
Abstract
Patients with a severe mental disorder report significantly higher levels of childhood trauma (CT) than healthy individuals. Studies have suggested that CT may affect brain plasticity through epigenetic mechanisms and contribute to developing various psychiatric disorders. We performed a blood-based epigenome-wide association study using the Childhood Trauma Questionnaire-short form in 602 patients with a current severe mental illness, investigating DNA methylation association separately for five trauma subtypes and the total trauma score. The median trauma score was set as the predefined cutoff for determining whether the trauma was present or not. Additionally, we compared our genome-wide results with methylation probes annotated to candidate genes previously associated with CT. Of the patients, 83.2% reported CT above the cutoff in one or more trauma subtypes, and emotional neglect was the trauma subtype most frequently reported. We identified one significant differently methylated position associated with the gene TANGO6 for physical neglect. Seventeen differentially methylated regions (DMRs) were associated with different trauma categories. Several of these DMRs were annotated to genes previously associated with neuropsychiatric disorders such as post-traumatic stress disorder and cognitive impairments. Our results support a biomolecular association between CT and severe mental disorders. Genes that were previously identified as differentially methylated in CT-exposed subjects with and without psychosis did not show methylation differences in our analysis. We discuss this inconsistency, the relevance of our findings, and the limitations of our study.
Similar content being viewed by others

Introduction
Childhood trauma (CT) is a well-established risk factor for developing a spectrum of severe mental disorders throughout life [1, 2], especially psychotic symptoms [3, 4]. Among patients with severe mental illness, reported prevalences of sexual and physical abuse are 26 and 39% [5], respectively, compared to 11 and 7% in healthy controls [6]. CT has diverse effects: children can show increased vulnerability to stressful events or resilience against adverse experiences [7, 8]. Both pre-clinical [9, 10] and clinical studies [8, 11, 12] suggest that adverse environmental risk factors, e.g., abuse and neglect, facilitate psychopathological processes in the brain during sensitive developmental periods. Over time, the accumulation of such environmental factors could modify distinct tissues and/or cell lineages and increase the risk of developing a psychiatric disease.
DNA methylation is an epigenetic mechanism that dynamically regulates gene expression by adjusting DNA accessibility to the transcriptional machinery. Environmental factors can cause long-lasting, altered DNA methylation patterns [13, 14]. DNA methylation may “embed” environmental factors in our genome, thus linking CT and stress-related neuropsychiatric disorders [15]. In rodent models, distress and adversities during development can induce DNA methylation changes in the brain, which can persist in adulthood and might even be transmitted through generations [16]. Several studies have aimed to replicate such findings in humans. However, heterogeneity in study designs (methylation platforms, tissues investigated, statistical methods, trauma definitions, study populations) complicates the direct comparison of findings and the interpretation of results [17]. A systematic review, looking at DNA methylation association to CT in individuals with and without a history of a psychotic episode, suggested BDNF, GCH1, MPB, NDEL1, AKT1, DICER1, DROSHA, COMT, DISC1, SLC6A4, NR3C1, KITLG, FKBP5, OXTR, IL-6, TNFa, IL1a, IL1B, IL8, and PTGS as candidate genes [18]. Presumably, epigenetic regulation of some of these genes might be involved in the psychopathology of severe mental disorders, while others may be vulnerability or resilience factors.
Large blood-based epigenome-wide association studies (EWAS) have been conducted for depression [19] and post-traumatic stress disorder (PTSD) [20]. Although the association between CT and DNA methylation has been widely studied (see review [21]), predominantly in healthy samples [22,23,24,25], no known research has investigated genome-wide DNA methylation associated with CT in severe mental disorders. Only one study investigated the association between CT and DNA methylation in first-episode schizophrenia by analysing specific cell lines. It found lower DNA methylation levels in patients with a history of CT [26]. Therefore, little is known of the epigenetic marks associated with CT in psychiatric illnesses. We now report an EWAS on 602 patients diagnosed with schizophrenia, bipolar disorder, or another psychotic disorder and have responded to the Childhood Trauma Questionnaire-short form (CTQ-SF). We aim to identify modulations in DNA methylation associated with CT in severe mental disorders.
Materials and methods
Sample
We included patients from the NORMENT study, also called the Thematically Organized Psychosis (TOP) study, Oslo (Norway). Participants were enrolled in the study between 2007 and 2018. A further description of the NORMENT sample has been published previously [27]. For our EWAS, we included 602 patients (no healthy controls). Previous studies have included smaller or equal samples sizes and detected moderate effects [22, 24]. In the lack of a standardized way to estimate the effect size of EWAS case studies (contrary to EWAS case-control studies), we considered our sample of 602 patients would provide enough power to detect moderate effects.
Patients
Patients were of European ancestry aged between 18 and 64 years (median = 28.0 years). All patients included have a DSM-IV diagnosis for a severe mental disorder based on a structured diagnostic interview (SCID-I for DSM-IV), which included the following: schizophrenia group (n = 268) [schizophrenia (n = 192), schizophreniform disorder (n = 26), schizoaffective disorder (n = 50)], bipolar disorder group (n = 229) [bipolar disorder I (n = 150), bipolar disorder II (n = 63), bipolar not otherwise specified (n = 16)] and the other psychosis group (n = 105) [psychotic disorder not otherwise specified (n = 55), major depressive disorder (n = 23), delusional disorder (n = 18), brief psychotic disorder (n = 9)]. All patients were outpatients or stable inpatients from psychiatric units. Previous studies have described patients' exclusion criteria and the clinical assessments of patients [27]. All patients were assessed with the five-factorial model of the Positive and Negative Syndrome Scale (PANSS) [28], the Inventory of Depressive Symptomatology (IDS) [29], and the Global Functioning Scale divided into function (GFS-F) and symptoms (GFS-S) [30]. A fasting blood sample was drawn in the morning within a narrow time range. All participants gave written informed consent. The Regional Committee for Medical Research Ethics and The Norwegian Data Inspectorate approved the study with ethical approval #2009/2485, #2013/1727.
Psychosis- and no-psychosis patient groups
To evaluate the association between DNA methylation and psychotic symptoms we divided patients into two groups, 514 with a history of psychosis and 81 (all with bipolar disorder, BP) negative for psychosis. Seven patients with no information on a history of psychosis were excluded. The analytic pipeline was run for psychosis/no psychosis separately, in addition to all 602 patients together. For further details, see the Supplementary Methods.
Childhood trauma questionnaire-short form (CTQ-SF)
Childhood trauma was retrospectively reported using the CTQ-SF (Norwegian version) [31, 32]. This 28-item self-report questionnaire includes five subtypes of trauma (emotional abuse, physical abuse, sexual abuse, emotional neglect, and physical neglect) and a total score of “all trauma”. We set a predefined cut-off score by the median per subtype and total score, and thereby a score above the median was defined as trauma. This cut-off is consistent with previous research [33]. Additionally, we report the percentage of individuals with scores above this cut-off. For further details, see the Supplementary Methods.
Imputation
From CTQ-SF data, we imputed missing values from participants with less than eight missing values in total, or less than two missing values per trauma subtype. Imputation was performed based on gender and the median value of the total score per sub-score. If more than two missing values were within one trauma subtype, but with not more than eight missing values in total, this domain was removed for the individual, and the rest kept. One participant did not meet this pre-established criteria and was removed. Of the remaining 602 participants, n = 31 (5.1%) had one or more imputed values from the CTQ-SF.
DNA methylation quantification
Methylation quantification was completed using the Illumina Infinium® Methylation EPIC BeadChip (Illumina, Inc. San Diego, USA).
Pre-processing and quality control
Samples were imported to the statistical programming software R (version 3.6.2). The Bioconductor R-package meffil [34] was used for quality assessment and data pre-processing. Samples were typed for methylation status in three separate batches. Initial principal component analysis (PCA) of the unprocessed data showed that these batches had a significant effect on the data; hence, the pre-processing steps were carried out separately for each batch before merging the samples into one large dataset. Samples and CpG sites that failed quality control checks were removed (see Supplementary Methods). Finally, samples were normalized using functional normalization. Based on plots generated by the meffil.plot.pc.fit function in meffil, a visual evaluation of residual variation by the principal components (PCs) was used to decide the number of PCs for normalization. Twenty PCs were included to normalize the first and second typing batches and 25 were included for the third batch.
The samples from the three batches were combined, and ComBat [35] was used to remove the batch effect from the typing. Technical replicates included in the different typing batches were used to evaluate whether the datasets could be merged. After visually inspecting a PCA plot (Supplementary Fig. 1), we concluded that the quality of data pre-processing was satisfactory, and the data was merged without further normalization. The samples from individuals who answered the CTQ-SF were extracted. The final dataset had 602 samples and 759,742 probes.
Statistical analysis
For the 602 patients, we applied a linear regression model where DNA methylation values were regressed against trauma scores to seek differentially methylated positions (DMPs). Six distinct trauma scores from the CTQ-SF—the total trauma score (“all trauma”) and the five trauma subtypes—were analysed separately. Because of the high correlation between the trauma subtypes, we did not further correct for the number of trauma subtypes tested. After testing three regression models (see Supplementary Methods), we selected Model A since it shows the best quantile-quantile plot (Q-Q plot). Model A includes trauma score, age, sex, smoking, and the five first PCs as covariates. Scree plots, Q-Q plots, and correlations of PCs with biological and technical covariates are shown in Supplementary Figs. 2–4.
We used limma to perform linear regression and thereby identification of DMPs [36], the comb-p algorithm to identify differentially methylated regions (DMRs) [37], and stats [38] for PCA. As recommended, M-values were applied for statistical analysis [39]. DMPs and DMRs were annotated to genes if CpGs were located in promoter regions, 5′/3′ UTRs, or in the gene itself through Illumina annotations [40]. For DMPs, nominal p values were converted to false discovery rate (FDR) values following the Benjamini and Hochberg approach [41]. For DMRs, Šidák correction at the 1% level was set as the multiple testing correction. Consistent with previous studies, a significance threshold was set to FDR <0.05 or Šidák p < 0.05 [14]. Previous studies have demonstrated that in the absence of a reference correlation map such as a linkage disequilibrium map, Šidák correction can account for some of the correlation being less conservative than Bonferroni correction [42]. Thus, in the absence of a correlation map reference as such in EWAS, we applied Šidák correction for DMRs rather than FDR correction as it assumes independence between all probes.
Gene pathway analysis
Through missMethyl [43], we performed a gene pathway analysis to identify possible mechanisms of genes identified in our EWAS.
Investigation of the association between CT and DNA methylation in candidate genes
Using the Bioconductor annotation package for Illumina annotations (reference genome hg19) [40], 1678 methylation probes were annotated to previously published candidate genes. We looked up the p values for these probes in the EWAS result lists (see Supplementary Dataset 1). Probes with p value < 2.98E-05 (0.05/1678) were taken to be significantly associated with CT.
Results
Childhood trauma data
After imputation, we analysed 602 patients with CTQ-SF data (Table 1). A total of 83.2% reported a trauma score over the pre-set cutoff in one or more trauma traits, and emotional neglect was the most frequently reported trauma. The median CTQ-SF total score for all trauma types was 39. The CT distribution was similar in the psychosis- and non-psychosis groups (Supplementary Fig. 5); thus, we merged these groups. The age distribution in the patient group reporting trauma and the non-trauma group was similar (Supplementary Fig. 6). We concluded that age would not be a confounding factor for further analysis when merging these groups to a patient group of n = 602. Supplementary Figs. 7,8 show score distributions for each trauma trait and trauma distributions for each diagnostic group.
Genome-wide identification of differentially methylated positions
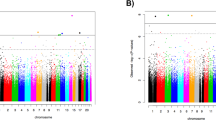
We applied six separate EWASs for the six trauma traits (five subtypes and total score) to the 602 patients. After multiple testing correction, one significant DMP (cg07625619) remained, which was associated with physical neglect (% DNA methylation difference = 2.54, p value = 7.74E-09, FDR = 0.0059). Figure 1 illustrates the genome-wide findings for physical neglect and Table 2 presents the 20 most significant DMPs for physical neglect. The same DMP was also significant when narrowing the analysis to the 514 patients with a history of psychosis (% DNA methylation difference = 2.67, p value = 3.25E-08, FDR = 0.025). This DMP was associated, at FDR < 0.1, with all trauma in the 602 patients (% DNA methylation difference = 2.14, p value = 1.02E-07, FDR = 0.077), and in the patients with psychosis (% DNA methylation difference = 2.15, p value = 8.78E-08, FDR = 0.067). For complete results, see Supplementary Dataset 1. No other single probes were associated at the FDR < 0.05 level for the other trauma traits.

The Manhattan plot illustrates associations between methylation probes and physical neglect in a group of patients with a severe mental disorder. All chromosomes (except sex chromosomes) are displayed on the x-axis, while p values (negative tenfold scale) are displayed on the y-axis. The red line on the y-axis indicates the pre-set cutoff for significance. The methylation probe cg07625619 on chromosome 16 survived multiple testing (% DNA methylation difference = 2.54, p value = 7.74E-09, FDR = 0.0059).
Identification of differentially methylated regions (DMRs)
DMRs accumulate the differences in methylation of consecutive probes in a region and can therefore capture the additional signal from single positions. For the six trauma traits, we identified seventeen DMRs at the significance level Šidák p < 0.05 (Table 3). Twelve of them were annotated to genes through Illumina annotations [40] according to hg19. The HM13; PSIMCT-1 genes were shared between two traits (sexual abuse and all trauma), while the remaining genes were unique to one trait.
Gene pathway analysis
The gene pathway analysis revealed no significant findings. This negative result was expected, as a limitation of the investigation was the low number of genes included.
Investigation of the association between CT and DNA methylation in candidate genes
A previous review listed 20 candidate genes associated with CT in patients with psychotic features and healthy subjects: BDNF, GCH1, MPB, NDEL1, AKT1, DICER1, DROSHA, COMT, DISC1, SLC6A4, NR3C1, KITLG, FKBP5, OXTR, IL-6, TNFa, IL1a, IL1B, IL8, and PTGS [18]. To investigate the association of single CpGs located within or near these genes, we annotated 1678 methylation probes to the genes through Ilumina annotations (hg19) [40], thus setting the experiment-wide significance threshold to 0.05/1678 = 2.98E-05.
We then investigated the genetic overlap with our results for all 602 patients, followed by the 514 patients with a history of psychosis for five trauma subtypes and total trauma score. None of the CpGs annotated to the candidate genes was associated with CT, i.e., no CpG had a p value < 2.98E-05.
Discussion
We performed an EWAS in 602 patients with a severe mental disorder for five trauma subtypes and a summative trauma score. One DMP was significantly associated with physical neglect in all patients, both with and without psychosis. For the remaining CT traits, we found no significantly associated DNA methylation sites with FDR <0.05. Analysis revealed seventeen DMRs (Šidák p < 0.05) associated with all trauma, sexual abuse, emotional abuse, physical abuse, and physical neglect. We identified no significant association to methylation sites located within or near candidate genes previously reported to be CT-associated in healthy individuals or patients.
The significant CpG (cg07625619) associated with physical neglect is located in the body of the gene TANGO6 (transport and Golgi organization protein 6 homolog), also called TMCO7 (transmembrane and coiled-coil domain-containing protein 7) (reference genome GRCHh37/hg19 [44]). TANGO6 encodes a known interactor of MACF1 (microtubule-actin crosslinking factor 1). MACF1 plays a major role in neural progenitor proliferation and neural migration through dynamic regulation of the cytoskeleton [45]. Neural migration is critical for constructing neuronal connections in brain development [46]. MACF1 interacts with the DISC1 (disrupted-in-schizophrenia 1) protein, reported as linked with psychosis [46, 47]. A large-scale GWAS conditioned on DISC1 found eight genes associated with susceptibility to psychosis, among them TANGO6 [48]. This is interesting in the context of repeated reports of synaptic pathophysiology in patients with schizophrenia [49, 50]. Further, differential expression of TANGO6 was reported in a mouse model of cognitive dysfunction [51]. However, no known studies have previously reported TANGO6 linked to trauma-related psychopathology. Additionally, yet not significant, we identified several interesting DMPs associated with physical neglect, including the PNOC (prepronoceptin) gene, previously found implicated in PTSD [52].
Several of the CT-associated DMRs were located near or in genes previously associated with severe mental disorders, cognitive impairment, and distress/trauma-related psychopathology. Two of the genes were previously linked to CT and PTSD Fig. 2 One of the physical neglect-associated DMRs is located in ALOX12 (arachnoid acid 12 lipoxygenase), which is involved in oxidative stress regulation and is associated with PTSD in adults, including a reduction in the right prefrontal cortex thickness [53]. Another DMR, associated with sexual abuse, was found in PM20D1 (peptidase M20 domain containing 1). Recently, a methylome-wide association study of saliva from 224 youths diagnosed with pediatric PTSD and a non-traumatized control group identified a DMR related to hypomethylation of PM20D1 [54]. This was replicated in another cohort and related to gray matter volume in the right fusiform gyrus [54]. In military servicemen, a DMR locating to PM20D1 was associated with longitudinal changes in PTSD symptoms [14]. A study investigating the relationship between CT and whole-blood methylation profiles in 45-year-old males with no specific health disorder found an association of PM20D1 with childhood abuse in two independent cohorts [25]. Thus, PM20D1 is associated with CT in older individuals with no specific phenotype, in youths and adults with PTSD, and, in our study, in CT-exposed adults with a severe mental disorder.

a, b The DMRs for ALOX12 and PM20D1, respectively. For each DMR, panel I reports the genomic location, gene organisation and location of the DMR relative to the gene and CpG Island. Panel II illustrates average methylation M-values for CpGs included in the DMR. Each CpG is represented by a dot. Purple represents the average methylation in individuals exposed to trauma (above the trauma cut-off) and the blue the average M-values for individuals not exposed to trauma (below the trauma cut-off). The DMR located in ALOX12 was associated with physical neglect and included 9 CpG probes (p-value = 3.38E−06; Šidák p-value = 1.58E−06, a.II). The DMR located in PM20D1 was associated with sexual abuse and included 7 CpGs (p-value 3.01E−05; Šidák p-value 5.14E−04; b.II).
Further, we identified genes related to neurodevelopment and psychiatric disorders. One DMR associated with physical neglect is located near C9orf66 (chromosome 9 open reading frame 66) and DOCK8 (dedicator of cytokinesis 8). DOCK8 is linked to altered neurodevelopment and intellectual disability [55, 56]. DOCK8 copy number variants occur in patients with psychotic features [57] and DOCK8 duplications were identified in patients with severe neuropsychiatric disorders, such as SCZ and BP, across five cohorts [58]. Another DMR associated with physical neglect was located within CREB5 (cAMP-responsive element-binding protein 5) [59]. CREB proteins participate in synaptic plasticity, neurotransmission, neurodevelopment, dopamine receptor signal transduction, metabolism, and adaptive response to stress [59,60,61]. Studies using postmortem brain tissue from patients diagnosed with SCZ and BP indicate alterations in CREB protein and gene expression [62]. CREB is a known regulator of BDNF signal transduction [59, 60], and BDNF has repeatedly been suggested as a candidate gene for CT [18] and as a marker of active severe mental disorder regardless of the diagnostic entity [63]. Mice with a genetic susceptibility to major affective disorders have altered CREB activity in hippocampal tissue and, consequently, activation of BDNF and reduced resilience to acute restraint stress [60]. Furthermore, mice with hippocampal CREB deficiency show increased resilience to long-term stress and altered affective behaviors [64].
We report an association between physical neglect and a DMR located in the gene RPTOR (regulatory associated protein of MTOR complex 1), which was previously associated with neglect, sexual abuse, and physical abuse in a buccal tissue-based DNA methylation study of CT in a healthy but at high-risk sample [24]. RPTOR was reported as hypomethylated in SCZ case-controls across blood and brain tissue [65]. These findings indicate an association of CT with RPTOR in individuals with and without a severe mental disorder.
We also found that emotional abuse and physical neglect are associated with DMRs located to ATG101 (autophagy-related 101) and RAET1L (retinoic acid early transcript L), respectively. ATG101 was identified in a large-scale study of gene expression in the dorsolateral prefrontal cortex of SCZ patients and controls [66]. RAET1L was associated with SCZ in a case-control DNA methylation study [67]. Our study also found some associations with developmental stress. One DMR was located in the PSIMCT-1 gene (MCTS2P) and the HM13 gene, which is involved in genomic imprinting. HM13 is essential for fetal development and is related to placental stress in pregnancy [68], and intrauterine growth restriction [69]. These relations indicate that the CT-associated epigenetic marks in severe mental disorders may also be related to stress in utero. Further studies are needed to investigate this association.
Since we investigated the association to CT in patients with a severe mental disorder, the observed DNA methylation alterations should be interpreted as possible epigenetic marks of CT in severe mental disorders. To our knowledge, no studies have investigated CT in such a group. Previous studies examining CT-DNA methylation associations have used heterogeneous study populations, including healthy individuals [22,23,24,25], first-episode psychosis [26], and PTSD phenotypes [54]. This makes it difficult to compare results. For example, we found no overlap between our results and a study investigating CT-associated DNA methylation patterns in an adult, healthy sample [22]. In patients, the identified CT-associated genetic regions could be involved in developing a psychosis phenotype or they may be regulated as a consequence of psychosis. When investigating healthy individuals with no psychosis phenotype, the identified epigenetic regions could (hypothetically) be involved in the genetic features of resilience. Genes involved in resilience do not necessarily have a protective effect on CT outcomes, but they may reduce the chances of adverse outcomes and may have an opposite effect on risk variants [70]. Ignoring these possible genetic hallmarks across study populations might lead to misleading interpretations of the effects of CT. Another important factor in CT is polyvictimization, which may limit the interpretation of results from individual trauma types.
In epigenetic research on neuropsychiatric disorders, it is critical to discuss whether blood is an adequate surrogate tissue for the brain. Significant differences in DNA methylation have been reported for schizophrenia [65, 71], PTSD [72], and autism [73] using blood samples. Research supports a high blood-brain concordance in DNA methylation levels [74]. In a study of SCZ patients, 94% of CpG-SNPs methylated in the brain overlapped with methylation in the blood [65]. However, some DNA methylation sites are variable in the blood and not in the brain, and vice versa [75]. Therefore, when investigating the effect of CT on brain phenotypes, we may possibly overlook disease-specific DNA methylation patterns when applying a blood-based EWAS.
A limitation of studying DNA methylation is its reversibility. CT has a spectrum of adverse health outcomes, which indicate systemic effects and give reasons to presume that there are stable DNA methylation changes in the blood. However, studies looking at cigarette smoking- and alcohol-associated DNA methylation patterns show that some DNA methylation changes are reversed in blood after the exposure ceases [76,77,78]. Peripheral blood frequently renews as opposed to brain cells—an essential difference between the two tissues since one of the mechanisms of reversing DNA methylation is thought to be dilution through cell division. Although white blood cells are post-mitotic [79], they are frequently renewed from the bone marrow. It is possible that CT-associated DNA methylation alterations might be reversed by the time adulthood is reached, affecting our results.
Our study reports DNA methylation changes at specific positions in several genomic regions associated with CT in severe mental disorders. These regions were located within genes observed to be dysregulated in mental illnesses like PTSD but also previously found to be associated with cognitive impairment and distress in utero. However, replication of our results is required in independent cohorts. We want to highlight the importance of carefully selecting the study design and methods when applying an EWAS to look at the effects of environmental factors, as previous studies in this field are highly heterogeneous. Further research should focus on a clear distinction between DNA methylation association for CT in healthy samples and affected individuals to separate genes involved in the psychopathology of severe mental disorders and genes playing a regulatory role in resilience mechanisms. Further research should also focus on the reversal of DNA methylation associated with trauma exposure and the timeframe for such reversal, which could be investigated using longitudinal data.
Data deposition
Summary statistics can be provided upon request.
Code availability
All code is available on request.
References
Kessler RC, McLaughlin KA, Green JG, Gruber MJ, Sampson NA, Zaslavsky AM, et al. Childhood adversities and adult psychopathology in the WHO World Mental Health Surveys. Br J Psychiatry. 2010;197:378–85.
Palmier-Claus JE, Berry K, Bucci S, Mansell W, Varese F. Relationship between childhood adversity and bipolar affective disorder: systematic review and meta-analysis. Br J Psychiatry. 2016;209:454–9.
Varese F, Smeets F, Drukker M, Lieverse R, Lataster T, Viechtbauer W, et al. Childhood adversities increase the risk of psychosis: a meta-analysis of patient-control, prospective- and cross-sectional cohort studies. Schizophr. Bull. 2012;38:661–71.
McGrath JJ, McLaughlin KA, Saha S, Aguilar-Gaxiola S, Al-Hamzawi A, Alonso J, et al. The association between childhood adversities and subsequent first onset of psychotic experiences: a cross-national analysis of 23 998 respondents from 17 countries. Psychol Med. 2017;47:1230–45.
Bonoldi I, Simeone E, Rocchetti M, Codjoe L, Rossi G, Gambi F, et al. Prevalence of self-reported childhood abuse in psychosis: a meta-analysis of retrospective studies. Psychiatry Res. 2013;210:8–15.
May-Chahal C, Cawson P. Measuring child maltreatment in the United Kingdom: a study of the prevalence of child abuse and neglect. Child Abus Negl. 2005;29:969–84.
Hughes K, Bellis MA, Hardcastle KA, Sethi D, Butchart A, Mikton C, et al. The effect of multiple adverse childhood experiences on health: a systematic review and meta-analysis. Lancet Public Health. 2017;2:e356–e66.
Abajobir AA, Kisely S, Scott JG, Williams G, Clavarino A, Strathearn L, et al. Childhood maltreatment and young adulthood hallucinations, delusional experiences, and psychosis: a longitudinal study. Schizophr Bull. 2017;43:1045–55.
Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–54.
McGowan PO, Suderman M, Sasaki A, Huang TC, Hallett M, Meaney MJ, et al. Broad epigenetic signature of maternal care in the brain of adult rats. PLoS ONE. 2011;6:e14739.
Arseneault L, Cannon M, Fisher HL, Polanczyk G, Moffitt TE, Caspi A. Childhood trauma and children’s emerging psychotic symptoms: a genetically sensitive longitudinal cohort study. Am J Psychiatry. 2011;168:65–72.
Croft J, Heron J, Teufel C, Cannon M, Wolke D, Thompson A, et al. Association of trauma type, age of exposure, and frequency in childhood and adolescence with psychotic experiences in early adulthood. JAMA Psychiatry. 2019;76:79–86.
Snijders C, Maihofer AX, Ratanatharathorn A, Baker DG, Boks MP, Geuze E, et al. Longitudinal epigenome-wide association studies of three male military cohorts reveal multiple CpG sites associated with post-traumatic stress disorder. Clin Epigenetics. 2020;12:11.
Rutten BPF, Vermetten E, Vinkers CH, Ursini G, Daskalakis NP, Pishva E, et al. Longitudinal analyses of the DNA methylome in deployed military servicemen identify susceptibility loci for post-traumatic stress disorder. Mol Psychiatry. 2018;23:1145–56.
Szyf M, Bick J. DNA methylation: a mechanism for embedding early life experiences in the genome. Child Dev. 2013;84:49–57.
Saab BJ, Mansuy IM. Neurobiological disease etiology and inheritance: an epigenetic perspective. J Exp Biol. 2014;217:94.
Cecil CAM, Zhang Y, Nolte T. Childhood maltreatment and DNA methylation: a systematic review. Neurosci Biobehav Rev. 2020;112:392–409.
Tomassi S, Tosato S. Epigenetics and gene expression profile in first-episode psychosis: the role of childhood trauma. Neurosci Biobehav Rev. 2017;83:226–37.
Jovanova OS, Nedeljkovic I, Spieler D, Walker RM, Liu C, Luciano M, et al. DNA methylation signatures of depressive symptoms in middle-aged and elderly persons: meta-analysis of multiethnic epigenome-wide studies. JAMA Psychiatry. 2018;75:949–59.
Smith AK, Ratanatharathorn A, Maihofer AX, Naviaux RK, Aiello AE, Amstadter AB, et al. Epigenome-wide meta-analysis of PTSD across 10 military and civilian cohorts identifies methylation changes in AHRR. Nat Commun. 2020;11:5965.
Parade SH, Huffhines L, Daniels TE, Stroud LR, Nugent NR, Tyrka AR. A systematic review of childhood maltreatment and DNA methylation: candidate gene and epigenome-wide approaches. Transl Psychiatry. 2021;11:134.
Houtepen LC, Hardy R, Maddock J, Kuh D, Anderson EL, Relton CL, et al. Childhood adversity and DNA methylation in two population-based cohorts. Transl Psychiatry. 2018;8:266.
Khulan B, Manning JR, Dunbar DR, Seckl JR, Raikkonen K, Eriksson JG, et al. Epigenomic profiling of men exposed to early-life stress reveals DNA methylation differences in association with current mental state. Transl Psychiatry. 2014;4:e448.
Cecil CA, Smith RG, Walton E, Mill J, McCrory EJ, Viding E. Epigenetic signatures of childhood abuse and neglect: Implications for psychiatric vulnerability. J Psychiatr Res. 2016;83:184–94.
Suderman M, Borghol N, Pappas JJ, Pinto Pereira SM, Pembrey M, Hertzman C, et al. Childhood abuse is associated with methylation of multiple loci in adult DNA. BMC Med Genomics 2014;7:13.
Misiak B, Szmida E, Karpiński P, Loska O, Sąsiadek M, Frydecka D. Lower LINE-1 methylation in first-episode schizophrenia patients with the history of childhood trauma. Epigenomics. 2015;7:1275–85.
Wortinger LA, Engen K, Barth C, Lonning V, Jørgensen KN, Andreassen OA, et al. Obstetric complications and intelligence in patients on the schizophrenia-bipolar spectrum and healthy participants. Psychol Med. 2020;50:1914–22.
Wallwork RS, Fortgang R, Hashimoto R, Weinberger DR, Dickinson D. Searching for a consensus five-factor model of the positive and negative syndrome scale for schizophrenia. Schizophr Res. 2012;137:246–50.
Rush A, Gullion C, Basco M, Jarrett R, Trivedi M. The inventory of depressive symptomatology (IDS): psychometric properties. Psychol Med. 1996;26:477–86.
Pedersen G, Hagtvet KA, Karterud S. Generalizability studies of the global assessment of functioning-split version. Compr Psychiatry. 2007;48:88–94.
Bernstein DP, Fink L. Childhood trauma questionnaire: a retrospective self-report manual.San Antonio: Harcourt Brace and Company; 1998. 68 p.
Bernstein DP, Stein JA, Newcomb MD, Walker E, Pogge D, Ahluvalia T, et al. Development and validation of a brief screening version of the childhood trauma questionnaire. Child Abus Negl. 2003;27:169–90.
Aas M, Andreassen OA, Aminoff SR, Færden A, Romm KL, Nesvåg R, et al. A history of childhood trauma is associated with slower improvement rates: findings from a one-year follow-up study of patients with a first-episode psychosis. BMC Psychiatry. 2016;16:126.
Min JL, Hemani G, Davey Smith G, Relton C, Suderman M. Meffil: efficient normalization and analysis of very large DNA methylation datasets. Bioinformatics. 2018;34:3983–9.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47-e.
Pedersen BS, Schwartz DA, Yang IV, Kechris KJ. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 2012;28:2986–8.
Core TR. R: A language and environment for statistical computing.: R Foundation for Statistical Computing; 2019.
Du P, Zhang X, Huang C-C, Jafari N, Kibbe WA, Hou L, et al. Comparison of beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587.
Hansen KD IlluminaHumanMethylationEPICanno.ilm10b2.hg19: Annotation for Illumina’s EPIC methylation arrays. R package version 0.6.0 ed 2016.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol). 1995;57:289–300.
Christoforou A, Dondrup M, Mattingsdal M, Mattheisen M, Giddaluru S, Nothen MM, et al. Linkage-disequilibrium-based binning affects the interpretation of GWASs. Am J Hum Genet. 2012;90:727–33.
Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics. 2015;32:286–8.
UCSC Genome Browser. Human Feb. 2009 (GRCh37/hg19) Assembly. Available from: https://genome.ucsc.edu/s/solbru/hg19_TANGO6.
Moffat JJ, Ka M, Jung E-M, Smith AL, Kim W-Y. The role of MACF1 in nervous system development and maintenance. Semin Cell Dev. Biol. 2017;69:9–17.
Camargo LM, Collura V, Rain JC, Mizuguchi K, Hermjakob H, Kerrien S, et al. Disrupted in schizophrenia 1 interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol Psychiatry. 2007;12:74–86.
Costas J, Suárez-Rama JJ, Carrera N, Paz E, Páramo M, Agra S, et al. Role of DISC1 interacting proteins in schizophrenia risk from genome-wide analysis of missense SNPs. Ann Hum Genet. 2013;77:504–12.
Tomppo L, Ekelund J, Lichtermann D, Veijola J, Järvelin M-R, Hennah W. DISC1 conditioned GWAS for psychosis proneness in a large Finnish birth cohort. PLoS ONE. 2012;7:e30643-e.
Osimo EF, Beck K, Reis Marques T, Howes OD. Synaptic loss in schizophrenia: a meta-analysis and systematic review of synaptic protein and mRNA measures. Mol Psychiatry. 2019;24:549–61.
Berdenis van Berlekom A, Muflihah CH. Snijders GJLJ, MacGillavry HD, Middeldorp J, Hol EM, et al. Synapse Pathology in Schizophrenia: A Meta-analysis of Postsynaptic Elements in Postmortem Brain Studies. Schizophrenia Bull 2019;46:374–86.
Wei C, Luo T, Zou S, Zhou X, Shen W, Ji X, et al. Differentially expressed lncRNAs and miRNAs with associated ceRNA networks in aged mice with postoperative cognitive dysfunction. Oncotarget. 2017;8:55901–14.
Girgenti MJ, Wang J, Ji D, Cruz DA, Traumatic Stress Brain Research G, Stein MB, et al. Transcriptomic organization of the human brain in post-traumatic stress disorder. Nat Neurosci. 2021;24:24–33.
Miller MW, Wolf EJ, Sadeh N, Logue M, Spielberg JM, Hayes JP, et al. A novel locus in the oxidative stress-related gene ALOX12 moderates the association between PTSD and thickness of the prefrontal cortex. Psychoneuroendocrinology. 2015;62:359–65.
Ensink JBM, Keding TJ, Henneman P, Venema A, Papale LA, Alisch RS, et al. Differential DNA methylation is associated with hippocampal abnormalities in pediatric posttraumatic stress disorder. Biol Psychiatry Cogn Neurosci Neuroimaging. 2021;6:1063–70.
Griggs BL, Ladd S, Saul RA, DuPont BR, Srivastava AK. Dedicator of cytokinesis 8 is disrupted in two patients with mental retardation and developmental disabilities. Genomics. 2008;91:195–202.
Krgovic D, Kokalj Vokac N, Zagorac A, Gregoric Kumperscak H. Rare structural variants in the DOCK8 gene identified in a cohort of 439 patients with neurodevelopmental disorders. Sci Rep. 2018;8:9449.
Martinez-Jacobo L, Ortiz-Lopez R, Rizo-Mendez A, Garcia-Molina V, Santuario-Facio SK, Rivas F, et al. Clinical and molecular delineation of duplication 9p24.3q21.11 in a patient with psychotic behavior. Gene. 2015;560:124–7.
Glessner JT, Li J, Wang D, March M, Lima L, Desai A, et al. Copy number variation meta-analysis reveals a novel duplication at 9p24 associated with multiple neurodevelopmental disorders. Genome Med. 2017;9:106.
Wang H, Xu J, Lazarovici P, Quirion R, Zheng W. cAMP response element-binding protein (CREB): a possible signaling molecule link in the pathophysiology of schizophrenia. Front Mol Neurosci. 2018;11:255.
Alboni S, Tascedda F, Corsini D, Benatti C, Caggia F, Capone G, et al. Stress induces altered CRE/CREB pathway activity and BDNF expression in the hippocampus of glucocorticoid receptor-impaired mice. Neuropharmacology. 2011;60:1337–46.
Fujino H, Kitaoka Y, Hayashi Y, Munemasa Y, Takeda H, Kumai T, et al. Axonal protection by brain-derived neurotrophic factor associated with CREB phosphorylation in tumor necrosis factor-α-induced optic nerve degeneration. Acta Neuropathologica. 2009;117:75–84.
Ren X, Rizavi HS, Khan MA, Bhaumik R, Dwivedi Y, Pandey GN. Alteration of cyclic-AMP response element binding protein in the postmortem brain of subjects with bipolar disorder and schizophrenia. J Affect Disord. 2014;152–14:326–33.
Nuernberg GL, Aguiar B, Bristot G, Fleck MP, Rocha NS. Brain-derived neurotrophic factor increase during treatment in severe mental illness inpatients. Transl Psychiatry. 2016;6:e985.
Manners MT, Brynildsen JK, Schechter M, Liu X, Eacret D, Blendy JA. CREB deletion increases resilience to stress and downregulates inflammatory gene expression in the hippocampus. Brain Behav Immun. 2019;81:388–98.
van den Oord EJ, Clark SL, Xie LY, Shabalin AA, Dozmorov MG, Kumar G, et al. A whole methylome CpG-SNP association study of psychosis in blood and brain tissue. Schizophr Bull. 2016;42:1018–26.
Huckins LM, Dobbyn A, Ruderfer DM, Hoffman G, Wang W, Pardinas AF, et al. Gene expression imputation across multiple brain regions provides insights into schizophrenia risk. Nat Genet. 2019;51:659–74.
Liu J, Siyahhan Julnes P, Chen J, Ehrlich S, Walton E, Calhoun VD. The association of DNA methylation and brain volume in healthy individuals and schizophrenia patients. Schizophr Res. 2015;169:447–52.
Lambertini L, Li Q, Ma Y, Zhang W, Hao K, Marsit C, et al. Placental imprinted gene expression mediates the effects of maternal psychosocial stress during pregnancy on fetal growth. J Dev Orig Health Dis. 2019;10:196–205.
Monteagudo-Sanchez A, Sanchez-Delgado M, Mora JRH, Santamaria NT, Gratacos E, Esteller M, et al. Differences in expression rather than methylation at placenta-specific imprinted loci is associated with intrauterine growth restriction. Clin Epigenetics. 2019;11:35.
Hess JL, Tylee DS, Mattheisen M. Schizophrenia Working Group of the Psychiatric Genomics C, Lundbeck Foundation Initiative for Integrative Psychiatric R, Borglum AD, et al. A polygenic resilience score moderates the genetic risk for schizophrenia. Mol Psychiatry. 2021;26:800–15.
Hannon E, Dempster E, Viana J, Burrage J, Smith AR, Macdonald R, et al. An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol. 2016;17:176.
Logue MW, Miller MW, Wolf EJ, Huber BR, Morrison FG, Zhou Z, et al. An epigenome-wide association study of posttraumatic stress disorder in US veterans implicates several new DNA methylation loci. Clin Epigenetics. 2020;12:46.
Andrews SV, Sheppard B, Windham GC, Schieve LA, Schendel DE, Croen LA, et al. Case-control meta-analysis of blood DNA methylation and autism spectrum disorder. Mol Autism. 2018;9:40.
Braun PR, Han S, Hing B, Nagahama Y, Gaul LN, Heinzman JT, et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl Psychiatry. 2019;9:47.
Hannon E, Lunnon K, Schalkwyk L, Mill J. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics. 2015;10:1024–32.
Dugué P-A, Jung C-H, Joo JE, Wang X, Wong EM, Makalic E, et al. Smoking and blood DNA methylation: an epigenome-wide association study and assessment of reversibility. Epigenetics. 2020;15:358–68.
McCartney DL, Stevenson AJ, Hillary RF, Walker RM, Bermingham ML, Morris SW, et al. Epigenetic signatures of starting and stopping smoking. EBioMedicine. 2018;37:214–20.
Bruckmann C, Islam SA, MacIsaac JL, Morin AM, Karle KN, Di Santo A, et al. DNA methylation signatures of chronic alcohol dependence in purified CD3(+) T-cells of patients undergoing alcohol treatment. Sci Rep. 2017;7:6605.
Scoccianti C, Ricceri F, Ferrari P, Cuenin C, Sacerdote C, Polidoro S, et al. Methylation patterns in sentinel genes in peripheral blood cells of heavy smokers: Influence of cruciferous vegetables in an intervention study. Epigenetics. 2011;6:1114–9.
Acknowledgements
We are very grateful to all patients who agreed to participate in the study and the NORMENT group for collecting the data. We thank the Research Council of Norway (#22327 and #273446), the KG Jebsen Stiftelsen, the South-Eastern Norway Health Authority (#2017060), and the Faculty of Medicine at the University of Bergen for a fellowship through the Medical Student Research Program for SL.
Author information
Authors and Affiliations
Contributions
A-KS, MA, AAO, OAA, IM, and SLH acquired the data. A-KS, TP, IM, and SLH designed the analysis. SL, A-KS, and TP performed the analysis. SL and SLH drafted the manuscript. All authors reviewed, edited, and finalized the manuscript before publication.
Corresponding author
Ethics declarations
Competing interests
OAA received a speaker’s honorarium from Lundbeck and Sunovion, and is a consultant for HealthLytix. The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Løkhammer, S., Stavrum, AK., Polushina, T. et al. An epigenetic association analysis of childhood trauma in psychosis reveals possible overlap with methylation changes associated with PTSD. Transl Psychiatry 12, 177 (2022). https://doi.org/10.1038/s41398-022-01936-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-01936-8
