
Abstract
Epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) have achieved satisfactory clinical effects in the therapy of non-small cell lung cancer (NSCLC), but acquired resistance limits their clinical application. NRF2 has been shown to enhance the resistance to apoptosis induced by radiotherapy and some chemotherapy. In this study, we investigated the role of NRF2 in resistance to EGFR-TKIs. We showed that NRF2 protein levels were markedly increased in a panel of EGFR-TKI-resistant NSCLC cell lines due to slow degradation of NRF2 protein. NRF2 knockdown overcame the resistance to EGFR-TKIs in HCC827ER and HCC827GR cells. Furthermore, we demonstrated that NRF2 imparted EGFR-TKIs resistance in HCC827 cells via upregulation of GPX4 and SOD2, and suppression of GPX4 and SOD2 reversed resistance to EGFR-TKIs. Thus, we conclude that targeting NRF2-GPX4/SOD2 pathway is a potential strategy for overcoming resistance to EGFR-TKIs.
Similar content being viewed by others


Introduction
Lung cancer is ranked first in terms of cancer-related deaths worldwide, and non-small-cell lung cancer (NSCLC) accounts for ~80%–90% of all lung cancers. Up to 50% of Asian NSCLC patients harbor EGFR mutations such as missense mutation in exon 18 (G719X), deletion of exon 19 (ex19del), or missense mutation in exon 21 (L858R) [1,2,3]. Although first-generation EGFR-TKIs (Epidermal growth factor receptor-tyrosine kinase inhibitors) such as gefitinib and erlotinib, show satisfactory curative effects on NSCLC patients harboring EGFR mutations, most patients develop resistance to EGFR-TKIs after treatment for 6~12 months. Resistance in ~50% of NSCLC cases is due to a secondary missense mutation in exon 20 (T790M). Recently, third-generation EGFR-TKI osimertinib, which aims to overcome resistance mediated by the T790M mutation, has been approved for first-line therapy [4, 5]. However, resistance to osimertinib is unavoidable, and the existing osimertinib resistance mechanisms include the C797S mutation, MET amplification, HER2 amplification, and SCLC transformation [6, 7]. However, many resistance mechanisms remain unclear and need to be identified.
NRF2, the master transcriptional regulator of the antioxidant program, is believed to be a double-edged sword for cancer depending on the stage. NRF2, on the one hand, prevents the initiation of cancer and, on the other hand, accelerates the progression of carcinogenesis [8, 9]. NRF2 overexpression has been observed in several cancers to inhibit apoptosis and contribute to chemoresistance [10, 11]. NRF2 also plays a critical role in eliminating lipid peroxidation and ferroptosis [12, 13]. Ferroptosis is a newly identified form of regulated cell death that may be triggered by the accumulation of reactive oxygen species (ROS) and lipid peroxidation products [14]. Hence, mutations that lead to defects in apoptosis and ferroptosis have been reported to result in resistance to chemotherapy [15,16,17]. However, the role of NRF2 in resistance to molecularly targeted drugs such as EGFR-TKIs is still not well understood.
Here, we found that NRF2 was upregulated in EGFR-TKI-resistant cells compared to control cells and that knockdown of NRF2 reversed the epithelial-mesenchymal transition (EMT) and inhibited the migration ability of the resistant cells. Inhibition of GPX4 and SOD2 also reversed EMT and impaired the migration ability of resistant cells. Moreover, NRF2 activation induced resistance to EGFR-TKIs, which was reversed by suppression of the GPX4 and SOD2. In addition, targeting the NRF2-GPX4/SOD2 pathway resensitized resistant cells to EGFR-TKIs. Our results suggest that NRF2 may be a potential therapeutic target to overcome resistance to EGFR TKIs.
Materials and methods
Reagents and antibodies
Erlotinib and gefitinib were purchased from LC Laboratories. Osimertinib and RSL3 were purchased from Selleck Chemicals. Dimethyl sulfoxide (DMSO) was obtained from Sigma. All chemicals were dissolved in DMSO for in vitro experiments.
Antibodies against NRF2, ALDH1A1, GPX4, and SOD2 were purchased from Abcam, and antibodies against E-cadherin, vimentin, β-actin, BCL2, and γH2AX were purchased from Cell Signaling Technology.
Cell lines and cell culture
Human NSCLC cell lines HCC827 and PC9, which harbor EGFR mutations and are sensitive to first- and third-generation EGFR TKIs, were obtained from the American Type Culture Collection (ATCC) and Dr. G. L. Zhuang (China State Key Laboratory of Oncogenes and Related Genes, Renji Hospital, Shanghai, China), respectively. H1975 cells, which harbor a p.T790M mutation and are resistant to first-generation EGFR-TKIs and sensitive to the third-generation TKI osimertinib, were obtained from ATCC. These cells were authenticated by short tandem repeat profiling. Isogenic cell lines resistant to erlotinib (HCC827ER), gefitinib (HCC827GR), and osimertinib (HCC827OR and H1975OR) were established, maintained, and authenticated as previously described [18, 19]. In brief, sensitive parental cells were cultured in medium containing escalating concentrations of erlotinib, gefitinib, and osimertinib. After continued culturing, the surviving cells proliferated in micromolar concentrations of TKI were considered resistant cells. Cells were cultured in RPMI-1640 medium (Thermo Fisher Scientific) containing 10% FBS (Gemini), 1% GlutaMAX (Thermo Fisher Scientific), and 1% penicillin-streptomycin (Thermo Fisher Scientific) at 37 °C and 5% CO2.
Cell viability assay
Cells were seeded in 96-well plates at 4000 cells/well. EGFR-TKIs, siRNAs, or other treatment reagents were added to the medium at the indicated concentrations in the legends for 72 h. Cell viability assays were performed with Cell Counting Kit-8 (Dojindo) according to the manufacturer’s instructions.
Transwell migration assay
Transwell migration assays were performed by using Transwell chambers (Corning Costar). Cells (1 × 104 per chamber) were plated into the upper chamber with serum-free medium after treatment with siRNA or reagents as described in the legends. The lower chambers were filled with medium containing 10% FBS. After incubation for 20 h, cells on the upper surface of the filter were scrubbed off. Cells adhering to the underside of the filter were stained with 0.1% crystal violet solution and imaged (Nikon). Then, stained cells were dissolved with 10% acetic acid (100 μL/chamber), and the optical density (OD) was detected at 600 nm using a microplate reader to quantify the cell migration ability.
Western blotting analysis
Cells were collected and lysed in RIPA buffer (Beyotime). Then, the samples were centrifuged at 13,800 × g for 20 min. The quantity of protein was determined by the BCA protein determination method. After that, the samples were separated in 10% SDS-polyacrylamide gel electrophoresis gel, transferred onto a polyvinylidene difluoride membrane (Millipore), blocked with 5% nonfat milk for 1 h, incubated with primary antibody overnight at 4 °C, incubated with secondary antibody at room temperature for 1 h and subsequently visualized by chemiluminescence (Odyssey FC, LI-COR Biosciences).
RNA extraction and real-time PCR
Total RNA of cells was extracted with an RNA extraction kit (Takara) according to the manuscript’s instructions. The isolated RNA was reverse-transcribed using the RevertAid First Strand cDNA Synthesis Kit (Qiagen). Real-time PCR was conducted with SYBR Premix Ex Taq using a Light Cycler 480 II system (Roche). The SOD2 primer sequences were 5′-CCTAACGGTGGTGGAGAACC-3′ (forward) and 5′-CTGAGCCTTGGACACCAACA-3′ (reverse). The β-actin primer sequences were 5′-TGGTGACGATGCACACGAAGC-3′(forward) and 5′-TGGTGACGATGCACACGAAGC-3′(reverse) and were used as internal controls. Relative quantification was determined by the ΔΔCT method.
RNA interference
Endogenous NRF2, GPX4, and SOD2 were transiently silenced using siRNAs (Genepharma) according to the manufacturer’s instructions. Scrambled-siRNAs were used as a mock control. Cells were seeded in 6-well plates at a density of 2 × 105/well, and 24 h later, cells were transfected with siRNAs. The silencing efficiency was detected using Western blotting. The sense sequences of siRNAs of NRF2 were as follows: #2 (TGCTCAGAATTGCAGAAAA) and #6 (CTCCTACTGTGATGTGAAA). The sense sequences of siRNAs of GPX4 were as follows: #1 (GGCAAGACCGAAGTAAACT) and #4 (CCGATACGCTGAGTGTGGT). The sense sequences of siRNAs of SOD2 were as follows: #1 (AAAAGCTATTTGGAATGTA), #2 (CAGCCTGCACTGAAGTTCA), and #3 (CTAATGATCCCAGCAAGAT).
Colony formation assay
Cells were seeded in six-well plates at 1000 cells/well and cultured at 37 °C and 5% CO2 for ~2–3 weeks. The colonies of cells were stained with crystal violet, and images were acquired by a scanner. The colonies consisting of >50 cells each were manually counted.
Cell apoptosis assay
Apoptosis, ferroptosis, and viable cells were detected using an annexin V/PI apoptosis detection kit (BD Pharmingen) according to the manufacturer’s instructions. A total of 10,000 cells were analyzed by flow cytometry (Accuri C6, BD Biosciences).
ROS detection
Intracellular ROS levels were determined by the fluorescent oxidation indicator 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) according to the manufacturer’s instructions (Beyotime). Briefly, the cells were collected at a density of 1 × 106, incubated with 10 μM DCFH-DA for 20 min at 37 °C, and washed with serum-free medium. The fluorescence intensity was examined by an Accuri C6 flow cytometer (BD Pharmingen).
Public clinical database analysis
Kaplan–Meier analysis of the association between the probability of overall survival (OS) of lung adenocarcinoma or NSCLC patients and studied gene expression profiles. The analysis was performed based on the TCGA and Gene Expression Omnibus (GEO) databases.
Statistical analysis
Quantitative data were analyzed by GraphPad Prism software and presented as the mean ± SEM. Differences were assessed accordingly with the two-tailed Student’s t test or ANOVA with Bonferroni posttest. P < 0.05 was considered statistically significant. Asterisks indicate significance at the following levels: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Results
NRF2 upregulation in EGFR-TKI-resistant NSCLC cells
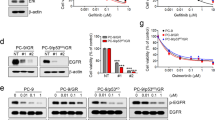
To investigate the mechanism of acquired resistance to EGFR TKIs, HCC827, and H1975 cells were chronically treated with escalating concentrations of EGFR-TKIs. Dose-response analysis with EGFR-TKIs demonstrated that the IC50 values of HCC827ER, HCC827GR, HCC827OR, and H1975OR cells were over 100-fold higher than those of their corresponding parental cells (Fig. 1a–d and Table S1). We have demonstrated that resistant cells had low ROS/RCS levels in a previous study [18]. We wondered whether NRF2, the master transcriptional regulator of the antioxidant program, played a role in EGFR-TKI resistance. Then, we examined the protein levels of NRF2 in resistant cells compared with their sensitive parental cells and in sensitive cells before and after TKI treatment. Western blot analysis showed upregulation of NRF2 in PC9ER, HCC827ER, HCC827GR, HCC827OR, and H1975OR cells (Fig. 1e–g). Cancer stem-like cells are resistant to chemotherapy drugs [20]. We found that NRF2 protein levels were also upregulated in ALDH1A1-overexpressing stem-like lung cancer cells (Fig. 1h). We proved that these cells were insensitive to erlotinib [18]. The upregulation of NRF2 was attributed to TKI challenge because erlotinib, gefitinib, or osimertinib treatment induced an increase in NRF2 levels in a time-dependent manner in HCC827 and H1975 cells (Fig. 1i, j). To explore the mechanism of upregulation of NRF2 in resistant cells, we measured the mRNA levels of NRF2 in parental and resistant cells. The results showed that NRF2 mRNA in resistant cells did not significantly change compared with that in parental cells (Fig. 1k), indicating that the upregulation of NRF2 levels may depend on the protein stability modulation mechanism. Keap1 is a substrate receptor of a Cul3-RING ubiquitin ligase (CRL3) that, under physiological conditions, constitutively binds and targets NRF2 for degradation [21]. Prompted by previous studies, we detected whether there are Keap1 mutations in resistant cells. No Keap1 mutations were detected in resistant cells by DNA sequencing analysis (Fig. S1). Then, we treated parental and resistant cells with the protein synthesis inhibitor CHX and found that NRF2 protein degradation in resistant cells was slower than that in parental cells, demonstrating that the upregulation of NRF2 in resistant cells is due to the longer half-life of the NRF2 protein (Fig. 1l). These data indicate that NRF2 is upregulated in EGFR-TKI-resistant NSCLC cells and that NRF2 may play an important role in EGFR-TKI resistance.

Cell viability of H1975OR (a), HCC827OR (b), HCC827GR (c), HCC827ER (d), and their corresponding parental cells was determined by CCK-8 assay after cells were treated with the indicated concentrations of TKIs. e–g NRF2 protein levels were detected in resistant cells compared with their parental cells by Western blotting analysis. h NRF2 protein levels were detected by Western blotting analysis in ALDH1A1-overexpressing stem-like cells after treatment with DOX for 48 h or 72 h. i, j NRF2 protein levels were detected by Western blotting analysis in HCC827 and H1975 cells after treatment with 1 μM erlotinib, gefitinib or osimertinib for 0, 24, 48, and 72 h. k NRF2 mRNA levels of HCC827, HCC827GR, HCC827ER, and HCC827OR cells were detected by RT-PCR. (l) Parental and resistant cells were treated with 100 μg/mL CHX, collected at the indicated times, lysed and immunoblotted.
NRF2 dependence in EGFR-TKI-resistant NSCLC cells
To evaluate whether EGFR-TKI-resistant cells depend more on upregulated NRF2, the growth, viability, colony formation, and apoptosis of the cells were measured after NRF2 was knocked down by siRNA. NRF2 knockdown markedly inhibited resistant cell growth but had only a minimal effect on sensitive parental cells (Fig. 2a–d). The selective effects of NRF2 knockdown on the resistant cells were recapitulated in cell viability and colony formation analysis. Both siNRF2 #2 and siNRF2 #6 inhibited cell viability more potently in HCC827ER and HCC827GR cells than in parental cells (Fig. 2e). Consistently, knockdown of NRF2 selectively inhibited the colony formation of H1975OR cells (Fig. 2f). Moreover, cell death was dramatically increased after NRF2 was silenced in HCC827GR cells (Fig. 2g). In addition, suppression of NRF2 by the selective inhibitor ML385 selectively inhibited the cell growth of resistant cells (Fig. 2h). These results indicate that compared to sensitive cells, EGFR-TKI-resistant cells are more dependent on NRF2 for growth and survival.

a HCC827 cells and HCC827ER cells were transfected with 20 nM siNRF2 or mock control for 72 h, and photographs of cells were captured by IncuCyte. Scale bar: 100 µm. b–d HCC827 cells, HCC827OR cells, and HCC827GR cells were transfected with 20 nM siNRF2 or mock control for 72 h, and cell growth curves were recorded by IncuCyte. e Cell viability was measured by CCK-8 assay as described in “Methods” after HCC827 cells, HCC827ER cells, and HCC827GR cells were transfected with 20 nM siNRF2 or mock control for 72 h. f Colony forming assay was performed as described in “Methods” after cells were transfected with 20 nM siNRF2 or mock control for 48 h. g Cell death was detected by flow cytometry after HCC827GR cells were transfected with 20 nM siNRF2 or mock control for 72 h. h HCC827 and HCC827GR cells were treated with 10 μM NRF2 inhibitor ML385 for 72 h, and cell growth curves were recorded by IncuCyte. Data are presented as the mean ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs Ctr.
EGFR-TKI-resistant cells depend on NRF2 to maintain elevated EMT/CSC properties
Epithelial-mesenchymal transition (EMT) is closely involved in drug resistance; [20, 22], therefore, we wondered whether EMT depends on NRF2. According to the morphological phase-contrast images of cells, resistant cells showed a spindle-like shape, including the loss of apical-basal polarity and cell–cell contacts, in contrast to their parental counterparts, which exhibited an epithelial morphology (Fig. 3a). We also found that expression of the epithelial marker E-cadherin was decreased, while expression of the mesenchymal marker vimentin was increased in HCC827ER and HCC827GR cells compared with HCC827 cells (Fig. 3b). Along with EMT, the CSC marker CD44 was also increased in resistant cells (Fig. 3b). Next, we found that the protein levels of E-cadherin were increased and those of vimentin were decreased after NRF2 knockdown (Fig. 3c). Moreover, knockdown of NRF2 also reversed the cell morphology change (Fig. 3a). Along with reversing EMT, NRF2 silencing downregulated the CSC marker ALDH1A1 (Fig. 3c). Cell migration assays also confirmed that knockdown of NRF2 impaired the elevated migration ability of HCC827ER cells (Fig. 3d). These data demonstrate that EMT and CSC-like characteristics of resistant cells rely on NRF2.

a Morphological phase-contrast images of cells after treatment with 20 nM siNRF2 for 48 h. b Western blot analysis of EMT biomarkers and CSC biomarker CD44 in HCC827, HCC827GR, and HCC827ER cells. c Western blot analysis of EMT biomarkers and CSC biomarker ALDH1A1 in HCC827ER, HCC827GR, and HCC827OR cells after cells were transfected with 20 nM siNRF2 or mock control for 72 h. d Cell migration ability was determined by Transwell migration assay after cells were transfected with 20 nM siNRF2 or mock control for 72 h. The quantification result was determined as described in the “Methods”. Scale bar: 100 µm. Data are presented as the mean ± SEM. *P < 0.05 vs mock, #P < 0.05 vs HCC827.
Upregulation of the antioxidant proteins GPX4 and SOD2 in EGFR-TKI-resistant cells
To further demonstrate the mechanism of resistance to EGFR-TKIs, we explored the cell death type insensitive cells and resistant cells with flow cytometry. We found that annexin V−/PI+ cell populations were decreased in HCC827ER cells (Fig. 4a). The iron chelator deferoxamine also reduced annexin V−/PI+ cell populations (Fig. 4b), which reminded us that annexin V−/PI+ cell populations might be ferroptotic. Ferroptosis is a newly recognized method of cell death involving iron-dependent accumulation of ROS and lipid peroxidation and depends on GPX4 [14, 23]. Consistent with that, HCC827ER and HCC827GR cells possessed lower basal levels of ROS than their sensitive parental cells (Fig. 4c). Studies have already demonstrated that the accumulation of ROS leads to the DNA damage response and apoptosis [24, 25]. As expected, HCC827ER expressed higher levels of antiapoptotic protein BCL2 and lower levels of DNA damage marker γH2AX (Fig. 4d). Lower levels of ROS may be caused by upregulation of some antioxidant protein scavenging ROS and mediated by NRF2. Indeed, we found upregulation of the antioxidants mitochondrial superoxide dismutase (SOD2) and ferroptosis-related glutathione peroxidase 4 (GPX4) in HCC827ER and HCC827GR cells (Fig. 4e, f). In parallel, short-term stimulation with erlotinib increased the expression of GPX4, SOD2, and NRF2 (Fig. 1e). Higher expression of GPX4 was also shown in human lung cancer tissues compared with normal lung tissues (Fig. 4g). Moreover, high expression of GPX4 (Fig. 4h) or SOD2 (Fig. 4i) correlated with poor prognosis in NSCLC patients. These results show that EGFR-TKI-resistant cells upregulate GPX4 and SOD2 to maintain cellular redox homeostasis, avoiding ROS stress-induced damage and also demonstrate that patients who express high GPX4 and SOD2 in lung cancers have poor prognoses.

a, b Cell death was determined by flow cytometry as described in the “Methods”. The cells were exposed to 100 μM DFO for 24 h. (c) ROS were analyzed by flow cytometry as described in the “Methods”. d Western blot analysis of the anti-apoptosis protein BCL2 and the DNA damage biomarker γH2AX in HCC827 and HCC827ER cells. e SOD2 mRNA levels of HCC827, HCC827ER, and HCC827GR cells were detected by RT-PCR. f Western blot analysis of SOD2 and GPX4 protein expression in HCC827 cells, HCC827ER cells, and HCC827GR cells. g Analysis of GPX4 expression in lung adenocarcinoma patient tumors and normal tissues. The analysis was based on TCGA and GEO datasets. h Analysis of the association between the probability of overall survival (OS) of lung adenocarcinoma patients (n = 720) and their GPX4 gene expression profiles. The analysis was performed based on TCGA and GEO datasets. Low or high levels of GPX4 were defined as higher or lower than the median value of 720 patients. i Analysis of the association between the probability of overall survival (OS) of lung cancer patients (n = 1010) and their SOD2 gene expression profiles. The analysis was performed based on the TCGA dataset. Low or high levels of SOD2 were defined as higher or lower than the median value of 1010 patients. Data are presented as the mean ± SEM. *P < 0.05 vs Ctr.
SOD2 and GPX4 dependence in EGFR-TKI-resistant NSCLC cells
Next, we explored the roles of SOD2 and GPX4 in EGFR-TKI resistance. We found that knockdown of SOD2 reversed EMT in HCC827GR cells (Fig. 5a) and selectively inhibited the cell migration ability of HCC827ER and HCC827GR cells (Fig. 5b). Moreover, transiently silencing SOD2 selectively inhibited the viability of HCC827ER and HCC827GR cells (Fig. 5c). The GPX4 inhibitor RSL3 induced the accumulation of ROS, which were scavenged by N-acetylcysteine (NAC) treatment (Fig. 5d). Consistently, RSL3 inhibited the cell migration ability of HCC827ER cells, and the effect was abrogated by NAC treatment (Fig. 5e). Genetic knockdown of GPX4 and pharmacological inhibition by RSL3 reversed EMT in HCC827ER cells as well (Fig. 5f). These results indicate that the EMT and migration of EGFR-TKI-resistant cells depend on GPX4 and SOD2.

a Western blot analysis of EMT biomarkers in HCC827GR cells transfected with 20 nM siSOD2 for 72 h. b Cell migration ability was determined by Transwell migration assay after cells were transfected with 20 nM siSOD2 for 72 h. Scale bar: 100 µm. The quantified result was determined as described in the “Methods”. c Cell viability was determined by CCK-8 assay. Cells were transfected with 20 nM siSOD2 for 72 h. d ROS were analyzed by flow cytometry as described in the “Methods”. HCC827ER cells were treated with 100 nM RSL3 and 10 mM NAC for 6 h. e Cell migration ability was determined by Transwell migration assay after cells were treated with 100 nM RSL3 and 10 mM NAC for 6 h. Scale bar: 100 µm. The quantified result was determined as described in the “Methods”. f Western blot analysis of EMT biomarkers in HCC827ER cells transfected with 20 nM siGPX4 for 72 h or exposed to 100 nM RSL3 for 6 h. Data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 vs Ctr.
NRF2-induced resistance to EGFR-TKIs depends on SOD2 and GPX4
To explore whether NRF2 induces resistance to EGFR-TKIs by upregulating SOD2 and GPX4, we treated HCC827 cells with KI696, an NRF2 activator, by inhibiting the Keap1-NRF2 interaction [26]. KI696 treatment led to an accumulation of NRF2 and an increase in the expression of SOD2 and GPX4 (Fig. 6a). Furthermore, knockdown of NRF2 decreased the expression of SOD2 and GPX4 in HCC827ER cells (Fig. 6a). In addition, NRF2 activation by KI696 promoted cell growth and induced resistance to EGFR-TKIs; these effects were abrogated by the GPX4 inhibitor RSL3 and by SOD2 knockdown (Fig. 6b–e). Moreover, patients with high expression of both GPX4 and SOD2 (GPX4high/SOD2high) tended to have poorer survival than those with the GPX4low/SOD2low signature (Fig. 6f). Taken together, these results demonstrate that SOD2 and GPX4 act as downstream molecules of NRF2 to mediate EGFR-TKI resistance.

a Western blot analysis of NRF2, GPX4, and SOD2 protein expression. HCC827 cells were exposed to 5 μM KI696 for 24 h or 48 h, and HCC827ER cells were transfected with 20 nM siNRF2 or mock control for 48 h. b–e Cell viability was determined by CCK-8 assay, and cell growth curves were determined by IncuCyte. HCC827 cells were treated with 5 μM KI696, 100 nM TKIs, 40 nM RSL3, and 20 nM siNRF2 as indicated. f Analysis of the association between the probability of overall survival (OS) of lung cancer patients (n = 496) and their GPX4 and SOD2 gene expression profiles. The analysis was performed based on the TCGA dataset. Low or high levels of GPX4 and SOD2 were defined as those higher or lower than the median value of 496 patients. Data are presented as the mean ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs Ctr.
Targeting the NRF2-GPX4/SOD2 pathway resensitizes resistant cells to EGFR-TKIs
Treatment with siNRF2 resensitized resistant cells to erlotinib and gefitinib but had little effect on sensitive parental cells (Fig. 7a–c). Knockdown of SOD2 also resensitized resistant cells to erlotinib and gefitinib (Fig. 7d, e). The GPX4 inhibitor RSL3 reversed resistance to erlotinib and osimertinib (Fig. 7f, g). Inhibition of NRF2 with ML385 also resensitized resistant cells to gefitinib and osimertinib (Fig. 7h, i). These results indicate that targeting the NRF2-GPX4/SOD2 pathway overcomes EGFR-TKI resistance.

a–c Cell viability was determined by CCK-8 assay. HCC827ER, HCC827GR, and HCC827 cells were treated with 100 nM EGFR-TKIs and 20 nM siNRF2 for 72 h. d, e Cell viability was determined by CCK-8 assay. HCC827ER and HCC827GR cells were treated with 100 nM EGFR-TKIs and 20 nM siSOD2 for 72 h. f, g Cell viability was determined by CCK-8 assay. The resistant cells were treated with 80 nM RSL3 and every concentration of TKIs by triple dilution from 100 μM for 72 h. h, i Cell viability was determined by CCK-8 assay; HCC827GR, HCC827OR were treated with 10 μM ML385 and every concentration of TKIs by triple dilution from 100 μM for 72 h. Data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs Ctr.
Discussion
EGFR-TKIs are recommended as first-line therapy for patients with EGFR mutations; however, resistance is the major obstacle to further clinical application [27]. Although the third-generation TKI osimertinib shows an encouraging improvement in OS, acquired resistance to this compound is still unavoidable [28]. NRF2, a transcription factor that induces the expression of genes bearing an antioxidant response element (ARE), is reported in conferring resistance to chemotherapy in several cancer cell lines [11, 29]. However, the role of NRF2 in acquired resistance mechanisms against EGFR-TKIs has not been thoroughly studied. Our study showed that resistant cells generated by chronic exposure to EGFR-TKIs at escalating concentrations had upregulated NRF2 levels. NRF2 mRNA levels did not increase in the resistant cells, suggesting that the protein stability of NRF2 may have changed. Then, we found that NRF2 protein degradation in resistant cells was slower than that in parental cells. Degradation of NRF2 is mediated by Keap1; therefore, Keap1 mutation can result in accumulation of NRF2. DNA sequencing analysis showed no Keap1 mutation in the resistant cells. Our previous studies found that EGFR-TKIs caused the accumulation of ROS/RCS in parental cells [18]. Bollong et al. demonstrated that ROS/RCS selectively modifies KEAP1 to form a methylimidazole crosslink between proximal cysteine and arginine residues (MICA). This posttranslational modification results in the dimerization of Keap1 so that Keap1 cannot complex with NRF2, which leads to the reduced NRF2 degradation and accumulation of NRF2 and activation of the NRF2 transcriptional program [30]. These findings may explain why short-term stimulation of EGFR-TKIs leads to upregulation of NRF2 in parental cells. In the process of gradual construction of resistant cells, the cells that survive under long-term drug pressure gradually upregulate NRF2 to fight against ROS/RCS induced by EGFR-TKI stimulation.
Many studies have indicated that EMT is closely involved in drug resistance [20, 22]; therefore, we explored the role of NRF2 in EMT. Indeed, knockdown of NRF2 reversed EMT in resistant cells and reduced their cell migration ability. Moreover, knockdown of NRF2 selectively inhibited cell viability, proliferation and colony formation. These results indicate that EGFR-TKI-resistant cells depend on NRF2.
Recently, several studies have shown that NRF2 activation can promote cell proliferation and inhibit ferroptosis, a newly recognized mechanism of cell death involving the ROS accumulation and lipid peroxidation [12, 13]. GPX4 can combat lipid peroxidation using reduced glutathione (GSH) as a substrate; thus, inhibition of GPX4 with pharmacological (RSL3) or genetic methods can induce ferroptosis [16]. In our study, we found that in resistant cells overexpressing GPX4, GPX4 inhibitor RSL3 overcame resistance to erlotinib, and knockdown of GPX4 also reversed EMT and inhibited the migration ability of resistant cells.
ROS can initiate the apoptotic cascade, and apoptosis mediated by death receptors also depends on the generation of ROS [24, 31]. SOD2 is reported to reduce apoptosis through scavenging ROS, thus mediating resistance to chemotherapy [31], but the role of SOD2 in EGFR-TKI resistance has not been investigated so far. We found that resistant cells upregulated the expression of SOD2 and that knockdown of SOD2 decreased the expression of the antiapoptotic protein BCL2 and inhibited the survival of resistant cells (data not shown). In addition, knockdown of SOD2 overcame EGFR-TKI resistance and reversed EMT in these cells. Moreover, NRF2 activation with KI696 induced the expression of GPX4 and SOD2, knockdown of NRF2 decreased the expression of GPX4 and SOD2, and accumulation of NRF2 by KI696 induced resistance to EGFR-TKIs; this resistance was reversed by RSL3 and siSOD2. All of the above results demonstrate that NRF2 suppresses cell death induced by EGFR-TKIs by upregulating SOD2 and GPX4 in NSCLC.
In conclusion, our findings reveal that the NRF2-GPX4/SOD2 axis imparts resistance to EGFR-TKIs in NSCLC. These results suggest a potential therapeutic strategy in which targeting NRF2 may overcome resistance to EGFR-TKIs.
Change history
01 August 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41401-023-01131-5
02 August 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41401-023-01130-6
References
Goldstraw P, Ball D, Jett JR, Le Chevalier T, Lim E, Nicholson AG, et al. Non-small-cell lung cancer. Lancet. 2011;378:1727–40.
Hirsch FR, Scagliotti GV, Mulshine JL, Kwon R, Curran WJ Jr., Wu YL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389:299–311.
Molina J. Molina. non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83:584–94.
Jänne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372:1689–99.
Ramalingam SS, Yang JC, Lee CK, Kurata T, Kim DW, John T, et al. Osimertinib as first-line treatment of egfr mutation-positive advanced non-small-cell lung cancer. J Clin Oncol. 2018,36:841–9.
Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, et al. EGFR mutations and resistance to Irreversible pyrimidine based EGFR inhibitors. Clin Cancer Res. 2015;21:3913–23.
Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560–2.
Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–91.
Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013;73:4158–68.
Tao S, Wang S, Moghaddam SJ, Ooi A, Chapman E, Wong PK, et al. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014;74:7430–41.
Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235–43.
Kerins MJ, Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Signal. 2018;29:1756–73.
Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. 2017;6:e371.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19:405–14.
Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–7.
Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–64.
Lei HM, Zhang KR, Wang CH, Wang Y, Zhuang GL, Lu LM, et al. Aldehyde dehydrogenase 1A1 confers erlotinib resistance via facilitating the reactive oxygen species-reactive carbonyl species metabolic pathway in lung adenocarcinomas. Theranostics. 2019;9:7122–39.
Ma P, Fu Y, Chen M, Jing Y, Wu J, Li K, et al. Adaptive and acquired resistance to EGFR inhibitors converge on the MAPK pathway. Theranostics. 2016;6:1232–43.
Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells—what challenges do they pose? Nat Rev Drug Discov. 2014;13:497–512.
Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34:21–43.
Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, et al. Identification of the tumour transition states occurring during EMT. Nature. 2018;556:463–8.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92.
Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–4.
Rodríguez-Vargas JM, Ruiz-Magaña MJ, Ruiz-Ruiz C, Majuelos-Melguizo J, Peralta-Leal A, Rodríguez MI, et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 2012;22:1181–98.
Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, et al. Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of Bach1. Cell. 2019;178:316–29.e18.
Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–54.
Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378:113–25.
Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017;11:254–62.
Bollong MJ, Lee G, Coukos JS, Yun H, Zambaldo C, Chang JW, et al. A metabolite-derived protein modification integrates glycolysis with KEAP1-NRF2 signalling. Nature. 2018;562:600–4.
Cheng Y, Dai C, Zhang J. SIRT3-SOD2-ROS pathway is involved in linalool-induced glioma cell apoptotic death. Acta Biochim Pol. 2017;64:343–50.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81872882, 81573018) and the Shanghai Municipal Science Foundation (No. 14YZ032, 2013-52).
Author information
Authors and Affiliations
Contributions
CSM and QML substantially contributed to designing the study, performing experiments, analyzing the data, and writing the manuscript. KRZ, YBT, and YFZ performed experiments and analyzed the data. LZ, HML, and YS conceived and designed the study and experiments, assembled and interpreted the data, wrote the manuscript, and approved the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
The original online version of this article was revised: A western blot image in Fig. 6a (NRF2 protein expression in HCC827ER cells) was inadvertently misplaced in the process of assembling figures.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ma, Cs., Lv, Qm., Zhang, Kr. et al. NRF2-GPX4/SOD2 axis imparts resistance to EGFR-tyrosine kinase inhibitors in non-small-cell lung cancer cells. Acta Pharmacol Sin 42, 613–623 (2021). https://doi.org/10.1038/s41401-020-0443-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0443-1
Keywords
This article is cited by
-
YES1 as a potential target to overcome drug resistance in EGFR-deregulated non-small cell lung cancer
Archives of Toxicology (2024)
-
Nanomedicine targeting ferroptosis to overcome anticancer therapeutic resistance
Science China Life Sciences (2024)
-
Circular RNA circRANGAP1/miR-512-5p/SOD2 Axis Regulates Cell Proliferation and Migration in Non-small Cell Lung Cancer (NSCLC)
Molecular Biotechnology (2023)
-
DNMT3A R882H mutation drives daunorubicin resistance in acute myeloid leukemia via regulating NRF2/NQO1 pathway
Cell Communication and Signaling (2022)
-
Cur@SF NPs alleviate Friedreich’s ataxia in a mouse model through synergistic iron chelation and antioxidation
Journal of Nanobiotechnology (2022)
