3, whereas the interaction was inhibited by the interference of β-arrestin2 (Fig. 5f). Immunofluorescence results also showed that LY171555 promoted the intracellular interaction between β-arrestin2 (Green) and NLRP3 (Red) (Fig. 5g). Taken together, these results indicate that β-arrestin2 interacting with NLRP3 is required for the inhibitory effect of Drd2 on NLRP3 inflammasome activation in astrocytes.

The effect of Drd2 activation on the interaction of β-arrestin2 and NLRP3. a Flag-tagged NLRP3 construct and β-arrestin2 pcDNA3.1 construct were co-transfected in HEK293T cells. Immunofluorescent histochemical staining for β-arrestin2, DAPI, and NLRP3 in HEK293T cells. Scale bar, 20 μm. b Flag-tagged NLRP3 construct and β-arrestin2 pcDNA3.1 construct were co-transfected in HEK293T cells. Cell lysates were immunoprecipitated with anti-Flag antibody and then the samples were analyzed by immunoblotting. c Cell lysates of primary astrocytes treated with LPS (100 ng/ml) + ATP (5 mM) and LY171555 or not were immunoprecipitated with anti-β-arrestin2 antibody, and then the samples were analyzed by immunoblotting. d Cell lysates of primary astrocytes treated with LPS (100 ng/ml) + ATP (5 mM) and LY171555 or not were immunoprecipitated with anti-NLRP3 antibody, and then the samples were analyzed by immunoblotting. e Flag-tagged NLRP3 construct was transfected in primary astrocytes. Cell lysates of primary astrocytes treated with LPS (100 ng/ml) + ATP (5 mM) and LY171555 or not were immunoprecipitated with anti-flag antibody, and then the samples were analyzed by immunoblotting. f Primary astrocytes were transfected with siRNA targeting the mRNA encoding β-arrestin2 or empty vector. Cytoplasm of primary astrocytes was isolated. Cell cytoplasm lysates of primary astrocytes treated with LPS (100 ng/ml) + ATP (5 mM) and LY171555 or not were immunoprecipitated with anti-Drd2 antibody, and then the samples were analyzed by immunoblotting. g Immunofluorescent histochemical staining for β-arrestin2, DAPI, and NLRP3 on the primary astrocytes treated with LY171555 compared with control. Scale bar represents 20 μm. Data are representative of at least three independent experiments
β-arrestin2 deficiency abolishes the anti-inflammatory and neuroprotective effects of Drd2 in MPTP-induced PD mouse model
To determine the role of β-arrestin2 in anti-inflammasome effect of Drd2 in vivo, wild type (WT) and β-arrestin2 knockout (β-arrestin2−/−) mice were treated with neurotoxin MPTP following LY171555 injection. As shown in Fig. 6a–e, the administration of LY171555 decreased MPTP-induced NLRP3 inflammasome activation in the midbrain of WT mice, as reflected by the levels of NLRP3, pro-IL-1β, caspase-1, IL-1β. The anti-inflammatory effect of LY171555 was abolished in β-arrestin2−/− mice. There was no significant difference in numbers of TH+ neurons in SNc in saline group between WT and β-arrestin2−/− mice, but β-arrestin2 deficiency aggravated SNc TH+ neurons loss after MPTP treatment (Fig. 6f). We then used nissl staining on adjacent sections to ensure that the loss of TH+ cell reflected cellular death instead of downregulation of TH expression (Fig. 6h). β-arrestin2 deficiency also aggravated astrocytes activation after MPTP challenge (Fig. 6j). These results indicated that β-arrestin2 plays a critical role in lowering MPTP-induced neuroinflammation via inhibition of NLRP3 inflammasome activation.

Knockout of β-arrestin2 has influence on the anti-inflammatory and neuroprotective effects of Drd2 in MPTP-induced PD mouse model. a WT and β-arrestin2−/− mice were made MPTP (20 mg/kg i.h., q.d., 5 d)-induced PD model with LY171555(5 mg/kg i.p., q.d.,11 d) administration or not. NLRP3, pro-caspase-1, caspase-1, pro-IL-1β and IL-1β from mouse mesencephalon homogenate were analyzed by immunoblotting. b–e Densitometric analysis of NLRP3, pro-IL-1β, caspase-1 and IL-1β. f Immunohistochemical staining of TH+ neuron in the SNc. g Counting TH+ neuron in the SNc. h Nissl staining of neuron. i Counting neuron. j Immunohistochemical staining of GFAP in the SNc. k Counting GFAP positive astrocytes in the SNc. Scale bar represents 200 μm. *p < 0.05, **p < 0.01 vs WT Con group; #p < 0.05 vs WT MPTP group; $p < 0.05, $$p < 0.01 vs β-arrestin2−/− Con group. Values are means ± SEM. Data are representative of six independent experiments
Besides to β-arrestin2−/− mice, we also used mice that are intraceretally injected with β-arrestin2 siRNA to verify the above results. Similarly, β-arrestin2 interference aggravated NLRP3 inflammasome activation (Figure S5a), SNc TH+ neurons loss (Figure S5f, S5h) and astrocytes activation (Figure S5j) after MPTP treatment. And the administration of LY171555 alleviated MPTP-induced pathological changes in WT mice instead of siRNA-injected mice. Interestingly, it is noted that β-arrestin2−/− mice were more susceptible to MPTP challenge than those receive β-arrestin2 interference only in the brain, suggesting that β-arrestin2 in peripheral system might also exerts anti-inflammatory effect to alleviate systemic damage and DA degeneration in MPTP-induced PD mouse model.
In addition, Drd2 knockout (Drd2−/−) mice showed increased neuroinflammation in normal conditions and aggravated SNc TH+ neurons loss, astrocytes activation, NLRP3 inflammasome activation in response to MPTP injections compared with those in WT mice (Figure S6). Together, our findings demonstrate that astrocytic Drd2 signaling is dependent on β-arrestin2, which may be a potential target for anti-inflammatory treatment of PD.
Furthermore, to investigate whether LY171555 injection or β-arrestin2 expression has effect on MPTP metabolism in vivo, MPP+ levels in blood, striatum, liver, and kidney were detected 90 min after MPTP injection (Fig. S7). We find that striatal MPP+ levels were not significantly different regardless of LY171555 treatment and β-arrestin2−/−.
Discussion
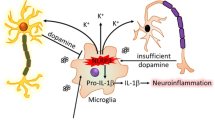
Drd2 agonist was first reported to suppress nitric oxide and tumor necrosis factor-α in peripheral neutrophils two decades ago [32]. Since then, more studies have showed that Drd2 is a potential candidate for amelioration of inflammatory diseases [33,34,35,36] and NDDs [22, 37–39]. Nevertheless, to date there are no reports on whether Drd2 participates in the suppression of inflammasome activation. In the present study, we found that LY171555, a selective Drd2 agonist, inhibited NLRP3 inflammasome activation in the SNc of MPTP-treated PD mice. Notably, the anti-inflammasome effect of Drd2 was abolished in β-arrestin2 deficient mice, suggesting the critical role of β-arrestin2 in Drd2-regulated NLRP3 inflammasome activation. LY171555 also blocked caspase-1 cleavage and IL-1β maturation in primary astrocytes stimulated by LPS and ATP. Moreover, we found that Drd2 enhanced the interaction of β-arrestin2 and NLRP3 to suppress the inflammasome assembling process. We demonstrate for the first time that astrocytic Drd2 exerts its anti-inflammasome effect through a β-arrestin2-dependent pathway as shown in Fig. 7 and this may prove a new target for the treatment of PD.

Schematic diagram of the anti-inflammasome effect of Drd2 via β-arrestin2. a proposed model for how β-arrestin2 regulates Drd2 signaling, thereby modulating Drd2 internalization and NLRP3 inflammasome activation in primary astrocytes under condition of neuroinflammation in Parkinson’s disease
In the CNS, astrocytes play a fundamental role in forming borders between neural parenchyma and non-neural cells to restrict access of leukocyte into brain parenchyma. They also play other important roles, such as providing energy sources for neurons, modulating synaptic activity and regulating extracellular glutamate levels etc. Multiple studies have shown that the pathogenesis and progression of PD are related to inflammation, which is accompanied by glia cell reaction [40,41,42]. We previously reported activation of NLRP3 inflammasome and upregulation of caspase-1 and IL-1β in the SNc of MPTP-injected mice, and that astrocytic Drd2 is an important component modulating inflammatory response and maintaining balance of innate immunity in CNS [22]. In the present study, we found that Drd2−/− mice showed increased astrocytes activation and neuroinflammation in normal conditions. After MPTP injection, Drd2−/− mice showed aggravated SNc TH+ neurons loss, astrocytes activation, and NLRP3 inflammasome activation. These results suggest that Drd2 serves protective effects on PD therapy. Previous studies also showed that Drd1 prevents inflammation via suppression of NLRP3 inflammasome in BMDMs [43]. With regards to whether Drd1 or Drd2 mediates the inhibition of NLRP3 inflammasome, we speculate that Drd1 and Drd2 may both have the same role in peripheral and central systems respectively. We found that Drd2 agonists inhibited NLRP3 inflammasome activation in mice primary astrocytes, and such inhibition was reversed by the interference of Drd2. Moreover, consecutive injection of LY171555 inhibited NLRP3 inflammasome activation in WT mice of PD model. Our results suggest that Drd2 may be a crucial anti-inflammasome element with protective effects on PD.
As GPCRs, Drd2 couples with heterotrimeric G proteins, generates second messengers once activated by agonists. Subsequently, GRKs phosphorylate the activated receptors, resulting in the recruitment of scaffolding proteins named β-arrestins (β-arrestin1 and β-arrestin2), which trigger downstream signaling or induce desensitization and internalization of receptors [28]. To clarify whether classical G protein pathways or β-arrestins-dependent pathways are involved in the Drd2 induced anti-inflammasome effect, the present study showed that forskolin, dbcAMP and PTX inhibited NLRP3 inflammasome, and Drd2 suppressed cAMP levels. If Gi protein pathway was involved, Drd2 agonists should enhance NLRP3 inflammasome activation. However, LY171555 was shown to decrease caspase-1 and IL-1β levels. This provides evidence to suggest that the inhibitory effect of Drd2 agonists on NLRP3 inflammasome may be dependent on the β-arrestins-dependent signaling instead of the classical G protein pathways.
In the past few years, numerous studies have demonstrated that β-arrestins play significant roles in inflammatory diseases. In particular, β-arrestin2 has been reported to have a protective role in many inflammatory diseases such as meningitis [44], asthma [45], endotoxemia [46], pulmonary fibrosis [47] etc. In contrast, β-arrestin1 is mainly reported having the opposite function in inflammatory diseases such as MS [48], myocardial infarction [49], colitis [50], cutaneous flushing [51] etc, perhaps because of its recently reported critical role in the formation of ASC pyroptosome and activation of the NLRP3 inflammasomes [52].
In the present study, we observed that interference of β-arrestin2 abrogated the downregulation of caspase-1 and IL-1β levels by LY171555 treatment, whereas deficiency of β-arrestin1 inhibited NLRP3 inflammasome activation in primary astrocytes after LPS and ATP stimulation. Similarly, in vivo studies show that β-arrestin2 interference aggravated SNc TH+ neurons loss, astrocytes activation and NLRP3 inflammasome activation after MPTP stimulation. The administration of LY171555 decreased MPTP-induced TH+ neurons loss, astrocytes activation and levels of NLRP3, pro-IL-1β, caspase-1, IL-1β in midbrain of WT mice instead of β-arrestin2−/− or β-arrestin2 siRNA-injected mice. Together, these results suggest that anti-inflammasome function of Drd2 is dependent on β-arrestin2-relevant signaling.
As a multifunctional joint protein, β-arrestins can interact with various molecules in cytoplasm. We observed that LY171555 promoted NLRP3 binding with β-arrestin2 and suppressed the binding between NLRP3 and ASC in mouse primary astrocytes as well as HEK293T cells. The results are consistent with what was reported previously [27]. In addition, we found that LY171555-induced Drd2 binding with NLRP3 and such interaction was inhibited when β-arrestin2 was interfered by siRNA. Herein, we first showed that β-arrestin2 mediates Drd2 to inhibit inflammasome activation via binding with NLRP3 in astrocytes, playing anti-inflammatory effects in PD model. A recent study [53] also shows that β-arrestins remain active after dissociation from receptors, allowing them to maintain at the cell surface and presumably triggering signaling independently. It will be interesting to explore if this brand new mechanism of β-arrestin2 is responsible for mediating the Drd2 anti-inflammasome effect.
In summary, our study reveals a novel anti-inflammatory function of Drd2 where the receptor is internalized and binds with NLRP3 to prevent inflammasome activation via a β-arrestin2-dependent pathway. However, there are some limitations of this study. First, NLRP3 inflammasome is only one of many inflammasomes and it is probable that the other inflammasomes are involved in the PD pathology. The effect of Drd2 on other kinds of inflammasomes remains to be studied. Second, although siRNA targeting on β-arrestin2 can interfere the its expression in SNc, it is imprecise that astrocytes instead of other kinds of cells, e.g., oligodendrocytes and neurons, are most essential to anti-inflammasome and neuroprotection through β-arrestin2-dependent pathways. To solve this issue, astrocytic β-arrestin2 conditional knockout mice will provide more conclusive evidence. Collectively, these results demonstrate a novel mechanism of Drd2 agonist inhibiting NLRP3 inflammasome in a β-arrestin2-dependent pathway, which extends our understanding on β-arrestin2-biased Drd2 signaling and sheds light on potential new therapeutic avenues for PD.
Materials and methods
Animals
C57BL/6J mice (male, 3-month old) were obtained from Comparative Medicine Centre of Yangzhou University. β-arrestin2−/− mice (male, 3-month old) were obtained from Gang Pei’s laboratory. Drd2 knockout (Drd2−/−) mice (male, 3-month old) were obtained from Jiawei Zhou’s laboratory. Mice were bred and maintained in the Animal Resource Centre of the Faculty of Medicine, Nanjing Medical University. Mice had free access to food and water in a room with an ambient temperature of 22 °C ± 2 °C and a 12:12–hour light/dark cycle. Neonatal mice within 3 d were used. All animal procedures were performed in strict accordance with the guideline of the Institutional Animal Care and Use Committee of Nanjing Medical University.
Reagents
LY171555, Quinelorane, and Bromocriptine were purchased from TOCRIS. LPS, ATP, MSU, nigericin, forskolin, dbcAMP, and PTX were purchased from Sigma-Aldrich (St. Louis, MO, USA). Anti-caspase-1 Ab (#06-503-1, 1:500) and anti-Drd2 Ab (#AB5084P, 1:500) were purchased from Millipore (Billerica, MA, USA). Anti-NLRP3 Ab (#AG-20B-0014-C100, 1:1000) was purchased from AdipoGen (San Diego, CA, USA). Anti-β-arrestin2 Ab (#3857, 1:800) and anti-β-arrestin1 Ab (#12697 S, 1:1000) were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-IL-1β Ab (#13767, 1:500) and anti-flag Ab (#F1804, 1:1000) were purchased from Sigma. Anti-ASC Ab (#sc-22514-R, 1:200) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-β-actin Ab (#BM0627, 1:4000) was purchased from Boster (Pleasanton, CA, USA).
Primary astrocyte cultures and treatments
Primary astrocyte cultures were conducted as described previously [54]. The midbrain and striatum of neonatal mice aged 1–3 d were removed and separated from meninges and basal ganglia. Tissue were dissociated with 0.25% trypase (Amresco, Solon, OH, USA) at 37 °C and terminated by Dulbecco’s modified Eagle’s medium (Gibco-BRL, Thermo Fisher Scientific, Rockford, IL, USA) supplemented with 10% fetal bovine serum (Gibco, Thermo Fisher Scientific) and 1% penicillin/streptomycin. Cells were plated on 25 cm2 T-flask flasks (Sigma). The culture medium was changed with fresh substratum 24 h later and then changed every 3 d. When 90% confluent, cells were split onto six-well culture plates at 1.5 × 105 per ml.
In the experiments, LPS (100 ng/ml, 6 h) primed-primary astrocytes were treated with different doses of LY171555 (10, 20, 40 μM), Quinelorane (10, 50, 100 μM) or Bromocriptine (10, 50, 100 μM) for 1 h and then stimulated with ATP (5 mM, 30 min), MSU (250 μg/ml, 6 h), or nigericin (20 nM, 30 min).
Cell transfection
siRNA targeting Drd2, β-arrestin2, β-arrestin1 or negative control (NC) siRNA (Jima, Shanghai, China) was transfected in mouse primary astrocytes using Lipofectamine 3000 reagent (Invitrogen, Life Technologies) in OPTI-MEM reduced serum medium (Gibco) according to the manufacturer’s instructions. siRNA duplexes used were as follows:
Drd2 (sense): GCAGGAUUCACUGUGACAUTT
Drd2 (antisense): AUGUCACAGUGAAUCCUGCTT
β-arrestin2 (sense): GCUUGUGGAGUAGACUUUGTT
β-arrestin2 (antisense): CAAAGUCUACUCCACAAGCTT
β-arrestin1 (sense): CCAGCUCAACAUUCUGCAATT
β-arrestin1 (antisense): UUGCAGAAUGUUGAGCUGGTT
NC (sense): UUCUCCGAACGUGUCACGUTT
NC (antisense): ACGUGACACGUUCGGAGAATT
Eight hours later, the transfection mixture was removed and cells were further incubated with normal medium for additional 72 h before LPS stimulation.
Plasmids of pcDNA3.1-GFP-β-arrestin2, and NLRP3-Flag were co-transfected to HEK293T cells or astrocytes using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) according to the instructions provided. Cells were collected for western blotting or Co-immunoprecipitation (Co-IP) analysis 48 h after transfection.
Western blotting analysis
Cells or mouse brain tissues were lysed in the buffer (Bio-Rad) or with Mem-PER™ Eukaryotic Membrane Protein Extraction Reagent Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Protein concentrations were determined with the Micro BCA Kit (Beyotime, Shanghai, China). Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis using polyacrylamide TGX gels (Bio-Rad, Hercules, California, USA) and then transferred to polyvinylidene difluoride (PVDF) membranes. After blocking, PVDF membranes were incubated with various specific primary antibodies as described above in Tris Buffered Saline with Tween 20 at 4 °C overnight. Membranes were washed and incubated in corresponding horseradish peroxidase-conjugated secondary antibodies (1:1000, KPL) for 1 h at room temperature. Proteins were visualized and detected by enhanced chemiluminescence western blot detection reagents (Pierce, Thermo Fisher Scientific) and analyzed with ImageQuant™ LAS 4000 imaging system (GE Healthcare, Pittsburgh, PA, USA).
Co-IP
Mouse primary astrocytes were lysed in the buffer (Bio-Rad). Proteins were immunoprecipitated with specific primary antibodies (1 μg antibody per 100 μg of total protein) followed by incubated with MagnaBindTM Protein G magnetic Beads (Thermo Fisher Scientific). Proteins were eluted from the beads prepared for western blotting.
cAMP Immunoassay
cAMP content in astrocytes was detected using a cAMP Direct Immunoassay Kit (BioVision, Milpitas, CA, USA) according to the manufacturer’s protocol. Absorbance of samples was detected by the Multiskan Spectrum (Thermo Fisher Scientific) at 450 nm.
Immunocytochemical staining
After co-transfection of pcDNA3.1-GFP-β-arrestin2 and NLRP3-Flag plasmids, astrocytes were rinsed with 0.1 M phosphate-buffered salin (PBS) and fixed with 4% paraformaldehyde, followed by block with PBS containing 5% bovine serum albumin (BSA), then incubated with the primary antibody (anti-NLRP3 Ab, 1:500, #AG-20B-0014-C100, AdipoGen) at 4 °C overnight. After washing, cells were exposed to secondary antibody (Alexa Fluor 555 goat anti-mouse IgG, 1:1000, #A21422, Invitrogen) for 1 h at room temperature. After washing and treatment with 4',6-diamidino-2-phenylindole (Life), cells were observed under stereomicroscope (Olympus, Tokyo, Japan).
In vivo experimental treatments
Mouse β-arrestin2 siRNA was transfected with packaging vectors (hU6-MCS-Ubiquitin-EGFP-IRES-puromycin) to generate lentivirus. For microinjection, intraperitoneal pentobarbital sodium (60 mg/kg, Sigma)-anesthetized mice are placed in a stereotaxic apparatus. They were injected with the lentivirus 2.5 μl by glass electrode aiming at SNc (AP: −3.0 mm; ML: ± 1.3 mm; DV: −4.2 mm) at a rate of 0.25 μl/min. Then it needed an additional 2-minute needle retention. Mouse subacute MPTP-induced PD model was conducted 10 d after virus microinjection.
Three-month-old WT C57BL6J, Drd2−/−, β-arrestin2−/−, and β-arrestin2 siRNA-injected mice were made mouse subacute MPTP-induced PD model. They were injected hypodermically (i.h) with MPTP (20 mg/kg i.h., Sigma) dissolved in saline, once a day for 5 consecutive days and left for 7 d. Some mice also received LY171555 (5 mg/kg) intraperitoneally (i.p) daily 3 d prior to treatment with MPTP and over the 5 MPTP-injectin days as well as 3 d afterwards. Control mice received saline only. Seven days after the last MPTP injection, mice were sacrificed for brain proteins detection or immunohistochemistry.
Immunohistochemical analysis
Brain slices were rinsed carefully in PBS followed by 3% H2O2 for 30 min to quench the endogenous peroxidase activity then incubated with 0.3 % Triton X-100 in PBS supplemented 5% BSA for 1 h. After that, the sections were incubated with specific primary antibodies (rabbit anti-GFAP Ab, 1:500, #MAB360, Millipore; mouse anti-TH Ab, 1:500, #ab6211, Abcam, Cambridge, MA, USA) in PBS containing 5% BSA at 4 °C overnight. After extensive washing, brain slices were incubated with secondary antibodies for 1 h at room temperature. Finally the slides were incubated with Diaminobenzidin for 5 min. For Nissl staining, the slides were soaked in CV solution (0.1 g cresyl violet, 99 ml H2O and 1% acetic acid 1 ml) for 30 min at room temperature then dehydrated with alcohol and xylene. The brain slices were observed under stereomicroscope (Olympus).
Cell counting
The method of cell counting was described in the previous publication of our laboratory [55]. In brief, for cell quantification in in vivo studies, the numbers of TH+, Nissl+, and GFAP+-immunoreactive cells in the SNc of the midbrain were assessed using the optical fractionator (Stereo Investigator software, Microbrightfield Bioscience, Williston, VT, USA). All stereological analyzes were performed under the × 200 magnification of an Olympus BX52 microscope (Olympus America Inc., Melville, NY, USA). The counting frame size was 50 μm × 50 μm and the sampling grid size was 100 μm × 100 μm. Within one counting frame, positive cells counted must show both immunostaining in the cell body and unstained cavity in the nuclei, and the nuclei does not touch or cross the red avoidance lines of the counting frame. Slices used for immunostaining were strictly collected at three intervals. For immunoreactive cells counting, each sample contains 12 serial sections. For brain area selection, we referred to the previous article [56]. The entire pars compacta was measured by using the medial terminal nucleus of the accessory optic tract as its medial border. The sections rostral and caudal to this anatomical landmark selected for quantitative analysis were representative for the entire pars compacta. The absolute numbers of positive cells in each group are automatic analyzed by the Stereo Investigator software.
High-performance liquid chromatography (HPLC)
For quantification of MPP+, WT, and β-arrestin2−/− mice were killed 90 min after a single dose of MPTP with or without a previous LY171555 injection. Blood plasma was mixed in 2 M perchloric acid (10 μl Blood plasma in 100 μl perchloric acid). Striatum, liver, and kidney samples were lysed in 0.1 M perchloric acid (1 mg samples in 100 μl perchloric acid). Samples were centrifuged and supernatant liquids were tested in Water HPLC-UV system with SHIMADZU reversed-phase column (5 μm, 250 × 4.6 mm). The mobile phase: 50 mmol/l KH2PO4 (pH 3.2): acetonitrile = 85:15. The flow rate is 1 ml/min. Samples were monitored by fluorescence detector at 245 nm wavelength. Peaks were quantified.
Statistical analysis
Data were presented as mean ± SEM. The significance of difference was determined by Student’s t test, One-factor analysis of variance or two-factor analysis of variance followed by Tukey’s post hoc test. Difference was considered significant at p < 0.05.
References
Ben Haim L, Carrillo-de Sauvage MA, Ceyzeriat K, Escartin C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front Cell Neurosci. 2015;9:278.
Davila D, Thibault K, Fiacco TA, Agulhon C. Recent molecular approaches to understanding astrocyte function in vivo. Front Cell Neurosci. 2013;7:272.
Cobb JA, O’Neill K, Milner J, Mahajan GJ, Lawrence TJ, May WL, et al. Density of GFAP-immunoreactive astrocytes is decreased in left hippocampi in major depressive disorder. Neuroscience. 2016;316:209–20.
Yang XL, Kim CK, Kim TJ, Sun J, Rim D, Kim YJ, et al. Anti-inflammatory effects of fimasartan via Akt, ERK, and NFkappaB pathways on astrocytes stimulated by hemolysate. Inflamm Res. 2016;65:115–23.
Hamby ME, Coppola G, Ao Y, Geschwind DH, Khakh BS, Sofroniew MV. Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple G-protein-coupled receptors. J Neurosci. 2012;32:14489–510.
Yun JW, Ahn JB, Kang BC. Modeling Parkinson’s disease in the common marmoset (Callithrix jacchus): overview of models, methods, and animal care. Lab Anim Res. 2015;31:155–65.
Mori F, Piao YS, Hayashi S, Fujiwara H, Hasegawa M, Yoshimoto M, et al. Alpha-synuclein accumulates in Purkinje cells in Lewy body disease but not in multiple system atrophy. J Neuropathol Exp Neurol. 2003;62:812–9.
Valera E, Masliah E. Therapeutic approaches in Parkinson’s disease and related disorders. J Neurochem. 2016;139:346–52.
Dong YF, Chen ZZ, Zhao Z, Yang DD, Yan H, Ji J, et al. Potential role of microRNA-7 in the anti-neuroinflammation effects of nicorandil in astrocytes induced by oxygen-glucose deprivation. J Neuroinflamm. 2016;13:60.
Shrivastava AN, Redeker V, Fritz N, Pieri L, Almeida LG, Spolidoro M, et al. Data in support of the identification of neuronal and astrocyte proteins interacting with extracellularly applied oligomeric and fibrillar alpha-synuclein assemblies by mass spectrometry. Data Brief. 2016;7:221–8.
Brites D, Fernandes A. Neuroinflammation and depression: microglia activation, extracellular microvesicles and microRNA dysregulation. Front Cell Neurosci. 2015;9:476.
Komine O, Yamanaka K. Neuroinflammation in motor neuron disease. Nagoya J Med Sci. 2015;77:537–49.
Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegener. 2015;4:19.
Song L, Pei L, Yao S, Wu Y, Shang Y. NLRP3 inflammasome in neurological diseases, from functions to therapies. Front Cell Neurosci. 2017;11:63.
Shi F, Kouadir M, Yang Y. NALP3 inflammasome activation in protein misfolding diseases. Life Sci. 2015;135:9–14.
Ozaki E, Campbell M, Doyle SL. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res. 2015;8:15–27.
Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6:262.
Krishnan SM, Sobey CG, Latz E, Mansell A, Drummond GR. IL-1beta nd IL-18: inflammatory markers or mediators of hypertension? Br J Pharmacol. 2014;171:5589–602.
More SV, Kumar H, Kim IS, Song SY, Choi DK. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediat Inflamm. 2013;2013:952375.
Mrak RE, Griffin WS. Common inflammatory mechanisms in Lewy body disease and Alzheimer disease. J Neuropathol Exp Neurol. 2007;66:683–6.
Rangel-Barajas C, Coronel I, Floran B. Dopamine receptors and neurodegeneration. Aging Dis. 2015;6:349–68.
Shao W, Zhang SZ, Tang M, Zhang XH, Zhou Z, Yin YQ, et al. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via alphaB-crystallin. Nature. 2013;494:90–94.
Hanlon CD, Andrew DJ. Outside-in signaling--a brief review of GPCR signaling with a focus on the Drosophila GPCR family. J Cell Sci. 2015;128:3533–42.
Sharma D, Parameswaran N. Multifaceted role of beta-arrestins in inflammation and disease. Genes Immun. 2015;16:576.
Gu YJ, Sun WY, Zhang S, Wu JJ, Wei W. The emerging roles of beta-arrestins in fibrotic diseases. Acta Pharmacol Sin. 2015;36:1277–87.
Jiang T, Yu JT, Tan MS, Zhu XC, Tan L. beta-Arrestins as potential therapeutic targets for Alzheimer’s disease. Mol Neurobiol. 2013;48:812–8.
Yan Y, Jiang W, Spinetti T, Tardivel A, Castillo R, Bourquin C, et al. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity. 2013;38:1154–63.
Srivastava A, Gupta B, Gupta C, Shukla AK. Emerging functional divergence of beta-arrestin isoforms in GPCR function. Trends Endocrinol Metab. 2015;26:628–42.
Peterson SM, Pack TF, Caron MG. Receptor, ligand and transducer contributions to dopamine D2 receptor functional selectivity. PLoS ONE. 2015;10:e0141637.
Jean-Charles PY, Rajiv V, Shenoy SK. Ubiquitin-related roles of beta-arrestins in endocytic trafficking and signal transduction. J Cell Physiol. 2016;231:2071–80.
Iacovelli L, Felicioni M, Nistico R, Nicoletti F, De Blasi A. Selective regulation of recombinantly expressed mGlu7 metabotropic glutamate receptors by G protein-coupled receptor kinases and arrestins. Neuropharmacology. 2014;77:303–12.
Meli R, Raso GM, Gualillo O, Pacilio M, Di Carlo R. Prolactin modulation of nitric oxide and TNF-alpha production by peripheral neutrophils in rats. Life Sci. 1997;61:1395–403.
Lu JH, Liu YQ, Deng QW, Peng YP, Qiu YH. Dopamine D2 receptor is involved in alleviation of type II collagen-induced arthritis in mice. Biomed Res Int. 2015;2015:496759.
Taylor BK, Joshi C, Uppal H. Stimulation of dopamine D2 receptors in the nucleus accumbens inhibits inflammatory pain. Brain Res. 2003;987:135–43.
Zhang Y, Cuevas S, Asico LD, Escano C, Yang Y, Pascua AM, et al. Deficient dopamine D2 receptor function causes renal inflammation independently of high blood pressure. PLoS ONE. 2012;7:e38745.
Laengle UW, Markstein R, Schneider V, Greiner B, Roman D. Effects of antiglaucoma drugs timolol and GLC756, a novel dopamine D2 agonist and D1 antagonist, on endotoxin-induced-uveitis in rats. Exp Eye Res. 2005;80:847–52.
Tanaka K, Kanno T, Yanagisawa Y, Yasutake K, Hadano S, Yoshii F, et al. Bromocriptine methylate suppresses glial inflammation and moderates disease progression in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2011;232:41–52.
Lieberknecht V, Junqueira SC, Cunha MP, Barbosa TA, de Souza LF, Coelho IS, et al. Pramipexole, a dopamine D2/D3 receptor-preferring agonist, prevents experimental autoimmune encephalomyelitis development in mice. Mol Neurobiol. 2017;54:1033–45.
Ezrokhi M, Luo S, Trubitsyna Y, Cincotta AH. Neuroendocrine and metabolic components of dopamine agonist amelioration of metabolic syndrome in SHR rats. Diabetol Metab Syndr. 2014;6:104.
Delattre AM, Carabelli B, Mori MA, Kempe PG, Rizzo de Souza LE, Zanata SM, et al. Maternal Omega-3 supplement improves dopaminergic system in pre- and postnatal inflammation-induced neurotoxicity in Parkinson’s disease model. Mol Neurobiol. 2017;54:2090–106.
Cao L, Li D, Feng P, Li L, Xue GF, Li G, et al. A novel dual GLP-1 and GIP incretin receptor agonist is neuroprotective in a mouse model of Parkinson’s disease by reducing chronic inflammation in the brain. Neuroreport. 2016;27:384–91.
Zhou P, Weng R, Chen Z, Wang R, Zou J, Liu X, et al. TLR4 signaling in MPP( + )-induced activation of BV-2 cells. Neural Plast. 2016;2016:5076740.
Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. 2015;160:62–73.
Xia R, Hu Z, Sun Y, Chen S, Gu M, Zhou Y, et al. Overexpression of beta-arrestin 2 in peripheral blood mononuclear cells of patients with cryptococcal meningitis. J Interferon Cytokine Res. 2010;30:155–62.
Walker JK, Fong AM, Lawson BL, Savov JD, Patel DD, Schwartz DA, et al. Beta-arrestin-2 regulates the development of allergic asthma. J Clin Invest. 2003;112:566–74.
Li H, Hu D, Fan H, Zhang Y, LeSage GD, Caudle Y, et al. Beta-arrestin 2 negatively regulates toll-like receptor 4 (TLR4)-triggered inflammatory signaling via targeting p38 MAPK and interleukin 10. J Biol Chem. 2014;289:23075–85.
Lovgren AK, Kovacs JJ, Xie T, Potts EN, Li Y, Foster WM, et al. beta-arrestin deficiency protects against pulmonary fibrosis in mice and prevents fibroblast invasion of extracellular matrix. Sci Transl Med. 2011;3:74ra23.
Shi Y, Feng Y, Kang J, Liu C, Li Z, Li D, et al. Critical regulation of CD4 + T cell survival and autoimmunity by beta-arrestin 1. Nat Immunol. 2007;8:817–24.
Watari K, Nakaya M, Nishida M, Kim KM, Kurose H. Beta-arrestin2 in infiltrated macrophages inhibits excessive inflammation after myocardial infarction. PLoS ONE. 2013;8:e68351.
Li J, Wei B, Guo A, Liu C, Huang S, Du F, et al. Deficiency of beta-arrestin1 ameliorates collagen-induced arthritis with impaired TH17 cell differentiation. Proc Natl Acad Sci USA. 2013;110:7395–400.
Walters RW, Shukla AK, Kovacs JJ, Violin JD, DeWire SM, Lam CM, et al. beta-Arrestin1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J Clin Invest. 2009;119:1312–21.
Mao K, Chen S, Wang Y, Zeng Y, Ma Y, Hu Y, et al. beta-arrestin1 is critical for the full activation of NLRP3 and NLRC4 inflammasomes. J Immunol. 2015;194:1867–73.
Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, Tobin AB, et al. Beta-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature. 2016;531:661–4.
Zeng XN, Sun XL, Gao L, Fan Y, Ding JH, Hu G. Aquaporin-4 deficiency down-regulates glutamate uptake and GLT-1 expression in astrocytes. Mol Cell Neurosci. 2007;34:34–39.
Lu M, Su C, Qiao C, Bian Y, Ding J, Hu G. Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ROS clearance. Int J Neuropsychopharmacol 2016, 19: pii: pyw047.
Sauer H, Rosenblad C, Bjorklund A. Glial cell line-derived neurotrophic factor but not transforming growth factor beta 3 prevents delayed degeneration of nigral dopaminergic neurons following striatal 6-hydroxydopamine lesion. Proc Natl Acad Sci USA. 1995;92:8935–9.
Acknowledgements
We would like to acknowledge Professor Gang Pei and Professor Jiawei Zhou for providing β-arrestin2−/− mice and Drd2 knockout (Drd2−/−) mice. We also thank Professor Guangyu Wu for critical reading of the manuscript. The work reported herein was supported by the grants from the National Natural Science Foundation of China (No. 81630099, No. 81473196, No. 81773706, No. 81573403 and No. 81473195), the key project of Natural Science Foundation of the Higher Education Institutions of Jiangsu Province (No. 15KJA310002), and Natural Science Foundation of Jiangsu Province (BK20151559).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Edited by A. Verkhratsky
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Zhu, J., Hu, Z., Han, X. et al. Dopamine D2 receptor restricts astrocytic NLRP3 inflammasome activation via enhancing the interaction of β-arrestin2 and NLRP3. Cell Death Differ 25, 2037–2049 (2018). https://doi.org/10.1038/s41418-018-0127-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0127-2
This article is cited by
-
The role of the brain renin-angiotensin system in Parkinson´s disease
Translational Neurodegeneration (2024)
-
β-arrestin1 regulates astrocytic reactivity via Drp1-dependent mitochondrial fission: implications in postoperative delirium
Journal of Neuroinflammation (2023)
-
Targeting CB2R in astrocytes for Parkinson's disease therapy: unraveling the Foxg1-mediated neuroprotective mechanism through autophagy-mediated NLRP3 degradation
Journal of Neuroinflammation (2023)
-
G protein-coupled receptors in neurodegenerative diseases and psychiatric disorders
Signal Transduction and Targeted Therapy (2023)
-
Retinal inner nuclear layer thickness in the diagnosis of cognitive impairment explored using a C57BL/6J mouse model
Scientific Reports (2023)
