
Abstract
Posttranslational modifications increase the complexity and functional diversity of proteins in response to complex external stimuli and internal changes. Among these, protein lipidations which refer to lipid attachment to proteins are prominent, which primarily encompassing five types including S-palmitoylation, N-myristoylation, S-prenylation, glycosylphosphatidylinositol (GPI) anchor and cholesterylation. Lipid attachment to proteins plays an essential role in the regulation of protein trafficking, localisation, stability, conformation, interactions and signal transduction by enhancing hydrophobicity. Accumulating evidence from genetic, structural, and biomedical studies has consistently shown that protein lipidation is pivotal in the regulation of broad physiological functions and is inextricably linked to a variety of diseases. Decades of dedicated research have driven the development of a wide range of drugs targeting protein lipidation, and several agents have been developed and tested in preclinical and clinical studies, some of which, such as asciminib and lonafarnib are FDA-approved for therapeutic use, indicating that targeting protein lipidations represents a promising therapeutic strategy. Here, we comprehensively review the known regulatory enzymes and catalytic mechanisms of various protein lipidation types, outline the impact of protein lipidations on physiology and disease, and highlight potential therapeutic targets and clinical research progress, aiming to provide a comprehensive reference for future protein lipidation research.
Similar content being viewed by others

Introduction
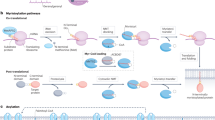
Over billions of years, organisms have acquired mechanisms, such as alternative splicing, cotranslational modifications and posttranslational modifications (PTMs), that allow genomes containing 20,000–25,000 genes to be converted into proteomes of more than one million proteins, thereby enhancing the complexity and functional diversity of proteins in response to complex external stimuli and internal changes.1 PTMs are covalent or enzymatic modifications that occur at the amino acid side chain of proteins after their biosynthesis. There are various types of PTMs, such as the addition of functional/chemical groups, the addition of polypeptide chains, the addition of complex molecules, and the modification of amino acids.2 To date, over 620 types of PTMs have been identified to regulate protein activity, and at least 20 PTMs have been extensively studied (Fig. 1) and found to be strongly associated with the pathogenesis of a variety of diseases.3

The main commonly studied posttranslational modifications (PTMs) that are associated with various diseases. PTMs play a critical role in a broad spectrum of biological processes. Here, we conclude that 25 common types of PTMs and dysregulation of those PTMs are strongly associated with the pathogenesis of a variety of diseases
Protein lipidation, one of the most common PTMs, can attach up to seven different types of lipids, including fatty acids (FA), lipoic acids, isoprenoids, sterols, phospholipids, glycosylphosphatidylinositol anchors, and lipid-derived electrophiles, to proteins.4 Based on the location of the modified proteins, protein lipidation can be divided into two categories. The first category comprises lipid modifications that occur in the cytoplasm or on the cytoplasmic side of membranes, including S-palmitoylation, N-myristoylation and S-prenylation. The other category is composed of lipid modifications that occur in the lumen of secretory organelles, such as glycosylphosphatidylinositol (GPI) anchor and cholesterylation. The 16-carbon fatty acid palmitate covalently linked to cysteine (Cys) residues of proteins via the labile thioester bond is defined as protein S-palmitoylation.5 Protein S-palmitoylation was first reported in 1979 and is catalysed by DHHC palmitoyl S-acyltransferases (DHHC-PATs), while depalmitoylation is mediated by depalmitoylases.6,7,8,9 Given the instability of the thioester bond, S-palmitoylation is characterised as the typical reversible type of lipidation.10 Techniques for detecting protein S-palmitoylation include radioactive isotope-labelled palmitic acid, click chemistry-based metabolic labelling with bioorthogonal palmitic acid probes, acyl-biotin exchange and others.11 In addition, there are other less frequently occurring palmitoylation types. O-palmitoylation is characterised by the attachment of palmitate to serine residues. N-palmitoylation is characterised by the attachment of palmitate to the N-terminus.12 The attachment of 14-carbon myristic acid to N-terminal glycine residues via an amide bond in a manner of co- or posttranslational modification is referred to as N-myristoylation.13,14 Protein N-myristoylation was first reported in 1982,15 and is catalysed by N-myristoyltransferase (NMT). Approaches for detecting protein N-myristoylation include radioactive-labelled fatty acids and bioorthogonal probes such as azido or alkynyl myristate analogues.14 Canonical N-myristoylation occurring on glycine residues is irreversible, but when myristic acid is attached to lysine residues, fatty acyl groups from Nε-modified Lys residues can be removed by sirtuins and histone deacetylases (HDACs),16,17 suggesting that lysine myristoylation is another reversible lipidation following S-palmitoylation. S-prenylation refers to the attachment of a 15-carbon farnesyl or a 20-carbon geranylgeranyl isoprenoid lipid onto Cys residues of proteins via a thioether bond.18 Protein S-prenylation was first reported in fungi in 1978 and later in mammals,19,20 and is mainly catalysed by protein farnesyltransferase (FTase) or protein geranylgeranyltransferase I (GGTase-I). Protein S-prenylation can be detected by bioorthogonal probes such as alkynyl isoprenoid probes and azido-isoprenoid probes.14 Most prenylated proteins belong to the small GTPase family, including the Ras subfamily, the Rab subfamily, the Rho subfamily and others.21 Generally, the C-terminal CAAX motif of substrate proteins is the structural basis for enzyme recognition, and after the Cys residue is prenylated, the -AAX residues are removed by RCE1, followed by a methyl group transfer to the isoprenoid-modified Cys residue by ICMT.22 The GPI anchor refers to the amino group of EtNP at the end of GPI attached to the carboxyl group at the C-terminus of the protein via an amide bond. During translocation from the endoplasmic reticulum (ER) to the plasma membrane (PM), nascent GPI-anchoring proteins (GPI-APs) undergo several remodelling steps on the immature GPI structure before maturation and finally anchor to the outer leaflet of the lipid bilayer.23 Detection of GPI-APs can be achieved by chemical probes such as a bifunctional analogue probe of the conserved glucosaminylphosphatidylinositol motif.24 To date, it is estimated that more than 250 GPI-APs are present in eukaryotes with various functions, including enzymes, receptors, antigens, and adhesion molecules, therefore mediating a myriad of biological functions.25 Cholesterylation refers to the covalent attachment of cholesterol to proteins in an autoprocessing manner,26 which can be labelled and detected by azido- and alkyne-modified cholesterol analogues.14 Currently known cholesterylated proteins include hedgehog (Hh) and smoothened (SMO), which are key molecules in the Hh signalling pathway and regulate a variety of important physiological activities such as embryonic development.27 The timeline of discovery of the five lipidation types is exhibited in Fig. 2.

The timeline of protein lipidation discoveries. The figure illustrates the timeline of protein lipidation research from their origin to the most advanced scientific discoveries, including regulatory enzymes, important protein substrates, clinical trials and the development of inhibitors
Lipid attachment to proteins plays a crucial role in regulating protein trafficking, localisation, stability, conformation, interactions, and signal transduction by enhancing the hydrophobicity of proteins, which serve as fundamental regulators of protein physiological functions. An increasing number of studies have revealed that aberrant regulation of lipidation is implicated in the pathogenesis of various diseases, such as cancer, neurological disorders, cardiovascular diseases, inflammatory diseases, metabolic disorders, infectious diseases and others, suggesting that elucidating the role of lipidation regulation in disease progression is promising for the development of potential therapeutic targets. In this review, we discuss the known regulatory enzymes and catalytic mechanisms of various lipidation types, outline the role of protein lipidation in physiology and disease, and highlight potential therapeutic targets and clinical research progress. We systematically review the relevant advances in protein lipidation with the aim of providing a comprehensive protein lipidation reference.
S-palmitoylation
Regulatory enzymes and catalytic mechanism
S-palmitoylation is catalysed by a specific class of enzymes called DHHC-PATs, which share a common structural feature of a highly conserved Asp–His–His–Cys tetrapeptide motif within a cysteine-rich region, which is where the catalytic reaction occurs.8,28 To date, 23 DHHC-PATs have been identified in Homo sapiens and several different DHHC proteins have been found in other species.29 As a type of polytopic integral membrane protein, DHHC-PATs possess 4-7 transmembrane domains (TMDs) and mainly localise to the Golgi and ER, while a minority localise to the PM, mitochondria and perineuclears regions.30,31 The localisation of DHHC-PATs partly depends on structure and motif. For example, lysine-based sorting signals on the sequences of DHHC4 and DHHC6 enable them to form KXX and KKXX motifs, thus guiding them to the ER membrane.32 DHHC1, 2, 4, 9, 12, 14, 20, and 22 can traffic between the ER and Golgi.33 Among DHHC-PATs, DHHC20 is most widely distributed in the cytosol, including PM, ER, Golgi and perineuclears regions.31,34,35
Although 23 DHHC-PATs are known to be responsible for catalysing protein S-palmitoylation, the exact process remains obscure. Previous studies have found that some DHHC-PATs go through two steps to catalyse substrate S-palmitoylation (ping-pong kinetic mechanisms)36 (Fig. 3). First, DHHC-PAT undergoes autopalmitoylation, in which the thiolate nucleophile of Cys156 attacks the carbonyl carbon in the palmitoyl-CoA thioester to form an acyl-enzyme transfer intermediate that links palmitoyl-CoA to Cys156 in the DHHC domain. Subsequently, the palmitoyl chain is transferred to the putative cysteine residue on the substrate protein to complete S-palmitoylation.35,37 However, autopalmitoylation of DHHC13, 17, 19 and 22 was not detectable by tritiated palmitate or alkyne fatty acid analogues,38,39,40 and more investigations are needed to explore whether the autopalmitoylation of these enzymes are not possible with other detection techniques or whether autopalmitoylation does not occur in all DHHC-PATs. Although the DHHC domain provides the key catalytic core and fatty acid binding cavity, understanding the mechanism of S-palmitoylation is clouded by exploring the role of the conserved DHHC domain alone. For instance, in addition to the DHHC domain, other conserved motifs such as Asp–Pro–Gly (DPG), Thr–Thr-x–Glu (TTxE), and PaCCT (palmitoyltransferase conserved C- terminus) have been reported to be adjacent to transmembrane domains.8 The second threonine of the TTxE motif could directly interact with the aspartic acid of the DHHC motif to form a hydrogen bond, and Asn266 in the PaCCT motif exploits its H-bond capabilities to assist the side chain in forming an amide, indicating that those conserved residues are potentially involved in substrate protein recognition and the catalytic process through some crucial contacts with the DHHC domain.35 In addition, some DHHC-PATs require interactions with cofactors to form complexes, such as the DHHC9/GCP16 complex41 and DHHC6/Selk complex,42 or need to be palmitoylated by other DHHC family members to provoke S-palmitoylation cascades before regulating substrate S-palmitoylation.43,44 Moreover, Robyn Stix et al. conducted an all-atom molecular dynamics simulation experiment and discovered that during the catalytic process, hDHHC20 induced local deformation of membranes, especially in the cytosolic site where cysteine is catalytic, allowing cysteine to be optimally positioned for autopalmitoylation with fatty acyl-CoA.45 However, whether this enzyme-induced membrane deformation and the adjustment of the catalytic site position are common phenomena in the palmitoylation process remain to be further explored in other DHHC-PATs. In addition, although the sequence for the S-palmitoylation site has not yet reached a consensus, some deep learning-based palmitoylation site prediction websites, such as GPS-Palm, can provide important clues for researchers to explore various palmitoylation sites,46 which immensely aids the study of protein S-palmitoylation.

Catalytic mechanisms of DHHC palmitoyl S-acyltransferases (DHHC-PATs) and depalmitoylases. Some DHHC-PATs go through two steps (ping-pong kinetic mechanisms) to catalyse substrate protein S-palmitoylation. First, DHHC-PAT undergoes autopalmitoylation, linking palmitoyl-CoA to Cys156 in the DHHC domain to form an acyl-enzyme transfer intermediate. Subsequently, the palmitoyl chain is transferred to the putative cysteine residue on the substrate protein to complete S-palmitoylation. For depalmitoylases, three categories of depalmitoylases are subject to being palmitoylated to arrive at proper positions to function. A portion of palmitoylated APT1 and APT2 localise to the Golgi, and APT1 is capable of depalmitoylating itself and APT2, which allows them to be released into the cytosol to ensure the steady-state distribution of APTs in the Golgi and the cytosol. A large proportion of palmitoylated proteins are depalmitoylated by APT1 and APT2 on the plasma membrane or in the cytosol. Some palmitoylated proteins on the plasma membrane could also be depalmitoylated by ABHDs. PPT1 and PPT2 are mainly responsible for depalmitoylation of substrate proteins that localise in lysosomes. There are two fates for substrate proteins after depalmitoylation. One of them returns to catalytic positions, such as the Golgi, to undergo repalmitoylation, and the other enters the lysosome to be degraded
The functional redundancy of DHHC-PATs as well as substrate redundancy complicates the resolution of individual DHHC characteristics.47,48 It remains unclear whether the recognition sites of the enzyme-substrate interaction, as well as the catalytic sites where S-palmitoylation occurs, are the same when the protein substrate can be palmitoylated by multiple DHHC-PATs. The degree of binding specificity between DHHC-PAT and its substrates has attracted the attention of researchers. Some unique protein‒protein interaction motifs, such as PDZ domains in substrate proteins and ankyrin repeats and the SH3 domain in DHHC-PATs, may provide docking capabilities to participate in substrate selectivity.49 For instance, the S-palmitoylation of the multi-PDZ domain-containing protein GRIP1b by DHHC5/8 requires a PDZ ligand-dependent recognition mechanism, as well as the S-palmitoylation of PSD93 by DHHC14.49,50 In addition, ankyrin repeats in the N-terminus of DHHC13 and DHHC17 and the type II PDZ-binding motif of DHHC6 may participate in substrate recruitment and regulation.51 The fusion of ankyrin repeats to the N-terminus of DHHC3 endows DHHC3 with the ability to regulate the S-palmitoylation of huntingtin proteins and their trafficking, which would otherwise not occur.51 Although enzyme functional and substrate redundancy has hindered the identification of individual DHHC-PATs to some extent, redundancy exists so that substrate proteins can be regulated by corresponding DHHC-PATs in different compartments or single DHHC-PATs can catalyse a fair amount of substrates to meet physiological needs.52 In turn, the localisation of DHHC-PATs in different organelles is also involved in substrate selectivity.
To date, there is still an absence of small-molecule probes or drugs that specifically inhibit DHHC-PATs. Among various lipid-based covalent inhibitors that paninhibit DHHC-PATs, 2-bromopalmitate (2-BP) is the most widely used and irreversibly inhibits S-palmitoylation by occupying the lipid binding cavity of the DHHC protein.35,53 Nevertheless, there are many concerns about using 2-BP to study the physiological or pathological role of S-palmitoylation, as in addition to inhibiting protein S-palmitoylation, 2-BP also regulates lipid synthesis, transport, and metabolism and even inhibits the depalmitoylases acyl protein thioesterase 1 (APT1) and APT2.48,54 To overcome the main shortcomings of 2-BP, Tong Lan et al. developed a novel broad-spectrum DHHC inhibitor cyanomethyl-N-myracrylamide (CMA), and they found that significant inhibition of the S-palmitoylation level in protein substrates was achieved by lower concentrations of CMA than 2-BP, and that CMA did not disturb the activities of APT1 and APT2.54 However, there is a paucity of direct experimental data on CMA that support these findings. Agents that selectively inhibit DHHC-PAT must be developed to overcome the current shortcomings in the study of S-palmitoylation.
Protein depalmitoylation is mainly achieved by cleavage of the thioester bond on palmitoylated proteins. Enzymes responsible for regulating depalmitoylation can be divided into three categories, namely APTs, palmitoyl‐protein thioesterases (PPTs) and ABHD17 family thioesterases.9 They all belong to the ‘metabolic’ serine hydrolase superfamily, with an active site serine for substrate hydrolysis and the ability to cleave ester, amide, or thioester bonds on small molecules, peptides and proteins.55,56 APT1 was initially recognised for its capability to hydrolyse a variety of lysophospholipids. It was not until 1998 that Duncan et al. discovered that it could remove radiolabelled palmitate from the α‐subunit of G‐proteins and other proteins, and it had a stronger depalmitoylation activity.57 It is generally believed that APT1 mainly localises to the cytosol, but it has also been reported that APT1 can localise to the PM, Golgi, ER, mitochondria and even the nuclear membrane to perform its catalytic function.58,59,60 APT2 is another depalmitoylase primarily localised in the cytosol and shares nearly 60% identity with APT1.55 Previous reports have shown that both APT1 and APT2 are palmitoylated at the Cys residue at position 2. APT1 is capable of depalmitoylating itself and APT2, rendering both APT1 and APT2 dynamically palmitoylated.61 The subcellular localisation and depalmitoylation activity of APTs are closely linked to the regulation of APT itself by dynamic palmitoylation.61 Vartak et al. proposed that due to dynamic palmitoylation regulation, a substantial fraction of palmitoylated APT1 and APT2 is recruited to the Golgi apparatus, whereas they undergo depalmitoylation in trans, resulting in their release into the cytosol to ensure the steady-state distribution of APTs in the Golgi and cytosol, and the depalmitoylation of palmitoylated proteins in various parts of the membrane is regulated by nonpalmitoylated APTs, which is crucial for substrate proteins to be repalmitoylated to execute their functions.61 Although these enzymes share approximately 60% identity, studies suggest that there are preferences for specific palmitoylated substrates between APT1 and APT2. For example, APT1, but not APT2, reverses agonist-dependent S-palmitoylation of β2-adrenergic receptors.62 The depalmitoylation of GAP-43 and DHHC6 depends on APT2 rather than APT1.43,63 The structure of apo-APT1 was first reported as early as 2000, but it was not until 2016 that the high-resolution structure of the complex formed by the binding of APT1/2 to their respective inhibitors ML348 and ML349 was resolved, contributing to resolving the functional differences between APT1 and APT2.64 These enzymes include unique structures such as the β5-α2 loop.64 The β5-α2 loop and residues around the catalytic triad contribute to the formation of hydrophobic channels that regulate the entry and engagement of hydrophobic substrates of different lengths by altering the openness, which is irrelevant to the selectivity of palmitoylated substrates.64 More in-depth studies are needed to explore the factors involved in the selectivity of palmitoylated substrates.
In addition to cytosolic APTs, another class of enzymes PPTs (PPT1 and PPT2), localise in lysosomes and are involved in protein depalmitoylation. PPT1 was the first enzyme found to have the function to depalmitoylate protein.65 Normally, PPT1 targets lysosomes via the mannose 6-phosphate receptor-mediated pathway and facilitates the degradation of substrate proteins by depalmitoylating them in the lysosomes.66,67,68 Previously, it was thought that PPT1 depalmitoylation activity plays a major part in protein degradation in lysosomes.69 However, Erica et al. found that in the PPT1 knockout mouse model, most substrate proteins exhibited an increase in palmitoylation, which was accompanied by unchanged or decreased protein levels, indicating that the depalmitoylation activity of PPT1 was not entirely for protein degradation.70 PPT2 is another lysosomal thioesterase homologous to PPT1 and shares 26% identity with PPT1.71 PPT2 does not have thioesterase activity for some substrates such as S-palmitoyl H-Ras. The crystal structures of PPT1 and PPT2 reveal that although both have similar architectural features, conformational differences near the active site partially explain substrate selectivity, as the entrance space of the lipid binding site consisting of β3-αA and β8-αF in PPT1 is larger than that in PPT2 and thus more inclusive of the substrates.71 Deficiency of PPT1 or PPT2 results in serious neurodegeneration, while overexpression of PPT1 or PPT2 may accelerate tumour growth.72,73,74,75
ABHDs including ABHD10, ABHD12 and ABHD17A/B/C have been identified as another category of cytosolic depalmitoylases.76 ABHD10 was found to modulate the depalmitoylation of the key antioxidant protein PRDX5, and ABHD12 could reduce PSD95 S-palmitoylation.9,77 ABHD17 proteins mainly regulate the depalmitoylation of proteins on the PM.78 With the subsequent identification of other substrate proteins, the possibility that ABHD17 regulates the depalmitoylation of substrates at other locations cannot be ruled out. ABHD17 also undergoes S-palmitoylation, which facilitates its targeting of postsynaptic membranes and enhancement of enzymatic activity.9 ABHD17 has a dual function. On the one hand, it promotes the depalmitoylation of the substrate protein PSD95 for dynamic palmitate turnover on proteins. On the other hand, it reduces the autopalmitoylation of DHHC proteins, increasing the complexity of the palmitoylation regulatory network.79 ABHD family proteins and APTs exhibit partial substrate redundancy. However, there are limited reports on the depalmitoylase activity, substrate-binding structure features, enzyme kinetics, substrate preferences and other physiological roles of ABHD family proteins. More efforts are needed to deepen the understanding of depalmitoylases for the development of corresponding small-molecule drugs for treatment.
Both broad-spectrum and selective inhibitors are used to inhibit the catalytic activity of despalmitoylases. Palmostatin B, a classical broad-spectrum inhibitor, has been shown to block the activities of APT1/2, PPT1, ABHD17A/B/C and other lipid-processing serine hydrolases with varying potency.9,80 Palmostatin M, another inhibitor structurally related to Palmostatin B, has similar biochemical IC50 values but is more active in cells.81 ML348 and ML349 are piperazine amide competitive inhibitors of APT1 and APT2, respectively. Structurally, ML348 is located above the catalytic triad and blocks it by contacting hydrophobic residues.64 ML349 blocks the function of APT2 by indirectly contacting the catalytic triad with the formation of a hydrogen bond network through the sulfur group and the water molecule at the catalytic active site.64 Selective inhibitors targeting the depalmitoylation activity of PPTs are not available. Recently, two autophagy inhibitors, GNS561 and hydroxychloroquine (HCQ), were shown to inhibit PPT1 and thus target lysosomes for cancer therapy.75,82 Intriguingly, inhibition of PPT1 with HCQ increased the palmitoylation level of AEG-1, which is regulated by PPT1/2, demonstrating that HCQ is a potential inhibitor of the depalmitoylation activity of PPT1.83 Regarding selective inhibitors that inhibit the ABHD17 enzyme, although ABD957 may act as a selective covalent inhibitor of the ABHD17 enzyme compared with traditional broad-spectrum inhibitors according to MS-ABPP data and in situ time-course ABHD17A/B/C inhibition experiments, it partially rescued the palmitoylation level of N-Ras.78 Future efforts are needed to elucidate the localisation and interaction patterns of ABHD17 enzymes and perform detailed biochemical and phenotypic analyses of each isoform, which could also benefit the development of relevant selective inhibitors.
Physiological function
Protein trafficking and membrane localisation
S-palmitoylation is capable of regulating protein trafficking and localisation, the hydrophobic palmitate covalently attached to the protein acts as a lipid anchor to increase protein lipophilicity and facilitate its interaction with the membrane.84 The dynamic palmitoylation-depalmitoylation cycle facilitates the trafficking of substrate proteins between the Golgi, PM, and endosomal recycling system by modulating their hydrophobicity.5 For example, the fatty acid transporter CD36 (also known as the scavenger receptor) is a widely expressed membrane glycoprotein with various metabolic functions, such as the regulation of fatty acid uptake.85 S-palmitoylation increases the incorporation of CD36 into the PM by enhancing its hydrophobicity, whereas inhibition of S-palmitoylation leads to CD36 accumulation on the ER.86 DHHC4 and DHHC5 were found to mediate CD36 S-palmitoylation, with DHHC4 responsible for CD36 S-palmitoylation at the Golgi, followed by vesicle-mediating trafficking of CD36 to the PM, while DHHC5 is responsible for keeping CD36 at the PM.87 Another study discovered that during the process of fatty acid uptake, CD36 was under the control of dynamic S-palmitoylation regulation.88 This was mainly reflected in the downstream kinase LYN being activated to phosphorylate DHHC5 when fatty acids interact with CD36.88 Phosphorylated DHHC5 loses its catalytic capacity, and CD36 is further depalmitoylated by APT1 to initiate endocytic uptake of FA.88
Proper localisation of proteins, mediated by S-palmitoylation in specific subcellular compartments, directly influences cell polarity. The apical‒basal polarity of epithelial cells regulates their proliferation, apoptosis, and migration.89 Loss of cell polarity leads to tissue disorganisation, abnormal proliferation and migration, and transformation of epithelial cells towards cancer.89 SCRIB, as a cell junction localisation protein, is a major regulator of epithelial cell polarity.89 A double mutation at Cys4 and Cys10 (C4/10S) completely blocked SCRIB S-palmitoylation and caused a diffuse distribution of SCRIB in the cytosol rather than its proper localisation in cell‒cell junctions, which resulted in luminal structure disruption and partial loss of cell polarity.90 In addition to epithelial cells, neurons are also highly polarised cells. In neurons, S-palmitoylation regulates synaptic activity and the targeted trafficking of cytosolic proteins to axons and dendrites.91 In regard to synaptic activity regulated by S-palmitoylation, as an example, research on the S-palmitoylation of AMPARs, an ionotropic glutamate receptor crucial to synaptic transmission and synaptic plasticity, gradually elucidated the complex regulation of S-palmitoylation.84 AMPAR has four subunits, GluA1-4, each with two palmitoylation sites in TMD2 and the C-terminus.92 S-palmitoylation at TMD2 increases receptor internalisation and accumulation in the Golgi, and depalmitoylation at this site promotes AMPAR trafficking from the Golgi to the cell surface.92 The S-palmitoylation of the C-terminal site regulates the interaction of AMPAR with 4.1 N, and depalmitoylation increases the affinity with AMPAR-associated proteins such as 4.1 N to maintain AMPAR on the cell surface.92 Interestingly, the majority of AMPAR-associated proteins, including PSD95, PICK1, GRIP/ABP, AKAP79/150 and others, are subject to S-palmitoylation, but S-palmitoylation of these proteins often produces opposite effects to those of the self-palmitoylation of AMPAR. Both AMPAR and PSD95 are substrates of DHHC3, indicating that S-palmitoylation regulates AMPAR trafficking and localisation in a dynamic and complex network. Any disturbances to this network can cause synaptic AMPAR disorders and lead to the occurrence of related diseases.84
While many proteins rely on S-palmitoylation for PM localisation, some proteins exhibit the opposite behaviour upon palmitoylation. FLT3 is a receptor tyrosine kinase that is mainly involved in haematopoietic development, and FLT3 internal tandem repeat (FLT3-ITD) is a common mutation in acute myeloid leukaemia (AML).93 FLT3-ITD is mainly localised to the ER rather than the PM, but increased PM localisation of FLT3-ITD was found after mutation of the palmitoylation site Cys563 and disruption of its S-palmitoylation, which was subsequently followed by the activation of Akt and ERK as well as sustained STAT5 activation, finally promoting the development of AML.93
While S-palmitoylation is known to facilitate the intracellular transport of substrate proteins by increasing their affinity to membranes, the underlying mechanism remains under investigation. Research indicates that after palmitoylation, substrate proteins aggregate in the curved region of the Golgi margin. Other domains of these proteins then interact with sorting adapters. From these interactions, transport vesicles form and mediate the movement of the substrate proteins to various intracellular compartments, and once they arrive at destinations such as newly formed axons, they will be separated from the vesicles and locally retained through depalmitoylation.5,94,95 For some proteins, additional lipidation is necessary for protein trafficking and PM localisation. For instance, H-Ras, N-Ras and K-Ras4a undergo S-palmitoylation and S-prenylation.96 Among them, H-Ras needs two palmitoyl groups for recycling endosome targeting and subsequent PM localisation. Such additional lipidation further enhances its hydrophobicity and sufficiently ensures the binding of the protein to the membrane.97
Protein stability and degradation
S-palmitoylation is also implicated in the regulation of protein stability and degradation, and the absence or blockade of S-palmitoylation usually leads to increased protein transport to lysosomes for degradation. The stability of PD-L1, a notable example, was found to be enhanced by S-palmitoylation.98 The development of checkpoint blockade therapies targeting PD-1 or PD-L1 is a major breakthrough in overcoming immune tolerance.99 Antibodies against PD-L1 block the transduction of inhibitory signals by binding to PD-L1 on the cell surface. However, these antibodies cannot inhibit the intracellular portion or its redistribution to the PM. Han et.al found that S-palmitoylation of PD-L1 is closely related to its stability.98 DHHC3 palmitoylated the Cys272 site in the transmembrane domain of PD-L1. Inhibition of PD-L1 depalmitoylation with ML348 or Palmostatin B resulted in an increase in PD-L1 intracellular expression levels, whereas inhibition of S-palmitoylation with 2-BP generated the opposite effect. Blocking the S-palmitoylation of PD-L1 did not affect its interaction with CMTM6, a molecule that facilitates the trafficking of PD-L1 to recycling endosomes, but rather promoted its lysosomal degradation by inducing PD-L1 ubiquitination through the ESCRT-MVB pathway.98 Therefore, targeting the S-palmitoylation of PD-L1 may represent a promising immunotherapy. Inhibition of PD-L1 S-palmitoylation can reduce its overall intracellular storage by promoting its degradation.98 Combined with antibodies against PD-L1 on the cell membrane, a strong and long-lasting antitumour synergistic effect may be achieved.
In addition to PD-L1, there are a number of proteins that are protected from lysosomal degradation by S-palmitoylation, including Oct4,100 NOD2,101 CLDN3,102 and Fas.103 Conversely, S-palmitoylation can also facilitate protein degradation under certain conditions. For instance, in hepatocellular carcinoma (HCC) cells, when the S-palmitoylation of AEG-1 was reduced by a point mutation in the palmitoylation site C75 or knockout of the corresponding palmitoyltransferase DHHC6, the protein degradation rate was significantly reduced. Subsequent mechanistic studies revealed that the AEG-1 variant was weakly bound to the E3 ubiquitin ligase FBXW7, resulting in decreased ubiquitination and increased stability of AEG-1.83 The negative regulation of protein stability by S-palmitoylation also occurs in NLRP3. As a component of the inflammasome, NLRP3 is degraded in lysosomes through chaperone-mediated autophagy after being palmitoylated.104 In addition, palmitoylated IFNGR1 is also sorted to lysosomes for degradation through binding to AP3D1, one of the subunits of AP3, which is important in cargo sorting and trafficking.105 S-palmitoylation exerts a dual effect on the stability of substrate proteins, either promoting or inhibiting their degradation. This duality might stem from how S-Palmitoylation influences protein–protein interactions. S-palmitoylation tends to promote degradation when it enhances the binding of substrate proteins to molecules associated with degradation processes. In contrast, S-palmitoylation exhibits antidegradation effects when it hinders the binding of substrates to degradation-related proteins.
Protein conformation and interaction
Beyond regulating protein trafficking, localisation, and stability, S-palmitoylation can also modify the accessibility of vital active sites, altering protein conformation and disrupting interactions with other proteins. A typical example of how S-palmitoylation meditates protein conformation is caspase-6 (CASP6). CASP6, a member of the cysteine protease family that is mainly involved in the regulation of cell apoptosis and the immune response.106 By constructing the crystal structure model of CASP6, it was found that CASP6 had four loops that formed the activation site with the central β-sheet.107 CASP6 is palmitoylated by DHHC17 at Cys264 on loop 4 and Cys277 near the start of the β-sheet.108 After palmitoylation at Cys264, the flexibility of loop 4 was increased, and the interaction between loop 4 and loop 2 became stronger, which led to the blockage of the activation site and increased the distance between the catalytic dyads, which prevented the activation of CASP6 via nucleophilic attack by C163 on D193.108 Moreover, during the palmitoylation at Cys277, Cys277 occupies the active site by linking palmitic acid, thereby blocking the activation and dimerisation of CASP6.108 In addition, PSD95 is another protein whose conformation and interactions are influenced by S-palmitoylation. S-palmitoylation induces a conformational change from extended to compact PSD95, which enables its direct binding to NMDAR and AMPAR subunits.109 DHHC-PATs and depalmitoylases are more readily aggregated into AMPAR nanodomains, making PSD95 S-palmitoylation and conformation more dynamic when interacting with AMPAR.109 Conversely, the state of PSD95 is more stable in the NMDAR nanodomains due to the strong interaction between PSD95 and SAP97, demonstrating that the precise regulation of PSD95 by S-palmitoylation in separate postsynaptic density domains plays an important role in synaptic plasticity.109 STING is a key sensor for cytosolic DNA-triggered immune responses, and it has been confirmed in multiple studies that S-palmitoylation is needed for STING activation in the Golgi and type I IFN responses.110,111 Recently, a study revealed that STING governs tumour growth through its interaction with VDAC2 on mitochondria, independent of classical innate immunity.112 Both STING and VDAV2 can be palmitoylated, while only cysteine mutations in STING but not VDAV2 significantly affect their interaction.112
Signal transduction
S-palmitoylation plays a role in regulating various cellular signal transduction pathways, primarily by influencing the aforementioned protein functions. At present, the related signalling pathways involved in tumours have received much attention, including the Ras signalling, EGFR signalling, Wnt signalling, Fas/FasL signalling, DNA damage repair, G protein-coupled receptor (GPCR) signalling and multiple death receptor signalling pathways.113,114 In addition, increasing evidence has found that S-palmitoylation also plays an important role in innate and adaptive immune signalling, such as the TLR signalling pathway, NLR signalling pathway, cGAS-STING signalling pathway, sensing DNA and RNA pathway, TNF α-TNF receptor 1 signalling pathway, T-cell coreceptor pathway, PD-1/PD-L1 signalling pathway, B-cell signalling and fragment crystalisable receptor (FcRs) signalling pathway.115 The role of S-palmitoylation in these signalling pathways has been previously discussed.113,114,115 Here, we discussed the functions of S-palmitoylation in some important signalling pathways to enrich our knowledge and understanding (Fig. 4). For instance, the cGAS-STING pathway is critical for triggering innate immune responses. For STING, palmitoylation is needed for its activation. However, palmitoylation of cGAS meditated by DHHC18 reduces the interaction between cGAS and double-stranded DNA, thereby negatively regulating cGAS-mediated innate immune responses, suggesting the dual effects of palmitoylation on signalling.116 Zhang et al. revealed that S-palmitoylation promotes Th17 differentiation during STAT3 signalling. STAT3 is involved in a palmitoylation-depalmitoylation cycle in colitis. DHHC7 palmitoylates STAT3 in the cytosol, which allows STAT3 to traffic and translocate to the PM for subsequent phosphorylation catalysed by JAK2, and then phosphorylated STAT3 is depalmitoylated by APT2, which leads to STAT3 detachment from the PM and transport to the nucleus to activate the downstream genes RORC and IL17A and initiate the differentiation of TH17 cells.117 This study demonstrated that S-palmitoylation regulated the trafficking and localisation of STAT3 as a prerequisite for STAT3 phosphorylation.

S-palmitoylation is involved in the regulation of multiple cellular signal transduction pathways. We summarised several crucial and well-established signalling pathways that are under the control of S-palmitoylation. The majority of proteins are subjected to S-palmitoylation at the ER or Golgi by the respective DHHC-PATs. Some protein substrates could traffic from the ER to the Golgi or Golgi to the plasma membrane after being palmitoylated to activate the corresponding signalling cascades (e.g., EGFR pathway, N-Ras/H-Ras pathway, GPCR pathway, AGK-Akt-mTORC1 pathway, STING pathway and LRP6 pathway). In some cases, protein substrates anchor at the original organelle after being palmitoylated. Once S-palmitoylation is blocked, these protein substrates detach from the original organelle to abnormally activate the corresponding signalling pathway (e.g., SCRIB pathway and FLT3-ITD pathway). In addition to regulating protein trafficking, some proteins are protected from degradation in lysosomes after being palmitoylated to further activate the corresponding signalling pathway (e.g., Fas pathway and PD-L1 pathway). In addition, S-palmitoylation can regulate the activation of signalling pathways by regulating protein‒protein interactions (e.g., the PCSK9-PI3K-Akt pathway). Furthermore, some protein substrates are under dynamic palmitoylation-depalmitoylation regulation, abnormally activating signalling pathways by increasing palmitate turnover (e.g., the STAT3 pathway). ER endoplasmic reticulum
The overactivation of the PI3K/Akt signalling pathway abnormally regulates cell-cycle progression, cell proliferation and metabolism, which is an important factor in the progression of a variety of tumours and other diseases.118 Recently, accumulating evidence demonstrated the role of S-palmitoylation in regulating the PI3K/Akt signalling pathway. For example, both AGK and GRK6 activate the PI3K/Akt signalling pathway by increasing Akt translocation to the PM through S-palmitoylation.119,120 In another study, PCSK9 was found to be related to cell proliferation and drug resistance in hepatocellular carcinoma, as the S-palmitoylation of PCSK9 increased its affinity with PTEN and induced it to target lysosomes for degradation, thereby removing the inhibitory effect of PTEN on Akt signalling and leading to abnormal activation of Akt signalling.121 These studies indicate that S-palmitoylation might influence signalling pathways indirectly, coordinating with other posttranslational modifications, such as protein phosphorylation. These findings offer new perspectives on the signalling role of S-palmitoylation.
Pathological implication
Cancer
Currently, multiple studies have highlighted the strong association between S-palmitoylation and cancer. This association can be seen in the retrieved results from SwissPalm, a protein S-palmitoylation database. By retrieving 299 previously identified tumour driver genes in SwissPalm,122,123 132 of them were detected at least once in multiple palmitoyl-proteomes from Homo sapiens, and 40 of them were detected at least once in palmitoyl-proteomes from other species. Moreover, a number of studies have also reported that S-palmitoylation also acts as a tumour suppressor in the process of tumour progression, and the protumour/antitumour effects of S-palmitoylation are highly variable in different cancers. However, the palmitoylation-regulating enzymes of these cancer-related proteins and how these protein substrates act in tumour development after being abnormally palmitoylated remain to be explored. Here, we discuss the role of S-palmitoylation in different cancer types to provide a glimpse into the relationship between S-palmitoylation and cancer.
Tumour-associated proteins exhibit enhanced S-palmitoylation in various cancer types, which is achieved through three primary mechanisms, including dysregulation of the PAT and APT enzymes, increased palmitic acid levels, or enhanced binding capacity between enzymes and substrate proteins. For example, it was found that DHHC2 was abnormally upregulated in tyrosine kinase inhibitor-resistant clear cell renal cell carcinoma tissues and cells, and the upregulation of DHHC2 increased the S-palmitoylation of AGK, which promoted the translocation of AGK to the PM for further activation of the PI3K/Akt/mTOR pathway, finally leading to TKI resistance.119 In addition, in colorectal cancer, the accumulation of palmitic acid caused by ACOX1 depletion contributes to the enhanced S-palmitoylation of β-catenin. S-palmitoylation protects β-catenin against β-Trcp mediated proteasomal degradation and activates DUSP14 by upregulating c-Myc, thereby boosting colorectal cancer progression.124 Moreover, an enhanced ability of enzymes to bind proteins was found in epithelial ovarian cancer. Epithelial ovarian cancer relies on the tricarboxylic acid cycle and oxidative phosphorylation to meet its anabolic growth requirements.125 S-palmitoylation increases the activity of the key tricarboxylic acid cycle enzyme MDH2 to support mitochondrial respiration and tumour cell proliferation, and the increased S-palmitoylation was attributed to the enhanced binding of DHHC18 to MDH2.125
Numerous tumour-associated proteins depend on S-palmitoylation for trafficking and proper localisation to the PM, enabling them to function effectively, either for signalling to activate various oncogenic signalling pathways, as mentioned above, or to assist tumour cells in taking up nutrients to meet their high metabolic demands. For instance, GLUT1 is essential for glucose uptake, and GLUT1 is palmitoylated by DHHC9 at Cys207 before it is accurately localised to the PM for glucose transport. Furthermore, disturbance of GLUT1 S-palmitoylation abrogated tumour glycolysis, thereby hindering the tumorigenesis of glioblastoma.126 The role of S-palmitoylation in supporting cellular glycolysis has also been found in hepatocellular carcinoma. In contrast, the S-palmitoylation of hexokinase 1 (HK1) occurs in hepatic stellate cells, followed by proper localisation to the PM. In the presence of TSG101, HK1 is secreted extracellularly via vesicles formed by PM budding and further taken up by hepatocellular carcinoma cells to accelerate hepatocellular carcinoma progression by promoting tumour cell glycolysis.127
Tumour suppression by protein S-palmitoylation can be achieved by promoting the activation of tumour suppressor signalling pathways or blocking the activation of oncogene signalling pathways. S-palmitoylation of melanocortin-1 receptor (MC1R) in melanoma is a classic example of tumour suppression by activation of tumour suppressor signals. Activation of MC1R by α-MSH reduces the damage caused by ultraviolet irradiation by stimulating cAMP signalling and melanin production, but MC1R function is impaired in individuals with the MC1R RHC variant.128 It has been found that the S-palmitoylation of MC1R exerts a protective effect on melanomagenesis, and the S-palmitoylation of the MC1R RHC variant by DHHC13 can restore its function and activate MC1R signalling, thereby promoting pigmentation and controlling melanomagenesis.128 Subsequent studies further revealed that APT2 is responsible for MC1R depalmitoylation, and the maintenance of MC1R S-palmitoylation with the APT2 inhibitor ML349 effectively enhanced its downstream signalling and inhibited melanomagenesis.129 In addition, another study tried to enhance the interaction between DHHC13 and MC1R by regulating the activation of DHHC13. They found that AMPK could phosphorylate DHHC13 at S208 to activate DHHC13, so targeting AMPK is also an effective strategy to promote MC1R S-palmitoylation.130
Blocking the activation of tumour-promoting signalling pathways by enhancing the degradation of protumor factors is another way for S-palmitoylation to exert its antitumour effect. In breast cancer, DHHC22 is abnormally downregulated. DHHC22 mediates the S-palmitoylation of mTOR and reduces its stability in a time-dependent manner, thereby disturbing the PI3K/Akt/mTOR signalling pathway, inhibiting breast cancer cell proliferation and decreasing its resistance to neratini.131 Therefore, targeting DHHC22 is a powerful strategy for the treatment of breast cancer. In addition, S-palmitoylation can also block the activation of tumour-promoting signalling pathways by restricting the localisation of tumour-associated proteins. The oncogenic FLT3 mutation FLT3-ITD is one of the most common genetic changes in AML, which puts patients at higher risk of relapse and poor prognosis. The S-palmitoylation of FLT3-ITD maintains it localisation at the ER, while disruption of FLT3-ITD S-palmitoylation by palmitoylation site C563S mutation leads to its translocation to the PM, which promotes the activation of Akt and ERK and ultimately leads to the proliferation of leukaemia cells and the progression of leukaemia.93 In the majority of tumours where S-palmitoylation exerts an antitumour effect on associated proteins, this process is typically hindered. Therefore, enhancing S-palmitoylation of these tumour-associated proteins, such as the activation of the associated PAT enzymes or inhibition of depalmitoylases, represents a promising therapeutic strategy.
Neurodegenerative diseases
Protein S-palmitoylation plays a regulatory role in the brain, and dysregulation of protein S-palmitoylation has been found to be closely related to a variety of neurodegenerative diseases.132 Using Parkinson’s disease (PD) as an example, we will briefly discuss how aberrant S-palmitoylation regulation contributes to the aggregation of nonfunctional proteins, affecting the pathogenesis and progression of PD. An analysis of the palmitoyl proteome in the cerebral cortex of PD patients found that compared to controls, altered palmitoylation resulted in at least 150 proteins, many of which could interact with α-synuclein, LRRK2, DJ-1 and other proteins closely linked to the pathogenesis of PD, suggesting that abnormal S-palmitoylation regulation is a ubiquitous phenomenon in PD.133 For example, α-synuclein cannot be palmitoylated due to the lack of cysteine, but hyperphosphorylation leads to its misfolding and aggregation.2 Moreover, pathological α-synuclein accelerates the palmitate turnover of membrane vesicle-trafficking-related protein MAP6 and thus disrupts the normal association of MAP6 with vesicles. Enhancement of MAP6 S-palmitoylation by inhibition of APT1 may alleviate α-synuclein phosphorylation and neurotoxicity.134 In addition, the vesicle-trafficking protein syt11 can be palmitoylated at Cys39 and Cys40 to localise on the intracellular membranes instead of being degraded in lysosomes, and this modification leads to enhanced binding of α-synuclein to the intracellular membranes and the abnormal aggregation of α-synuclein in PD neurons,135 suggesting that the intervention of S-palmitoylation of proteins involved in the abnormal aggregation of α-synuclein may be a feasible measure for the treatment of PD.
Cardiovascular diseases
Studies have shown that S-palmitoylation can influence the function of cardiac myocytes by regulating ion channels and signal transduction.136 The important role of S-palmitoylation regulation in ion channel homeostasis and excitation-contraction coupling of cardiac myocytes has been discussed in detail in previous reviews.136,137 Using the S-palmitoylation of voltage-gated sodium channels (NaVs) as an example, we will briefly discuss its influence on cardiac electrical activity and its implications in heart diseases, highlighting the role of S-palmitoylation in maintaining cardiac homeostasis. Nav tightly controls the generation and propagation of sodium-dependent action potentials.136 As a heterotrimeric protein, S-palmitoylation often occurs in the early stages of Nav channel biosynthesis and induces different outcomes by palmitoylating cysteine sites in different regions.136 For example, the four palmitoylation sites of Nav1.5 are located between structural domains II and III, and S-palmitoylation may modulate the movement of the domain III voltage sensor and channel availability.138,139 Increased Nav1.5 S-palmitoylation induces enhanced cardiac excitability,138 which may be a potential mechanism of arrhythmia.
Inflammatory and autoimmune diseases
S-palmitoylation is implicated in the pathogenesis of several inflammatory and autoimmune diseases, such as inflammatory bowel disease, psoriasis, nonalcoholic steatohepatitis, and systemic lupus erythematosus.86,117,140,141 For instance, in colitis, the DHHC7 and APT2-mediated palmitoylation-depalmitoylation cycle of STAT3 enhances its activation, which in turn promotes TH17 cell differentiation to aggravate colitis.117 The excessive activation of TH17 cells is also linked with a variety of inflammatory and autoimmune diseases.142 Thus, targeting S-palmitoylation regulation of STAT3 to inhibit TH17 cells serves as a potential therapy for the treatment of these diseases. Moreover, in addition to regulating protein S-palmitoylation in TH17 cells, the pro-inflammatory activity of macrophages is also a potential target for intervention in colitis. After being palmitoylated in a FASN-dependent manner, Akt in colonic lamina propria mononuclear cells is recruited and localised to the PM and subsequently phosphorylated, which further activates downstream MAPK signalling to activate the pro-inflammatory effect of macrophages.143 Metformin, a traditional hypoglycaemic drug, disturbs the S-palmitoylation of Akt by inhibiting FASN, thereby suppressing pro-inflammatory responses.143 In addition, it was found that DHHC5-mediated S-palmitoylation of NOD1/2 is indispensable for their proper membrane recruitment and immune signalling, as defective S-palmitoylation of NOD1/2 was found to be associated with NOD-driven inflammatory diseases.144 Therefore, it is possible to balance immune activation by regulating each link of the protein S-palmitoylation in various immune cells involved in the inflammatory response and thus alleviate inflammatory and autoimmune diseases.
Infectious diseases
Emerging evidence has revealed the inextricable link between protein S-palmitoylation and infectious diseases, and a previous review has summarised the function of S-palmitoylation on key molecules of the innate immune response in infectious diseases.10 Using the S-palmitoylation regulation of SARS-CoV-2 proteins as an example, we will briefly explore its role in the viral infection process. SARS-CoV-2 relies on spike proteins to target cell binding and viral fusion. During viral infection, DHHC20 and DHHC9 were found to be responsible for S-palmitoylation of spike proteins in the ER and Golgi, which is followed by viral budding to mediate viral fusion and infection of host cells.34,145 Another study discovered that DHHC2, 3, 4, 5, 8, 9, 11, 14, 16, 19 and 20 all promoted the palmitoylation of the spike protein, and the palmitoylated spike protein promoted viral infection of host cells by mediating syncytium formation and the entry of SARS-CoV-2 pseudovirus particles into host cells.146 In addition, ACE2, the cell surface receptor interacting with the viral spike protein, is also regulated by DHHC3 and APT1 for its PM targeting and extracellular vesicle secretion.147 Therefore, intervention with the viral spike protein or ACE2 S-palmitoylation to block their interaction could inhibit viral fusion and infection to prevent and treat SARS-CoV-2 infection.
Therapeutic targets and clinical research progress
Various agents targeting S-palmitoylation have been evaluated in preclinical studies (Table 1). However, these agents failed to enter clinical trials. In cancer research, strategies to inhibit S-palmitoylation in tumour-bearing animal models typically involve administering 2-BP or knocking down the associated DHHC-PATs. For example, inhibition of PD-L1 S-palmitoylation with 2-BP activates antitumour immunity in MC38 tumour-bearing mice.98 Nevertheless, the broad spectrum and toxicity of 2-BP are serious shortcomings that limit its wider application. Consequently, Han Yao and his team developed a competitive peptide inhibitor derived from the palmitoylation motif of PD-L1. This selective inhibitor was found to effectively downregulate PD-L1 expression.98 Interestingly, researchers have found that some popular clinical drugs can also inhibit protein S-palmitoylation. For example, the classical antimalarial drug artemisinin has been discovered to covalently bind DHHC6 and thus disturb its catalytic function, resulting in reduced S-palmitoylation of N-Ras, which could impair its downstream oncogenic signalling and thereby exert a potential anticancer effect by inhibiting protein S-palmitoylation.148 In addition, local anaesthetics, such as proparacaine, have been shown to decrease GP130 S-palmitoylation levels by suppressing DHHC15 transcripts. This action disrupts the IL-6/STAT3 signalling pathway and ultimately inhibits the growth and self-renewal of glioblastoma stem cells.149 In preclinical studies of other diseases, targeting S-palmitoylation remains a promising strategy. In CMA-treated mice, a small-molecule compound known as H-151 was identified to suppress STING palmitoylation, subsequently reducing systemic cytokine responses.150 The lipid peroxidation product 4-HNE is usually increased during viral infection. Interestingly, STING palmitoylation could be inhibited by 4-HNE, which induces carbonylation of STING at Cys88, indicating a new therapeutic target for the treatment of diseases with improper STING activation.151
N-myristoylation
Regulatory enzymes and catalytic mechanism
NMTs belong to the GCN5-related N-acetyltransferase superfamily.152 While lower eukaryotes typically have a single NMT gene, higher organisms possess both NMT1 and NMT2, which share nearly 77% peptide sequence identity in Homo sapiens.153 To some extent, NMT1 and NMT2 exhibit partial substrate overlap and functional redundancy. This is evident as depleting either isozyme can trigger apoptosis.154 However, the physiological complexity predicts that the two isozymes also have functional specificity and substrate preference, which could be reflected in the different roles of NMT1 and NMT2 during embryogenesis and tumour cell proliferation, with NMT1 playing a critical role in these physiological and pathological processes.154,155,156 During apoptosis, caspases cleave NMTs, causing NMT1 to move from the PM to the cytosol and relocate NMT2 in the opposite direction,157 and this positional change may influence its substrate specificity.
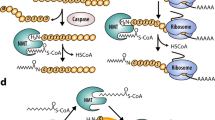
Through kinetic and X-ray crystallographic research, the molecular mechanisms underlying NMT catalysis have been revealed.13,158,159,160,161 Briefly, both cotranslational and posttranslational N-myristoylation obey a well-organised Bi−Bi mechanism involving a direct nucleophilic addition-elimination reaction (Fig. 5). First, NMT binds the fatty acid chain of myristoyl-CoA to form the myristoyl-CoA-NMT complex and induces conformational changes to expose the substrate-binding site. Subsequently, the enzyme-substrate complex is converted to the enzyme product complex via a myristoyl transfer reaction to release CoA and myristoylpeptide. Finally, NMT returns to its original conformation, hiding its substrate-binding pocket.

The catalytic mechanism and process of protein N-myristoylation. a Cotranslational myristoylation usually occurs on the glycine at the N-terminal end of nascent proteins after the methionine initiator has been removed by MetAP2. The catalytic mechanism follows the Bi–Bi mechanism. Conformational changes were induced after NMTs bound to the fatty acid chain of the myristoyl-CoA binding site. Then, the substrate binds to the NMTs and produces myristoylpeptide via a myristoyl transfer reaction. Finally, the NMTs release the myristoylpeptide and restore its conformation. b Posttranslational protein N-myristoylation often occurs during apoptosis. After the internal glycine of the substrate protein is exposed by caspase cleavage, NMTs catalyse the attachment of myristic acid to the glycine residue of the substrate. c Reversible protein N-myrisotylation occurs on the Nε-side chain of lysine residues instead of glycine residues, which is reversed by sirtuins or HDACs
Cotranslational myristoylation usually occurs on the glycine at the N-terminal end of nascent proteins after the methionine initiator has been removed by MetAP2, and the N-terminal region of NMTs is necessary for isozyme-specific binding to the ribosome.14,159,162,163 The majority of myristoylated proteins responsible for basal cell function undergo cotranslational modifications, whereas during apoptosis, a portion of myristoylated proteins undergo posttranslational modifications when the internal glycine is exposed by caspase cleavage.164 Myristoylation usually occurs at the protein NH2-terminal glycine and is considered irreversible. However, research has found that myristate can covalently attach to some proteins that lack NH2-terminal glycine. Further studies revealed that the Nε-side chain on Lys residues undergoes myristoylation, and NMTs are the enzymes responsible for catalysing the myristoylation of lysine.16,165 Intriguingly, fatty acyl groups on Nε-modified lysine residues can be removed by sirtuins and HDACs, suggesting that in addition to palmitoylation, lysine myristoylation is also dynamic and reversible17,166,167 (Fig. 5). As a deacetylase, SIRT6 not only hydrolyses lysine myristoylation of proteins such as TNF-α, but its activity as a demyristoylation enzyme is much higher than its deacetylation activity, indicating that the regulation of intracellular deacylation activity is extremely delicate and complex.167 Lysine myristoylation of gravin-α is needed for GPCR signalling, and demyristoylation of gravin-α by HDAC11 inhibits this pathway, suggesting that reversible lysine myristoylation is involved in the delicate regulation of GPCR signalling.168
Physiological function
Intracellular trafficking and protein-lipid interactions
Similar to S-palmitoylation, attaching myristoyl acids to proteins enhances their ability for intracellular trafficking and increases membrane-binding affinity. Nevertheless, this increased affinity by N-myristoylation is essential but not sufficient for protein anchoring to the membrane.169,170 Unlike S-palmitoylation, the myristoyl component of proteins undergoes more dynamic changes, as reflected by the fact that the myristoyl moiety can either be hidden in the hydrophobic pocket of the protein or exposed for membrane binding, and it was previously speculated that these two conformational changes are regulated under the assistance of several secondary signals, termed the myristoyl switch.171,172,173,174 The first type of myristoyl switch is the myristoyl-electrostatic switch. The myristoyl component works in synergy with electrostatic interactions, especially between the protein’s positively charged effector domain and phospholipids, facilitating protein binding to the membrane.174,175 Serines on such domains are highly susceptible to phosphorylation thereby altering electrostatic attraction. For instance, myristoylated MARCKS would detach from the membrane and translocate to the cytosol when phosphorylated by protein kinase C, while it could reattach to the membrane once it was dephosphorylated by phosphatases.176,177 Interestingly, even without assisting motifs, a portion of PKA-C can focus PKA activity once its electrically neutral N-terminus undergoes myristoylation, indicating that myristoylation alone is sufficient for this portion of PKA-C to interact with the membrane under certain circumstances.178 The second type of myristoyl switch is the myristoyl-ligand switch. A typical example of being subject to such a switch is the retinal calcium-binding protein Recoverin, whose myristoyl moiety needs to be exposed upon Ca (2 + )-binding sites of N-terminal binding to calcium, and this binding induces not only the myristoyl moiety but also many hydrophobic residues for membrane localisation.179,180 The third type is the myristoyl-palmitoyl switch. Here, in addition to N-myristoylation, proteins undergo an additional modification, usually S-palmitoylation, to ensure robust membrane binding. Proteins that require these two lipidations for anchoring to the membrane include FCaBP of Trypanosoma cruzi,181 H-Ras,182 FRS2α,183 RNF11184 and others. Intriguingly, S-palmitoylation alters the localisation preferences of some myristoylated proteins. For example, N -myristoylated but not palmitoylated Gαi1 predominantly bound to ordered lipid microstructural domains with a net negative charge. In contrast, Gαi1 containing both FAs preferentially interacted with nonnegatively charged raft-like lamellar membranes.185
Protein stability and degradation
Like S-palmitoylation, N-myristoylation plays a role in controlling protein stability. Quite a few proteins can be altered not only in their intracellular localisation but also in their fate of being degraded after undergoing myristoylation. For instance, myristoylation not only facilitates FSP1 membrane translocation but also promotes its stability by evading the proteasome degradation pathway.186 Another study discovered that myristoylation also protects VILIP3 from lysosomal pathway-mediated degradation.187 In some cases, myristoylation only enhances protein stability without causing significant changes in membrane binding. The presence or absence of myristoylation only affects the thermal stability of calcineurin but not its binding to phospholipid monolayers.188 As early as 1986, the presence of N-degrons that exert degradation signals at the N-terminus of proteins was discovered,189 and theories relevant to N-degron degradation have been continuously updated.190 A subsequent study revealed that protein myristoylation prevented the N-terminal glycine from being exposed, thereby eclipsing the selective proteasomal degradation mediated by Cul2ZYG11B and Cul2ZER1191 However, myristoylation does not invariably shield proteins from degradation. For instance, compared to myristoylated c-Src, nonmyristoylated c-Src resists proteasome-mediated degradation, which is achieved by reducing its association with the ubiquitin E3 ligase Cbl.192 These studies demonstrated that the regulation of the conformation changes of N-terminal sites by myristoylation is sophisticated and thus delicately regulates protein degradation.
Protein–protein interactions
Apart from modulating intracellular trafficking, membrane attachment and protein stability, myristoylation also caters to cellular and viral needs via its interactions with other proteins. A prime example of how myristoylation participates in protein–protein interactions is Src.193 p60v-src of Rous sarcoma virus is essential for the transformation activity of the virus, and studies have shown that this transforming activity is largely dependent on the interaction of p60v-src with the membrane. Specifically, this interaction relies on the binding of myristoylated p60v-src to receptor SLC25A5.193,194 Another well-studied example illustrating the impact of myristoylation on protein–protein interactions is Gag. As a major coordinator of the assembly process of many retroviruses, its N-terminal matrix domain is myristoylated to target the PM and anchor.195,196,197 Yiping Zhu et al. found that the host factor HO-2 could impede HIV replication by specifically binding to the myristic acid portion of Gag; such binding requires the myristoylation of HO-2, and blocking HO-2 myristoylation would lead to an increase in viral replication.198
Signal transduction
N-myristoylation plays a pivotal role in cellular signalling pathways, primarily by modifying protein intracellular trafficking, membrane association, and protein–protein interactions, among other functions. Accumulating evidence suggests that myristoylation has a substantial impact on various signalling pathways that directly or indirectly regulate cancer, metabolism, immunity and others, such as the Src signalling pathway,199 AMPK signalling pathway,200 Wnt signalling pathway,201 PI3K/Akt signalling pathway,202 Notch signalling pathway,203 the LPS-induced TLR4 inflammatory response,204 the cGAS-STING signalling pathway,205 the B-cell receptor pathway206 and pathways involved in T-cell development207 (Fig. 6). For instance, AMPK, an αβγ heterotrimer, is a crucial regulator of cellular energy homeostasis. It plays a vital role in cellular energy sensing and bioenergetics and is linked with the regulation and progression of numerous diseases.208 In rheumatoid arthritis, due to defective NMT1 function in T cells, the inability of AMPK to be myristoylated impedes its lysosomal recruitment and activation, which leads to the overactivation of the mTORC1 pathway and promotes T-cell differentiation into pro-inflammatory TH1 and TH17 helper T cells, resulting in severe synovial tissue inflammation.209 Recently, it has been found that myristoylation is involved in the regulation of the STING pathway mainly through the modulation of ARF1, which is a major regulator of STING membrane trafficking, and its function is dependent on N-myristoylation.205 Therefore, the myristoylation of ARF1 is an important target for balancing STING-dependent autophagy and IFN responses to promote immune homeostasis by regulating STING pathway activation. For Notch signalling, myristoylation of Neurl-1 is indispensable for endocytosis and relocalization of the PM of the Notch ligand jagged 1.203

N-myristoylation is involved in the regulation of multiple cellular signal transduction pathways. We briefly summarised several well-established signalling pathways that are under the control of N-myristoylation, including the Scr-meditating oncogenetic pathway, Wnt pathway, Akt pathway, STING-autophagy pathway, Notch signalling pathway, TCR activation signalling pathway, TLR4 inflammatory responses, AMPK signalling pathway and B-cell receptor pathway
Pathological implication
Cancer
Numerous studies indicate that while NMT1 is crucial for organismal growth and development, it is also an oncogenic protein that is aberrantly expressed in many human cancers, suggesting that NMT1-mediated protein myristoylation is a potential mechanism of tumorigenesis and development. In lung cancer, EZH2, which undergoes myristoylation, is notably highly expressed. The resulting hydrophobic interaction can drive and stabilise liquid-liquid phase separation. Myristoylated EZH2 binds STAT3 to recruit it to phase-separated droplets, thereby overactivating the STAT3 pathway to promote lung cancer progression.210 In bladder cancer, upregulated NMT1 causes the lysosomal anchoring protein LAMTOR1 to be myristoylated at Gly2, enhancing its protein stability and lysosomal localisation, which leads to the activation of the mTORC1 pathway and mediates bladder cancer progression.211 In HCC, upregulated NMT1-mediated myristoylation of the VILIP3 protein enhances its stability and subsequent NFκB/Bcl-2 signalling, thereby contributing to the progression of HCC.187 In ovarian cancer, upregulated ACSL1 is involved in metabolic reprogramming, which enhances fatty acid beta-oxidation in tumour cells by promoting the myristoylation of a series of substrate proteins, such as AMPKβ, Src, and FSP1, thereby regulating the progression and metastasis of ovarian cancer.186,212 In addition, myristoylation also plays an essential regulatory role in certain tumours driven by specific oncogenes, such as EGFR-dependent tumours. For EGFR, translocation from the Golgi to the PM is a prerequisite for its pro-oncogenic role, and this process requires the recognition and binding of EGFR by myristoylated ARF6, which facilitates its budding from the Golgi and its translocation in the GTP-bound form.213
Metabolic disorders
Myristoylation dysregulation is a frequent occurrence in metabolic disorders. A 2-fold increase in NMT activity was observed in a rat model of insulin-dependent diabetes mellitus induced by STZ compared to controls, whereas a nearly 5-fold decrease in NMT activity was observed in a rat model of non-insulin-dependent diabetes mellitus compared to controls.214,215 NMT activity appears to be negatively correlated with plasma insulin levels.215 Furthermore, saturated FAs were found to enhance the membrane localisation of c-Src by promoting its myristoylation, therefore causing insulin resistance through JNK pathway activation, while unsaturated FAs had the opposite effect.216 Furthermore, obesity, another prevalent metabolic disorder, often accompanies the abnormal regulation of myristoylation in several pathogenesis-linked proteins. Similar to type 2 diabetes, obesity is affected by saturated FA-mediated activation of the JNK pathway.216 In addition, abnormal regulation of leptin signalling is quite common in the development of obesity, and studies have shown that it is myristoylated Akt but not nonmyristoylated Akt that induces increased leptin levels in 3T3-L1 adipocytes,217 which indicates that targeting Akt myristoylation may present a promising way to improve obesity.
Autoimmune disorders
In rheumatoid arthritis (RA), aberrant regulation of AMPK myristoylation leads to abnormal CD4 T-cell differentiation and triggers inflammation. Specifically, low expression of NMT1 inhibited myristoylation-dependent AMPK lysosomal trafficking and activation, which resulted in the overactivation of mTORC1 signalling in CD4 T cells and contributed to the differentiation of CD4 T cells into pro-inflammatory Th1 and Th17 cells209,218 Moreover, a strong anti-inflammatory effect could be achieved by overexpressing NMT1 in RA T cells with low NMT1 expression.
Infectious diseases
Research indicates that the survival and invasion of several pathogens, including viruses and parasites, hinge on protein myristoylation. In the absence of NMTs, several viruses exploit the host’s NMTs to myristoylate essential assembly proteins, facilitating its assembly and replication. A typical example is HIV. Myristoylated Gag guides the virus and anchors it to the PM, demonstrating that this structural protein has a critical effect on viral assembly.195 In the absence of Gag or when the myristoylation site is mutated, HIV-1 RNA is highly dynamic and is unable to either anchor to the membrane or complete the assembly of virions correctly.197 As a myristate-binding protein, HO-2 of the host blocks HIV membrane localisation by selectively binding to the myristate moiety of Gag, thereby impeding viral assembly and replication.198 The consequence of HIV infection is that it causes the progressive loss of CD4 T cells and widespread immune abnormalities in the host, and the myristoyl protein Nef has been found to have a pivotal function in compromising host immunity.219 Binding of Nef to the PM through myristoylation induces rapid internalisation of CD4 on the surface and MHC-1 of T cells, leading to its degradation in lysosomes.220,221 In addition, Nef negatively regulates phagocytosis in macrophages. Through interaction with AP-1, myristoylated Nef disrupts the membrane delivery of VAMP3- and TNFα positive endosome compartments and impairs optimal phagosome formation, thereby inhibiting macrophage phagocytosis.222 Overall, myristoylation is involved in multiple aspects of HIV infection of the host, supporting viral replication and immune evasion by regulating the membrane binding and protein–protein interactions of multiple proteins.
Therapeutic targets and clinical research progress
Growing evidence strongly supports the development of therapeutic targets against NMT activity and myristoylation. Currently, the development of related drugs mainly focuses on antitumour and anti-infection activities (Tables 1 and 2). For tumours with elevated NMT expression and activity, several compounds are available as NMT inhibitors. The usage of myristoyl-CoA analogues to inhibit NMT dates back as far as 1990, when researchers compared the inhibitory effects of three myristic acid derivatives: 2-fluoromyristic acid, 2-bromomyristic acid and 2-hydroxymyristic acid.223 All three compounds exerted weak inhibitory effects on NMT. Subsequent synthesis of these myristic acid derivatives into 2-substituted acyl-CoA analogues by acyl-CoA synthase significantly enhanced the inhibitory effect on NMT.223 Investigating how structural changes in acyl-CoA derivatives alter the binding of protein substrates will provide remarkably useful information for the design of potent antitumour compounds targeting NMT. Another myristoyl-CoA analogue, B13, also known as D-NMAPPD, potently inhibited NMT1 enzymatic activity against prostate cancer cells.199 By competing with the myristoyl-CoA binding site of NMT1, B13 impaired the myristoylation of Src, leading to the blocking of relevant oncogenic signalling and thereby promoting the antitumour effect. Furthermore, by optimising the structure of B13, the derivative LCL204 exhibited a lower IC50 for NMT1 enzymatic activity.199 However, a later study obtained an inconsistent conclusion that B13 was not an NMT inhibitor, as no changes in N-myristoylation in c-Src were observed when MDA-MB-231 or HeLa cells were treated with B13 at 30 μM.224 Tris dipalladium (Tris DBA) is another NMT inhibitor with debatable efficacy. A previous study demonstrated that the expression of NMT1 at the mRNA and protein levels and cell proliferation were inhibited after Tris-DBA treatment of B16 and A375 cells.225 However, there is no direct evidence for the interaction of Tris DBA with NMT1 and its effect on protein myristoylation.225 Wouter W Kallemeijn et al. further discovered that Tris DBA causes cytotoxicity independent of NMT and myristoylation,224 suggesting that the characterisation of Tris DBA as an NMT1 inhibitor needs to be revisited. In addition to developing small-molecule compounds such as myristoyl-CoA analogue, targeting biological NMT inhibitors including NIP71 and HSC70 as well as enolase is also a promising approach. Studies initially found that NIP71 was negatively correlated with the expression of the NMT substrate pp60Src, and subsequently revealed that purified NIP71 and HSC70 could inhibit human NMT in a dose-dependent manner.226,227 Similarly, enolase was found to inhibit human NMT activity in a dose-dependent manner.228 The discovery of these biological NMT inhibitors has broadened the strategy of targeting NMT for the treatment of tumours, but more studies are needed to confirm the effectiveness of their application.
To date, clinical studies of NMT inhibitors targeting tumours have focused on haematologic malignancy. For instance, a phase I clinical trial showed that asciminib, by binding to the myristoyl site of the BCR-ABL1 protein, effectively inactivates it, benefiting CML patients with TKI resistance or T315I mutations.229 In a phase III clinical study evaluating the major molecular remission (MMR) rate after treatment with asciminib or bosutinib in patients with CML-CP who were previously resistant/intolerant to at least two TKIs, the MMR of asciminib was significantly higher than that of bosutinib at week 24.230 Another clinical study at a more distant observation point (week 96) also discovered that the MMR of asciminib was superior to that of bosutinib.231 In 2021, asciminib received FDA approval for the treatment of adults with Ph+ CML in the chronic phase (Ph+ CML-CP), previously treated with ≥ 2 TKIs, and Ph+ CML-CP with the T315I mutation.232 These encouraging clinical data suggest that targeting protein myristoylation is highly promising, motivating researchers to explore further development of relevant drugs to expand their clinical applications in the future.
In addition to anti-tumours, studies targeting protein myristoylation for the development of anti-infective drugs are also in full swing. For instance, the recently identified compound IMP-1088 acts as a novel NMT inhibitor. It effectively hinders the assembly and replication of various viruses, including rhinoviruses and vaccinia virus, by blocking the myristoylation of their proteins,233,234 thus highlighting the potential of protein myristoylation as a drug target for a wide range of viral infections. In addition, selective inhibitors targeting NMT, including myristate analogues, myristoylpeptide derivatives, histidine analogues (peptidomimetics), aminobenzothiazoles, quinolines and benzofurans showed broad-spectrum antifungal activity.235,236,237 In addition, given the difference in NMT between humans and microorganisms, NMT is an attractive target for antiparasitic drugs, and several compounds that have demonstrated antiparasitic activity against Trypanosoma brucei, Plasmodium falciparum and Plasmodium vivax238,239,240,241 could be used in the development of anti-malaria and anti-Human African trypanosomiasis drugs. These findings further validate NMTs and myristoylation as exciting drug targets for the treatment of a myriad of diseases, and related drugs can be further optimised to become more effective and safer.
S-prenylation
Regulatory enzymes and catalytic mechanism
Currently, four known heterodimeric prenyltransferases are responsible for the prenylation of proteins: protein aryltransferase (FTase), protein gammaglutamyltransferase type I (GGTase-I), Rab geranylgeranyltransferase (GGTase-II) and protein gammaglutamyltransferase type III (GGTase-III).21,242 The first two prenyltransferases are responsible for adding a farnesyl or geranylgeranyl group to the C-terminus of the substrate protein. FTase and GGTase-I are both heterodimers and share the common α subunit FNTA or PTAR2, but their β subunit is different ((FNTB in FTase and PGGT1B in GGTase-I), which shapes their reactivity and substrate specificity.243,244 Structural analyses indicate that most prenylated proteins have a CAAX motif at their C-terminus, serving as the enzyme recognition site, where “C” denotes a cysteine residue, “A” usually indicates hydrophobic amino acids, and “X” denotes any amino acid that determines whether the protein is farnesylated or geranylgeranylated.245 Generally, farnesylation selectively occurs in proteins with alanine, methionine, serine, or glutamate at the “X” position, whereas fam prefers proteins with leucine, methionine, phenylalanine, isoleucine and valine at the “X” position.246,247,248 Despite the well-recognised substrate selectivity of these two prenyltransferases, some substrate proteins can be regulated by both FTase and GGTase-I. For example, an earlier study in yeast showed that Ras is a substrate for FTase, but overexpression of GGTase-I in an FTase-deficient yeast strain compensated for the farnesylation of Ras and rescued growth defects caused by FTase deficiency.249 In addition, the farnesylation of K-Ras could not be completely inhibited when using an FTase inhibitor, as it would be catalysed by GGPTase-I.250 To some extent, this cross-prenylation process adequately ensures that some genes, such as Ras perform physiological functions. However, for the RhoB protein, different farnesylation modifications yield different functions. The farnesylated form of the RhoB protein is mainly involved in cell growth, whereas the geranylgeranylated form of the RhoB protein mainly induces apoptosis.251 GGTase-II, the third protein prenyltransferase, comprises the α subunit PTAR3 and the β subunit RabGGTB. For GGTase-II, whose subunits consist of RabGGTA (PTAR3) and RabGGTB, the catalytic feature is that it can contribute to both monogeranylgeranylation of cysteine residues near the C-terminus of the substrate protein and dual geranylgeranylations of the two cysteine residues near the C-terminus. Specifically, monogeranylgeranylation occurs when the C-terminus of the substrate protein contains the CAAX or CXXX motif, while double geranylgeranylation occurs when the C-terminus of the substrate protein contains the CC, CXC, CCX, CCXX or CCXXX motif.21 Dual geranylgeranylation is not redundant, as the substrates of GGTase-II Rab proteins require dual geranylgeranylation to properly mediate vesicular trafficking.252 GGTase-III is a fourth protein prenyltransferase composed of the α subunit PTAR1 and the β subunit RabGGTB, whose β subunit is the same as that of GGTase-II.21 FBXL2 was the first identified substrate to undergo dual geranylgeranylation catalysed by GGTase-III, although it can also be regulated by GGTase-I to undergo monogeranylgeranylation.253 A crystal structure analysis revealed an extensive multivalent interface specifically shaped between FBXL2 and PTAR1, demonstrating that the recognition of FBXL2 by GGTase-III was mediated by PTAR1 and substrate–enzyme specificity.253 In addition, the Golgi SNARE protein Ykt6 is another substrate for GGTase-III, which is responsible for shifting a geranylgeranyl group to mono-farnesylated Ykt6 to generate geranylgeranyl-farnesyl Ykt6 for proper function.254 Mono-farnesylated Ykt6 prevents the Golgi SNARE complex from assembling properly, thus compromising Golgi structure and function.254
Before trafficking to the PM, most CAAX proteins undergo three stages: initial prenylation, endoproteolysis, and final methylation (Fig. 7). Some proteins may also undergo additional S-palmitoylation (see the S-palmitoylation section). Endoproteolysis refers to the removal of AAX amino acids from proteins on the surface of the ER after the attachment of isoprene to its C-terminal cystine residue, and the enzyme responsible for this process is RCE1, which was initially discovered in yeast.255 Regarding the proteolytic mechanism, earlier studies have suggested that RCE1 belongs to the cysteine protease family, and some have hypothesised that RCE1 is a membrane-bound metalloproteinase, but none of these theories are able to adequately explain its hydrolysis mechanism.256,257 The crystal structure of RCE1 orthologues from Methanococcus maripaludis (MmRce1) shows the involvement of glutamate-activated water and an oxyanion hole,258 which provides a glimpse into the mechanism of protein hydrolysis in Homo sapiens RCE1. However, crystal structural data on eukaryotic enzymes are also urgent to elucidate their mechanisms since the primary structure and topology of RCE1 differ among species.258 Methylation refers to the ICMT-mediated methylation of the exposed carboxyl terminus of farnesylated proteins after the removal of AAX amino acids, a process that increases protein membrane affinity by neutralising the negative charge of prenylcysteine.22,259 Similar to RCE1, ICMT was first discovered in yeast as STE14, and ICMT is restricted to the ER.22,260 Topology and mutational analysis of human ICMT revealed that some of the conserved amino acid clusters located on transmembrane fragments altered enzyme activity upon mutation, suggesting that they may be involved in substrate binding and/or catalysis.259,261 In the whole process of protein prenylation, it has been hypothesised that the final ICMT-catalysed methylation is reversible. Much of the speculation links to the discovery that unmethylated CAAX proteins have been observed in different cells.262,263,264 Furthermore, the carboxylesterase CES1 was identified in MDA-MB-231 cells, and knockdown of CES1 significantly reduced the amount of unmethylated RhoA protein, suggesting that CES1 is a prenylcysteine-specific methylesterase.265 However, the methylation status of other Rho family members was not affected by CES1, implying that either CES1 is highly selective for substrates or that only a fraction of prenylated proteins can achieve reversible methylation, which is a question worthy of further investigation.

The catalytic process of protein S-prenylation and postprenylation reactions. Most substrate proteins have a characteristic CAAX motif, which is the enzyme recognition site. First, a 15-carbon farnesyl or a 20-carbon geranylgeranyl isoprenoid lipid is attached to the cysteine residues on the CAAX motif of the substrate by FTase or GGTase-I, respectively. Then, the prenylated proteins translocate to the ER and undergo removal of the AAX motif by RCE1 and methylation by ICMT. After that, proteins such as Ras may be directly trafficked to the plasma membrane or undergo additional S-palmitoylation. Other proteins, such as RHO GTPases, bind to RHO guanine nucleotide dissociation inhibitors (GDIs) to assist in their trafficking. GDIs guanine nucleotide dissociation inhibitors, ER endoplasmic reticulum
Physiological function
Intracellular trafficking and membrane localisation
For proteins exemplified by the Ras protein, accurate localisation on the PM is crucial. Cytosolic proteins, which start as soluble entities lacking hydrophobic sequences, rely on prenylation to effectively associate with the membrane. The Ras proteins have extensively showcased this property. Once prenylated, proteins such as N-Ras, H-Ras, and K-Ras4a undergo palmitoylation, they are anchored to the PM via two hydrophobic moieties. K-Ras4b, on the other hand, anchors to the PM using its farnesyl group combined with a lysine-rich polybasic sequence.266 Both nonfarnesylated mutant Ras and blocked isoprenoid biosynthesis can impair protein PM anchoring.96,242 In addition to targeting the PM, directing the positioning of proteins to different compartments is also one of the effects of prenylation. For instance, upon viral infection, Rac1 of the Rho family undergoes geranylgeranylation and subsequent palmitoylation and is subsequently transported to the mitochondria-associated endoplasmic reticulum membrane (MAM), where Rac1 continues to limit the interaction of mitochondrial antiviral signalling proteins with the E3 ligase Trim31, thereby inhibiting MAVS ubiquitination, aggregation and activation to prime the antiviral immune response.267 In addition, NGF-induced geranylgeranylated Rac1, along with other prenylated proteins in sympathetic axons, is recruited to TrkA-harbouring endosomes to assist receptor trafficking, which is critical for axonal growth during neuronal development.268 Previous studies have also assessed the importance of the three processing steps (prenylation, protein hydrolysis and methylation) of CAAX proteins for targeting the PM. Studies of K-Ras4b from rabbit reticulocytes revealed that compared with farnesylation alone, a twofold increase in membrane binding from the protein hydrolysis step and a further twofold increase in membrane binding from methylation.269 In addition, it was also found that prenylation alone was not sufficient for rab4 and Rab5 to achieve membrane association compared to their maturation forms which underwent the full three modification stages.270 For these proteins, hydrolysis-induced conformational changes that modify membrane binding and neutralise the negatively charged ionised carboxyl group induced by methylation are important factors responsible for increasing membrane affinity.
Protein–protein interactions
While the primary focus of many studies has been on the anchoring of proteins to membranes, protein prenylation plays a pivotal role in mediating protein–protein interactions as well. A classic example is that prenylation affects the binding of K-ras and SOS. SOS forms a complex with prenylated K-Ras4b and catalyses its guanine nucleotide exchange, whereas non-pernylated K-Ras4b is not able to form a complex with SOS.271 Furthermore, replacing the farnesyl with a geranylgeranyl moiety does not influence the binding between SOS and K-Ras. This observation indicates that the length of the attached lipid is not a limiting factor in their interaction.271 Among the various proteins that can interact with Ras, PDEδ is also one of the most extensively studied. The farnesylated Ras protein on the PM is bound and solubilized by PDEδ through its GDI-like pocket and unloaded at the perinuclear membrane with the assistance of Arl2. It is then captured in the recycling endosome (RE), from which it recovers its localisation on the PM via vesicular transport, which results in a PDEδ-regulated dynamic membrane distribution of Ras, therefore enhancing its signalling capacity.272,273 The crystal structure of the human K-Ras4b-PDEδ complex reveals the details of the interaction, and a 5-amino-acid-long sequence motif (Lys–Ser–Lys–Thr–Lys) in K-Ras4b was found to be particularly important for the interaction.274 In certain scenarios, nonprenylated proteins might exhibit a higher affinity for interactions than their prenylated counterparts. One example of this is Rac1. Nonprenylated Rac1 actively interacts with its effectors Iqgap1 and Tiam1 to support GTP loading and cytokine production, while the prenylation of Rac1 limits these interactions.275 Thus, promoting Rac1 prenylation could restrain pro-inflammatory signalling and innate immunity by hindering the interactions of Rac1 with Iqgap1 and Tiam1.
Stability and protein degradation
Prenylation also plays a pivotal role in modulating protein stability. For example, farnesylation enhanced the stability of Ykt6, as a faster degradation rate was observed in nonfarnesylated Ykt6 than in farnesylated Ykt6, and this increased stability is closely related to the more compact and stable structure induced by farnesylation.276 In certain instances, the influence of prenylation on protein stability is more indirect. For instance, geranylgeranyation of the Rho protein CDC42 is crucial for the interaction with its chaperone protein RHOGDI.277 The binding of RHOGDI to Rho protein not only facilitates correct membrane association of the Rho protein but also protects it from degradation.278 In the case of CAAX proteins, methylation of prenylated cysteine groups at their C-terminus significantly impacts their stability. Loss of methylation exerts distinct effects on different methyl-accepting proteins. An experiment conducted in the RAW264.7 macrophage line showed that both the RhoA and CDC42 half-lives were shortened to varying degrees when methylation was inhibited.279 Another experiment in fibroblasts showed that when ICMT was inactivated, the levels of total RhoA and GTP-bound RhoA protein were only 5–10% of those of controls.280 However, another experiment performed in human TM cells appears to be contradictory to the previous observation, as promoting RhoA geranylgeranylation by the addition of geranylgeranyl pyrophosphate (GGPP) decreases its half-life and promotes its degradation through the 20 S proteasome pathway.281 Given that this study focused on initial geranylgeranylation and did not explore the role of methylation, experiments involving prenylated cysteine methylation changes should be included to determine whether methylation affects RhoA degradation. Notably, the lack of ICMT indeed increased the K-Ras half-life, demonstrating that the absence of methylation contributes to K-Ras stability.280 More investigations are needed to explore the effect of the three catalytic steps of CAAX protein maturation on protein stability.
Signal transduction
Given the widespread occurrence of prenylation among cellular signalling proteins, a multitude of signal transduction pathways are regulated by protein prenylation (Fig. 8). For instance, after being prenylated, Ras properly localises on the PM and binds to the receptor protein Grb2 to form a complex, which subsequently recruits SOS to cause the conformation change of Ras, allowing it to dissociate from GDP and binding to GTP and be activated. Subsequently, activated Ras activates various effectors, such as PI3K, Raf, and PLC, resulting in the activation of distinct signalling cascades.282 Maintaining a balance between farnesylation and geranylgeranylation is pivotal for optimal cellular function. Any imbalance can induce abnormal signal transduction, subsequently leading to pathological cellular alterations. For example, GGPPS is a key enzyme in the mevalonate pathway catalysing the synthesis of GGPP from FPP.283,284 Knocking down or pharmacologically inhibiting GGPPS results in reduced GGPP synthesis, and excess farnesyl pyrophosphate (FPP) can enhance the farnesylation of Rheb, which in turn overactivates mTORC1 signalling, thus leading to cardiomyocyte hypertrophy.285 In addition, the selective shaping of downstream pathways by farnesylation and geranylgeranylation is reflected in the regulation of the immune system. Fntb-mediated protein farnesylation regulates eTreg cell maintenance through activation of mTORC1 and ICOS signalling, while Pggt1b-mediated protein geranylgeranylation regulates eTreg cell differentiation through activation of Rac signalling.286 In addition to the regulation of Treg cells, farnesylation and geranylgeranylation also play their respective roles in regulating peripheral T cells. It has been found that Fntb-mediated protein farnesylation is indispensable for peripheral T-cell homeostasis, as peripheral T cells lacking Fntb exhibited a less activated phenotype and were susceptible to apoptosis. Pggt1b-mediated protein geranylgeranylation is responsible for the egress and trafficking of thymocytes, as Pggt1b deficiency blocks S1P1 signalling and Cdc42-Pak signalling, which are essential for thymocyte egress and trafficking.287 For the Rab protein, its dual geranylgeranylation, facilitated by GGTase-II, is vital for the proper trafficking and positioning of Notch signalling elements, such as Delta. Mistrafficking of Notch signalling components caused by the mislocalization of Rabs leads to Notch signalling defects.288

S-prenylation is involved in the regulation of multiple cellular signal transduction pathways. We briefly summarised several well-established signalling pathways of the main small GTPase family. The prenylation and post prenylation of these proteins often occur on the ER. K-Ras and N-Ras can be either farnesylated or geranylgeranylated, while H-Ras can only be farnesylated. N-Ras, H-Ras and K-Ras4a undergo S-palmitoylation before anchoring at the plasma membrane and activating downstream signalling. Other proteins, such as Rho, can switch membrane-bound states through binding to RhoGDI. Overall, protein prenylation mediates downstream signalling by regulating protein trafficking and protein‒protein interactions. ER endoplasmic reticulum, MAM mitochondria-associated endoplasmic reticulum membrane, RE recycling endosome
Pathological implication
Cancer
The involvement of small GTPases, particularly Ras GTPases, in the carcinogenesis of diverse cancers is well-documented. Approximately 20% of human cancers harbour Ras subfamily mutations, which maintain Ras in a constitutively active GTP-binding conformation and subsequently overactivate downstream signalling to mediate tumorigenesis.266 In addition, other small G proteins, such as Rho, Rab and Arf subfamilies, are overexpressed in a variety of cancers and promote tumorigenesis by influencing intracellular regulatory processes.266 These molecules need to undergo prenylation to ensure their proper subcellular localisation to the PM and/or endomembranes before performing their oncogenic functions. For example, H-Ras can be farnesylated.289 K-Ras, N-Ras and RhoB can be either farnesylated or geranylgeranylated.290 RhoA, RhoC and CDC42 can be geranylgeranylated.247 RAB1 and RAB3b can be double geranylgeranylated.291 Disrupting the prenylation process can significantly hinder the oncogenic role of Ras. For example, inhibiting FTase activity with a selective inhibitor of FTase tipifarnib to suppress H-Ras prenylation showed that the proliferation, survival, and spheroid formation of H-Ras-mutant cells were impaired, and tumour stasis or regression was observed in head and neck squamous cell carcinoma H-Ras-mutant xenografts after treatment with tipifarnib.292 Similarly, treatment with tipifarnib in H-Ras-mutant fusion-negative rhabdomyosarcoma xenografts could reduce tumour growth by reducing H-Ras PM localisation and downstream signalling transduction.289 Overall, the discovery of the critical role of prenylation in regulating Ras function sheds light on the intricate mechanisms and regulatory networks in cancer development driven by Ras and provides a potential strategy targeting key links in protein prenylation.
Neurological disorders
The pathophysiology of numerous neurological disorders has been associated with protein prenylation. For example, elevated levels of FPP and GGPP were observed in the brains of AD patients, and suppression of FTase or GGTase-I could mitigate Aβ-associated neuropathology in an AD mouse model.293 In another study, upregulated expression of FT and H-Ras farnesylation was observed in AD brains and the upregulation of FT and H-Ras farnesylation was associated with mild cognitive impairment, indicating that abnormal regulation of protein prenylation is involved in the AD pathogenic cascade. Targeting protein prenylation by deleting FT to inhibit amyloid generation and overactivation of mTORC1 signalling could effectively alleviate memory impairment in AD model mice.294 In addition to AD, other neurological disorders, including Hutchinson-Gilford progeria syndrome (HGPS),295 multiple sclerosis,296 Niemann-Pick disease type C disease293 and Parkinson’s disease297 are subject to aberrant regulation of protein prenylation. As a rare genetic disease, the pathogenesis of HGPS is closely linked to point mutations in LMNA, which encodes lamin A.298 The mutant lamin A lacks its Zmpste24 site, preventing it from undergoing endoproteolysis. Instead, it remains consistently farnesylated and carboxymethylated, which leads to limited remodelling of the nuclear scaffold and increased blebbing, thereby accelerating premature ageing.295 Given that aberrant protein prenylation is involved in the pathology of many neurological disorders, in-depth investigations of the regulatory mechanisms of prenylation will provide new avenues for the development of effective therapeutic approaches for these neurological diseases.
Infectious diseases
A growing body of evidence underscores the important role of protein prenylation in the infection processes of multiple pathogens. As one of the pathogens causing pneumonia, Legionella pneumophila enters cells by phagocytosis and subsequently secretes effector proteins, which are prenylated by host FTase, anchor into the PM and escape from lysosomal fusion for sustained infection.299 Beyond bacteria, prenylation has been found in various viruses, including type 1 adenovirus, hepatitis D virus (HDV), influenza virus, and HIV, among others.300 In the hepatitis D virus, the delta virus large antigen is prenylated, which is essential to anchor to HBsAg and be packaged into virus particles for HDV particle formation.301 HDV replication can be attenuated by inhibiting geranylgeranylation.302,303 Furthermore, protein prenylation is also involved in the regulation of the survival and activity of several parasites, such as Plasmodium falciparum.304 Researchers found that Plasmodium falciparum survival during heat or cold shock is dependent on the regulation of protein prenylation, which governs the membrane binding of HSP40 to the ER and interaction with its client proteins, thereby controlling thermotolerance. Inhibition of protein prenylation would result in Plasmodium falciparum being sensitive to otherwise nonlethal temperature stresses.304 Taken together, these research findings highlight the crucial role of protein prenylation regulation in ensuring the viability of an extensive array of pathogens, including bacteria, viruses, and parasites.
Other diseases
A variety of diseases affecting several systems have been linked to abnormalities in protein prenylation, such as diabetes, obesity, fatty liver disease, male infertility, chronic heart failure, and inflammatory bowel disease.305,306,307 Aberrant expression of GGPPS was observed in diabetes, fatty liver disease, male infertility and cardiac hypertrophy, which disrupted the balance of protein farnesylation and geranylgeranylation. For instance, highly expressed GGPPS in adipocytes enhanced K-Ras geranylgeranylation to activate Ras/MAPK/Erk1/2 signalling, which inhibited insulin signalling and led to insulin resistance.308 However, downregulated GGPPS in the heart increases Rheb farnesylation to activate mTORC1 signalling, which further leads to cardiac hypertrophy and heart failure.285 By adjusting the levels of GGPP and FPP, GGPPS can play a pivotal role in governing both physiological and pathological cellular processes. Therefore, targeting the aberrant expression of GGPPS in diseases is a promising therapeutic direction for the direct inhibition of protein prenylation.
Therapeutic targets and clinical research progress
Targeting the mevalonate pathway
Given that the lipid substrates needed for protein prenylation, FPP or GGPP, are synthesised through the mevalonate pathway, inhibition of key catalase enzymes in the mevalonate pathway to limit the synthesis of FPP or GGPP is expected to be a therapeutic target for diseases (Tables 1 and 2). Statins are renowned agents that selectively inhibit HMG-CoA reductase, consequently diminishing the biosynthesis of downstream molecules such as FPP and GGPP.283 Although statins were initially used to control plasma cholesterol levels and prevent cardiovascular disease, studies have revealed that statins also potentially exert a therapeutic effect on cancer, neurological disorders and other diseases.309,310 Regarding antitumour effects, considerable evidence suggests that statins can inhibit tumour proliferation and metastasis by disrupting farnesylation and geranylgeranylation of small GTPases.311,312 For instance, statin-mediated inhibition of K-Ras prenylation induces immunogenic cell death by enhancing ER stress, thereby stimulating a strong CD8 T-cell immune response against K-Ras mutant tumours.313 However, statins mediate antitumour effects not only by disrupting protein prenylation but also by lowering cholesterol levels, modulating autophagy and inducing ferroptosis and pyroptosis to promote the apoptosis of tumour cells and inhibit their proliferation and invasion.314,315,316,317 Despite preclinical and epidemiological data indicating that statins may have antitumour properties, clinical trial results show that statins exert varying therapeutic effects on different malignancies. For instance, a prospective cohort study of breast cancer patients found that lipophilic statin use was associated with a reduced risk of recurrence in breast cancer patients.318,319 However, a randomised adjuvant chemotherapy clinical trial in stage III colon cancer patients showed that compared to nonusers, patients who received statin treatment during and after adjuvant chemotherapy had similar survival outcomes. The survival outcomes were similar among users regardless of K-Ras mutation.320 Similarly, other clinical trials in advanced gastric cancer patients found that the addition of simvastatin or pravastatin to chemotherapy does not improve survival outcomes.321,322 In conclusion, careful consideration of tumour specificity and indications, as well as multiple pharmacological properties, is needed before applying statins in the clinical treatment of cancer. Another class of drugs that target FPP synthase within the mevalonate pathway includes bisphosphonates, with zoledronic acid being a notable example. It is widely used to treat benign and malignant bone diseases by disrupting protein prenylation.323 In addition to bone disease, bisphosphonates have been explored for the treatment of cancer, Hutchins-Gilford progeria, AD and cerebral cavernous malformations, and to some extent, such therapeutic potential is dependent on inhibiting protein prenylation, although it also induces apoptosis and exerts antiangiogenic and antimigratory effects.324,325 One noteworthy aspect for bisphosphonates is that bisphosphonates prefer to inhibit protein geranylgeranylation rather than protein farnesylation, although the synthesis of both GGPP and FPP is reduced,326,327 suggesting that bisphosphonates may have greater efficacy in diseases triggered by protein geranylgeranylation. Clinical evaluation of bisphosphonates mainly focuses on bone diseases or cancers with a high tendency to develop bone metastasis, and some clinical trials have also been conducted to test bisphosphonates for the treatment of progeria. The therapeutic applications of bisphosphonates in other diseases are equally worthy of exploration.
Targeting protein prenylation
Methods for targeting prenylation include inhibiting FTase for protein farnesylation or GGTase for protein geranylgeranylation. The former is generally achieved by FTase inhibitors (FTIs), which are broadly classified into four categories. The first category is FPP analogues that bind with FTase.328 The second category of FTIs includes BMS-184467 and BMS-185878, which are able to bind the structural motifs of farnesyl pyrophosphate and CAAX tetrapeptide.329 The third category is peptidomimetic compounds that mimic the CAAX box, such as GGTI-2418.330 The last category is nonpeptidomimetic enzyme-specific inhibitors, which are identified from high-throughput screening efforts such as tipifarnib, lonafarnib and BMS-214662.331,332 While FTIs showed promising antitumour effects in preclinical studies, their performance in clinical trials was less than satisfactory.333 Most clinical trials have shown that monotherapy with FTIs has no or little efficacy in the treatment of haematopoietic cancers or solid tumours.333 One of the reasons for the discrepancy between laboratory findings and clinical trial results is that in tumours with predominantly K-Ras and N-Ras mutations, K-Ras and N-Ras can escape FTI-mediated inhibition through alternative geranylgeranylation and thus continue to retain signalling activity.334 This finding suggests that tumours driven by H-Ras mutations might be more susceptible to FTIs, given that H-Ras undergoes only farnesylation. To support this hypothesis, a phase II clinical trial in patients with recurrent and/or metastatic head and neck squamous cell carcinoma (HNSCC) showed that HNSCC patients with H-Ras mutations had a 55% objective response after receiving tipifarnib.335 Another phase II clinical trial in advanced refractory uroepithelial cancer patients with H-Ras mutations also found that response was observed in 5 out of the 12 evaluated patients after treatment with tipifarnib.336 For other tumours, FTIs in combination with other treatments can improve treatment efficacy to some degree. For example, the combination of the FTI tipifarnib with gemcitabine and cisplatin was observed to be well-tolerated and showed antitumour activity in a phase I clinical trial in patients with advanced solid tumours.337 However, the side effects of FTI application should not be ignored. Common adverse events include gastrointestinal toxicity, myelosuppression, fever, hypokalaemia, hyperbilirubinemia, peripheral neuropathy, skin rash and others,338,339,340,341 and the majority of adverse events are associated with the FTI dose. Even though clinical trials for cancer treatment were less promising, the application of FTIs showed encouraging results in treating HDV infections, as well as progeria and progeroid laminopathies. In 2020, lonafarnib was approved by the FDA for the treatment of HGPS and processing-deficient progeroid laminopathies, and clinical development of lonafarnib for the treatment of HDV infection is also underway.342 As protein geranylgeranylation is catalysed by GGTases, the development of GGTase inhibitors (GGTIs) represents a promising strategy for diseases with protein geranylgeranylation. For instance, a genetic study demonstrated that inactivation of GGTase-I activity could reduce lung tumour formation and improve the survival of mice with K-Ras-induced cancer.343 Combined treatment with FTI and GGTI produces a synergistic cytotoxic effect, further promoting an increase in cancer cell apoptosis, which is expected to improve treatment efficacy in FTI-resistant cancers.344,345 Although multiple GGTIs have shown antitumour activity in preclinical studies, only the maximum tolerated dose of GGTI-2418 was assessed in patients with advanced solid tumours.330,346 Beyond cancer treatment, GGTIs have potential applications in addressing certain infectious and inflammatory diseases.267,347 In addition to the combination of FTIs and GGTIs, agents inhibiting both FTase and GGTase-I, such as FGTI-2734 and L-778,123, have been developed. The dual inhibitor FGTI-2734 impedes tumour cell growth and is expected to overcome the barrier of K-ras resistance.348 In addition, FGTI-2734 inhibits mast cell-dependent allergic reactions, which is a promising therapeutic strategy for allergic diseases.349 L-778,123 has been assessed in preclinical and clinical studies. The results showed that L-778,123 has a radiosensitizing effect in cell lines with Ras activation and produces a clinical response in patients with advanced solid tumours.350,351
Targeting protein post prenylation
Researchers have been increasingly interested in targeting postprenylation processes such as endoproteolysis and methylation, given that complete catalysis is vital for the functioning of most CAAX proteins. When RCE1 is conditionally deleted, Ras loses its capability to participate in endoproteolysis and methylation, leading to the partial mislocalization of K-Ras and H-Ras, which retards cell growth, reduces Ras-induced transformation, and sensitises tumour cells to farnesyltransferase inhibitors.352 Inactivating ICMT activity has stronger implications in disrupting oncogenesis than activating RCE1. Inactivation of ICMT inhibits not only cell growth and K-Ras-induced oncogenic transformation but also the transformation of the oncogenic form B-Raf.280 In addition, ICMT inhibition by the compound UCM-1336 suppresses the activity of the four Ras isoforms, which further suggests that ICMT is a valuable target.353 Tumours induced by Ras and Rho GTPases are targets for RCE1 and ICMT inhibitors,247 but the efficacy of such inhibitors in Rheb-induced tumours is likely to be limited, as RCE1 and ICMT inhibitors do not block Rheb function.354 Although several small-molecule inhibitors targeting RCE1 have been identified, with some causing Ras mislocalization, studies on their therapeutic applications remain limited.258 A variety of ICMT inhibitors have been developed targeting Ras-related cancers, positioning them as potential anticancer drugs.355 Cysmethynil, a representative ICMT inhibitor, has demonstrated antitumour activity in a wide range of tumours. For example, the treatment of colon cancer cells with cysmethynil resulted in Ras mislocalization and impaired epidermal growth factor signalling, thereby inhibiting cell growth.356 Treatment of prostate cancer cells with cysmethynil leads to the accumulation of cells in the G (1) phase and cell death and hinders tumour growth.357 Subsequent development of cysmethynil derivatives such as analogue 15 and Compound 8.12 further improved its solubility and PAMPA permeability, increasing the potency of the compounds and making them more suitable for preclinical and clinical development.358,359 Beyond cancer treatment, ICMT knockout enhances the survival rates of HGPS model mice. Furthermore, using the ICMT inhibitor C75 has been found to delay ageing and stimulate the proliferation of HGPS cells and Zmpste24-deficient mouse fibroblasts, suggesting that ICMT inhibitors also have therapeutic potential in the treatment of HGPS.360 Although ICMT inhibitors are not yet in clinical studies, their therapeutic potential should not be underestimated.
Glycosylphosphatidylinositol (GPI) anchor
Biosynthesis and maturation of GPI-anchored proteins (GPI-APs)
GPI is primarily synthesised in the ER through a series of 11 consecutive reactions.23 The biosynthesis and maturation of GPI-APs is also a sophisticated process that mainly involves the major steps of precursor protein localisation to the ER, GPI attachment, and GPI maturation (Fig. 9). All GPI-anchored proteins possess a signal sequence, with ~20 hydrophobic amino acids at the N-terminus, that facilitates translocation across the ER membrane.361,362 After localisation of the precursor protein to the ER, GPI attaches to the C-terminus of the precursor protein, and the ω site refers to the amino acid residue to which GPI attaches, usually Ser, Asn, Asp, Ala, Gly, Cys and Thr.363,364 The signal peptide that mediates C-terminal GPI attachment is a nonconserved sequence of 20–30 amino acids starting from the ω + 1 amino acid, which includes a segment of ~10 hydrophilic amino acids and a segment of ~20 hydrophobic amino acids.361

Biosynthesis and maturation of GPI-anchored proteins (GPI-APs). N Nascent proteins first translocate to the ER following the N-terminal signal. After arriving at the ER, the N-terminal signal is removed. Subsequently, GPI-T recognises the amino acid at the ω position and catalyses the attachment of GPI at the ω position, followed by fatty acid remodelling of GPI with or without GalNAc side chain modification of the ER and Golgi. Finally, mature GPI-APs are trafficked to the plasma membrane and associate with raft microdomains. ER endoplasmic reticulum
The GPI-transamidase (GPI-T) complex, residing in the ER and comprising five subunits (PIGK, GPAA1, PIGT, PIGS and PIG-U), attaches the GPI anchor to proteins.365 PIGK is considered to be the catalytic subunit.366,367 Structural analysis of the GPI-T complex suggests that PIGK exhibits caspase-like folding and forms an extensive interaction with the other subunits, GPAA1, PIGT and PIGS, and an open cavity located directly beneath the PIGK subunit accommodates a lipid moiety of the GPI molecule and supports its binding to the GPI substrate.368 Regarding the biosynthesis process of nascent GPI-AP, the GPI-T complex initially cleaves the peptide bond between ω and ω + 1 amino acid of the precursor protein via its PIGK subunit, removing the GPI attachment signal peptide, and then connects the ω amino acids via a thioester bond to form a substrate–enzyme intermediate. Subsequently, the amino group of the terminal EtN of GPI attacks the thioester bond and completes the transfer of GPI by transamidation.23,369 A recently identified subsite featuring human GPI-T structures further revealed its broad proprotein specificity and activation mechanis.370 The selectivity of GPI-T mainly depends on the ω-site, which is restricted by a deep pocket consisting of various residues to define ω-residue preferences. The autoinhibitory loop of PIGK regulates GPI-T activation through a drastic conformational rearrangement. Prior to maturation, GPI molecules of nascent GPI-APs need to undergo remodelling reactions during transport from the ER to the PM. Generally, in the ER, the acyl chain attached to the inositol ring of GPI is removed by PGAP1,371 and the EtNP attached to the second mannose is removed by PGAP5.372 Following this, GPI-APs are recruited to COPII-coated transport vesicles at the ER exit in the presence of p24 family proteins and then transported to the Golgi.373 Upon arrival at the Golgi, the PGI molecule undergoes fatty acid remodelling, which is characterised by the removal of the unsaturated 2-acyl chain by PGAP3, followed by the transfer of a saturated chain back to the sn2-position and acylation by PGAP2.374,375 The two long saturated lipid chains formed by fatty acid remodelling of GPI-APs contribute to trans-bilayer interactions with long acyl-chain-containing phosphatidylserine and are essential for GPI-APs incorporation into lipid rafts.374,376 After FA remodelling, some GPI-APs also undergo GalNAc side chain modifications on the Golgi before trafficking to the PM and associating with lipid rafts. For example, βGalNAc is transferred to the 4 position of Man1 as a side chain and is further extended by β1-3Gal and Sia.23 The physiological roles of such side chain modifications remain largely unknown and need to be further elucidated.
Physiological function
Protein trafficking and lipid raft association
The attachment of GPI anchors gives GPI-APs unique trafficking characteristics and offers a solid PM anchor, setting the stage for GPI-APs to play various biological roles. GPI-APs undergo numerous regulatory changes and modifications throughout the ER-Golgi-PM transport process. If any of the key modifications are abnormal, their transport and lipid raft associations are disrupted. For example, when PGAP3 is deficient, unsaturated FAs remain on PGI-APs, preventing GPI-APs from associating with membrane microdomains and thereby reducing PGI-AP levels on the cell surface.374 Similarly, defects in PGAP3 decrease stable surface expression of a variety of GPI-APs.377 For proteins that naturally have both GPI-anchored and transmembrane isoforms, there is no functional difference between them.378 However, for other GPI-APs, GPI anchoring is an important determinant of GPI-AP lipid raft association, and when replacing GPI with the transmembrane region from a nonraft protein, the majority of GPI-APs lose their raft association ability.379,380
Signal transduction
In mammalian cells, in addition to acting as membrane anchors, GPI-APs are capable of transmitting a variety of signals and regulating multiple intracellular and extracellular responses such as immune responses, cell proliferation and metastasis (Fig. 10). One of the most typical representatives of mediated signal transduction is CD molecules. For instance, CD109 is a GPI glycoprotein that is widely expressed on immune cells, myoepithelial cells, and basal cells in normal tissues as well as various tumour cell lines.381,382,383,384 CD109 can be expressed on the cell surface and enriched in lipid rafts or extracellularly released with exosomes.385 It can also be a secreted protein presented as a truncated form harbouring the 180-kDa CD109 subunit.386 It was shown that both GPI-anchored CD109 and soluble CD109 could bind to the TGF-β receptor, thus inhibiting TGF-β-induced SMAD2/3 phosphorylation and further hindering its regulatory function in the cell nucleus.387 In addition to negatively regulating TGF‐β signalling, CD109 is involved in EGFR-Akt-mTOR signalling, JAK-STAT3 signalling and YAP/TAZ signalling to mediate tumour proliferation and metastasis.388,389,390 Another typical GPI-anchored glycoprotein, CD14, which is widely expressed on the cell membrane surface of myeloid lineage cells, is the first pattern recognition receptor (PRR) identified to bind directly to LPS and is an important regulator of innate immunity.391,392 After recognising and binding LPS, CD14 transfers LPS to MD-2 of the TLR4/MD-2 complex to promote TLR4 dimerisation. TLR4 dimerisation further induces the formation of Myddosomes with MyD88, which triggers a signalling cascade leading to activation of the NF-κB and MAPK pathways.392,393

Glycosylphosphatidylinositol anchor proteins (GPI-APs) play a role in several cellular signal transduction pathways. In this section, we describe several classical GPI-APs and their roles in cellular signal transduction. CD109 negatively regulates TGF‐β signalling, while it can activate the Jak-STAT3 pathway to drive tumour metastasis. CD14 can activate TLR4 to trigger signalling pathways, including INF, tnf-α and IL-6 signalling, to initiate the immune response. Folate binds to FOLR1 to activate MEK/ERK signalling and the Jak-STAT3 pathway. ART could trigger NAD + -induced cell death. uPAR binds to Upa to stimulate the association of EGFR and β1 integrin to activate MEK/ERK signalling. ER endoplasmic reticulum
Other physiological functions
Given the diversity of GPI-anchored proteins, which involve enzymes, adhesion molecules, receptors, antigens and others, each GPI protein performs its respective physiological functions. For instance, ADP-ribosyltransferase ART2 is a GPI-anchored exonuclease that uses free NAD+ to mono-ADP-ribosylate the P2X7 receptor on CD8 T cells, leading to NAD-induced cell death and a reduction in the P2X7R + CD8 + T-cell subpopulation in the TME.394,395 NCAM is a typical adhesion molecule that regulates cell–cell adhesion by homophilic and heterophilic interactions and is involved in the regulation of the development and plasticity of the nervous system.396 The receptors expressed on the cell membrane surface, in addition to mediating signal transduction, are also involved in nutrient uptake. Folate is one of the essential vitamins for cell growth, and its uptake is mediated through GPI-anchored folate receptors (FOLR1). When the GPI anchor of the folate receptors was replaced with transmembrane and cytosolic portions, the uptake efficiency of folate was significantly reduced, indicating that PGI-anchored protein-mediated nutrient uptake is indispensable and plays an important role in modulating JAK-STAT3 signalling and ERK1/2 signalling.397,398,399
Pathological implication
Cancer
Research indicates that GPI-anchored proteins are crucial in various cancers, influencing cancer cell adhesion, migration, angiogenesis, extracellular matrix (ECM) remodelling, and immune responses. GPAA1, a type of GPI-AP, is overexpressed in various cancers.400,401,402 Thorough exploration of its tumorigenic function and underlying mechanism in gastric cancer revealed that GPAA1 enhanced lipid raft formation to support the interaction between EGFR and ERBB2, followed by further activation of downstream Akt signalling to enhance the proliferation of cancer cells.403 Another GPI-AP, uPAR, is prevalent in numerous cancers, encompassing solid tumours, leukaemias, and lymphomas. One factor that drives tumour progression by uPAR is its ability to regulate ECM proteolysis. After binding to uPAR, uPA cleaves plasminogen to generate the active protease plasmin, which further activates MMPs to degrade ECM components and promote tumour cell migration and invasion.404 In addition, uPAR regulates intracellular signalling pathways to control cell proliferation. uPAR–α5β1 integrin interaction signals to FAK and then activates EGFR, and uPAR–β1 integrin–EGFR signalling enhances ERK activation to promote cancer cell proliferation.405 The importance of these GPI-APs in cancer also implies that GPI-T may act as an oncogene in tumour development. Abnormal expression of each GPI-T subunit has been observed in diverse cancer types to be involved in the regulation of GPI-AP expression on the cell surface,365 indicating the value of GPI-T as a potential biomarker and therapeutic target.
Haematological disorders
Paroxysmal nocturnal haemoglobinuria (PNH) is one of the most representative haematological disorders due to GPI-AP defects, with the main clinical manifestations of haemolytic anaemia, thrombosis and bone marrow failure in some cases. In PNH, the most common cause of GPI-AP defects is somatic mutations in the X-linked gene PIGA, as it is a critical gene encoding one of several enzymes that is needed for the biosynthesis of GPI anchors.406 Such mutations lead to defective surface expression of various GPI-anchored proteins, including CD55 and CD59, which function as complement regulators to prevent activated complement toxicity.407 In contrast to normal red blood cells, CD55-defective and CD59‑defective PNH red blood cells lack self-protective complement regulatory factors and thus are highly sensitive to complement activation, which results in intravascular haemolysis.408
Neurological disorders
GPI for GPI-APs, such as PrPC, plays a crucial role in protecting neurons, which is reflected in the fact that GPI is indispensable for PrPC to protect and repair the neuronal cytoskeleton and neuronal communication.409 Many neurological disorders exhibit pathological shifts stemming from downregulated cell surface GPI-AP expression, often linked to mutations in genes responsible for GPI-AP biosynthesis and remodelling. For instance, mutations in PGAP3 impair the GPI-anchor fatty acid-remodelling step, resulting in GPI-AP deficiency, which affects normal neural and embryonic development, leading to intellectual disabilities.410 Sequencing analysis of a family with encephalopathy and nonspecific autosomal-recessive forms of intellectual disability revealed the homozygous 3 bp deletion of another PGI-APs remodelling gene, PGAP1.411 In addition, biallelic splice mutations in ARV1 have been detected in patients with early infantile epileptic encephalopathy, as a reduction in ARV1 levels leads to impaired GPI-anchor synthesis, which hampers normal neurological development and leads to symptoms such as early-onset epilepsy.412 Furthermore, mutations in other genes regulating the biosynthesis of GPI-APs, such as GPI-T,413 PGAP2,414 PIGB,415 C18orf32416 and others, have been detected in a variety of patients with neurodevelopmental anomalies, indicating that the downregulated expression of cell surface GPI-APs due to mutations in genes regulating GPI-AP remodelling is a common phenomenon in neurological disorders.
Infectious diseases
Many of the GPI-APs in pathogenic protozoans and fungi act as virulence factors involved in survival and infection within distinct host environments. A prime example is the GPI-anchored variant surface glycoprotein (VSG) in Trypanosoma brucei, aiding in immune evasion.417 Other GPI-APs of Trypanosoma brucei, such as Procyclin, are implicated in protease resistance, while GPI-PLC and GP63 are implicated in VSG shedding. TfR is implicated in transferrin uptake.418 Similarly, various GPI-APs, such as MSP-1, MSP-2, MSP-4, MSP-5 and MSP-10, are spread over the surface of Plasmodium merozoites, contributing to parasite survival and invasion.419 In addition to protozoans, fungi also require GPI-APs to support biological processes such as cell wall construction,420 selective adhesion and invasion.421,422
Therapeutic targets and clinical research progress
Due to the intricate processes from the initiation to maturation of GPI-APs, targeting their biosynthesis for therapy is both promising yet challenging (Tables 1 and 2). MSLN is a GPI-AP overexpressed in multiple tumour tissues and an attractive target for CAR T-cell therapy. Deficiencies in GPI-anchor biosynthesis by downregulating MSLN expression resulted in MSLN CAR T-cell resistance in pancreatic cancer.423 Among the many regulatory enzymes, the GPI-T subunit PIG-U was pinpointed as an oncogene for bladder cancer in 2004,424 which sparked the interest of researchers to explore whether other subunits also play oncogene roles and whether they have therapeutic value for cancer treatment. Moreover, most GPI-T subunits overexpressed in a wide range of tumours and have potential as anticancer targets.425 Unfortunately, the development of relevant inhibitors has not progressed remarkably. In addition to targeting the modification of GPI attachment to proteins, targeting GPI itself is also a therapeutic strategy. Although the GPI core structure is conserved among organisms, species-specific differences have been observed in the additional modifications to GPI structures and biosynthetic pathways, suggesting that it can be exploited for the development of anti-infectious drugs. GPI structures and GPI-APs play critical roles in the virulence of pathogenic protozoans and fungi. Research on hindering the protozoan GPI pathway has chiefly focused on GlcNAc-PI de-Nacetylase, which is essential for the second step of GPI biosynthesis. Salicylic hydroxamic acid (SHAM) has been identified as a nonsubstrate analogue inhibitor of the trypanosomal de-N-acetylase with high ligand efficiency.426 Regulatory enzymes Gwt1 and Mcd4 have been exploited for the development of antifungal inhibitors. Gwt1 is responsible for catalysing inositol acylation, while Mcd4 is responsible for catalysing phosphoethanolamine addition to first mannose. The identified Gwt1 inhibitors include BIQ, E1210 (APX001), Gepinacin, G365, G884 and Compound A1.427 Mcd4 inhibitors include M743 and M720.428 Among those inhibitors, APX001 which showed profound in vitro broad-spectrum antifungal activity, has entered phase I/II clinical trials and the results show that APX001 is safe, well-tolerated, and efficacious in participants with candidemia caused by C. auris.429,430 Species-specific differences in the pathogen GPI pathway create opportunities for the development of anti-infective drugs, and further optimisation of the potency, as well as the specificity of inhibitors, will be beneficial and expand their application.
Cholesterylation
Mechanism of cholesterylation
Hedgehog (Hh) and smoothened (SMO) are proteins that undergo cholesterylation through an autoprocessing reaction (Fig. 11). In the case of Hh, its protein precursor undergoes autocatalytic cleavage, generating a 19 kDa N-terminal (N-Hh) and a 25 kDa C-terminal fragment (C-Hh). The C-terminus of N-Hh undergoes cholesterylation through a two-step autoprocessing reaction.431 First, the thiol side chain of Cys257 initiates a nucleophilic attack on the carbonyl carbon of Gly,256 triggering the cleavage of the Hh protein precursor and the formation of a labile thioester intermediate. Subsequently, cholesterol attacks the same carbon in the thioester intermediate, resulting in the cleavage of the intermediate as well as the formation of cholesterol-modified HhN and free HhC.26 During the process of cholesterylation, certain conserved amino acids of Hh act as vital factors. For instance, His,328 Thr325 and Cys399 are involved in thioester formation by forming hydrogen bonds with the α-amino group of Cys258 or by activating free thiols.431,432 Asp302 is needed for cholesterol attachment and 63 C-terminal amino acids are needed for the formation of a hydrophobic pocket to mediate cholesterol binding.433 In addition to the specific structure of HhC, the characteristics of cholesterol itself also contribute to the successful sterol attachment of Hh. The portion of the sterol that most likely acts as an attacking nucleophile is the 3β hydroxyl, and replacing the 3β hydroxyl with other groups, such as epicholesterol, would abolish autoprocessing, and the addition of hydroxy groups to the side chain or ring structures would result in decreased efficiency of the reaction.433,434 For SMO, the cholesterylation mechanism similarly involves a two-step process. The aspartic acid 95 (D95) and tyrosine 130 (Y130) of SMO join by an ester bond to form a high-energy intermediate, followed by ester exchange from D95–Y130 to D95–cholesterol.435,436 In addition to D95 and Y130, eight other residues, including E160, W109, W119, W163 and F166, are also essential for SMO cholesterylation. Replacing alanine with E160, F166, W109, W119, and W163 has been shown to entirely eliminate SMO cholesterylation.437 Moreover, the cation-π pair between Y85 and K133 is necessary for cholesterol conjugation to SMO.437 In addition to the specific structure of SMO, the N-Hh–PTCH1 axis increases [Ca2 + ] in SMO-localised endosomes, which further enhances SMO cholesterylation by promoting ester exchange from D95–Y130 to D95–cholesterol.436

Mechanism of hedgehog (Hh) and smoothened (SMO) cholesterylation. a Autoprocessing nucleophilic attack between C258 of Hh and G257 induces the formation of a labile thioester intermediate. Subsequently, the activated intermediate undergoes cleavage, and cholesterol attaches to HhH to complete HhH cholesterylation. b The autoprocessing nucleophilic attack between D95 and Y130 of SMO induces the formation of a high-energy intermediate, followed by ester exchange from D95–Y130 to D95–cholesterol. C258: Cystine 258; G257: Glycine 257. D95: Aspartic acid 95; Y130: Tyrosine 130
Role of cholesterylation
Hh signalling is a signal transduction pathway that regulates vital physiological activities such as embryonic development.27 Both N-Hh and SMO are critical components of the Hh signalling pathway, and cholesterylation has a pronounced effect on their functions (Fig. 12). In Hh-producing cells, N-Hh targets the PM after being covalently modified by cholesterol at the C-terminus and palmitate at the N-terminus, which is irreversibly catalysed by HAAT.438 Then, Dispatched, a multipass transmembrane protein located on the cell surface, controls the release of Hh into the extracellular space in a cholesterol-dependent binding event.439 The release of Hh is also dependent on Scube2, which is an extracellular protein assisting in the extracellular space trafficking of N-Hh.439 Although Dispatched and Scube2 cooperate to dramatically facilitate N-Hh release, the functional groups of cholesterol for recognition are different, and Dispatched is crosslinked 25-azicholesterol, while Scube2 is crosslinked with 6-azicholestanol.439 In Hh-receiving cells, the transmembrane protein PTCH1 is enriched in primary cilia and inhibits SMO activity before binding of Hh.440 Hh binding to PTCH1 facilitates endocytotic internalisation and the activation of N-Hh-PTCH1 complex degradation in lysosomes.441 Consequently, the inhibitory effect of PTCH1 on SMO is disarmed, and SMO becomes activated and relocates to the cilia, leading to activation of GLI transcription factors.442 Regarding SMO activation, exposure to cholesterol caused an enrichment of SMO at the cell surface, indicating that cholesterol is a candidate endogenous activator of SMO and needed for Hh signalling,443 as abolishment of cholesterylation by mutation of the D95 residue on SMO results in reduced Hh-stimulated ciliary localisation and compromised Hh signal transmission. Of the PM cholesterol pool consisting of accessible cholesterol (free cholesterol), sphingomyelin (SM) chelated cholesterol, and essential cholesterol, only free cholesterol is available for activation of Hh signalling.444 To further explore the relationship between PTCH1 and cholesterol concentration, it was found that fibroblast treatment with the Hh signalling antagonist cyclobenzaprine downregulated PTCH1 expression and reduced BODIPY cholesterol efflux, while the opposite phenomenon was observed when treated with the Hh signalling agonist SAG,445 suggesting that PTCH1 regulates intracellular cholesterol concentration by controlling cholesterol efflux from cells. In the absence of N-Hh, PTCH1 lowers the intracellular cholesterol concentration to inhibit the activation of SMO. In contrast, the interaction between N-Hh and PTCH1 creates a PTCH1-free ciliary environment for the further accumulation of free cholesterol, thereby contributing to SMO cholesterylation.

The role of cholesterylation in Hedgehog (Hh) signalling. The Hh protein undergoes cholesterylation and O-palmitoylation on the ER and Golgi membrane before targeting the plasma membrane (PM). Subsequently, it is secreted into the extracellular space and trafficked with the help of Dispathed and Scube. After arriving at the Hh-receiving cell, Hh binds to PATCH1, resulting in the degradation of PATCH1, which reduces cholesterol efflux and activates smoothened (SMO). Cholesterylated SMO traffics to the PM and then activates the downstream oncogenic GLI signalling pathway. ER endoplasmic reticulum
Pathological implication
The Hh signalling pathway is pivotal in embryonic development.441 Regarding the impact of cholesterylation loss on development, abolishing cholesterylation of SMO was found to lead to embryonic death and severe developmental delay in homozygous SMOD99E/D99E knockin mice.436 In addition, ShhN/- mutant mice unable to bind cholesterol exhibit developmental defects with phenotypic features similar to holoprosencephaly.446 Moreover, cholesterylation of N-Hh is essential to the development of limb buds; the role of the cholesterol moiety is to limit the spread of Shh across the anteroposterior axis, and it was observed that ShhN− limbs lacking cholesterol modification have defective digit development.447 Taken together, the cholesterylation of N-Hh and SMO plays a pivotal role in regulating Hh signalling, and loss of cholesterylation disrupts Hh signalling and thus severely affects developmental patterning in various systems.
Therapeutic targets and clinical research progress
Increased Hh pathway activity has pathological consequences in several cancer types,448 and targeting cholesterylation for the development of therapies against HH-related cancers maintains a promising outlook (Table 1). One of the strategies is targeting the autoprocessing of the Hh protein to block Hh cholesterylation and downstream signalling. Two compounds have been identified that effectively inhibit the cholesterol-dependent autocleavage of Hh proteins by targeting nucleophilic attack by cholesterol or cholesterol binding.449 CID 5717, also known as zafirlukast, inhibits cholesterol-dependent autocleavage in a time-dependent manner, which means that the inhibitory effect is reversible. CID 72303 was inhibited in a time-independent manner. The activity and safety of these compounds need to be further validated in vivo. In addition, reducing the biosynthesis of cholesterol is another strategy. Inhibition of HMG-CoA reductase by statins reduces cholesterol biosynthesis, markedly decreases Hh pathway activity and represses the proliferation of medulloblastoma cells.314 In addition, it is speculated that inhibition of sterol biosynthetic enzymes to divert sterol flux away from cholesterol into a “shunt” pathway may impair SMO activation by depleting cellular cholesterol, while more preclinical and clinical data are needed to support its application for SMO inhibition. Furthermore, SMO cholesterylation is regulated by SMO-interacting protein EBP. Overexpression of EBP suppresses SMO cholesterolization and downstream signalling, and the opposite phenomenon was observed after genetic disruption of EBP.450 Therefore, it may be considered therapeutically useful to upregulate EBP expression in diseases with enhanced Hh pathway activity and inhibit EBP expression in diseases with reduced Hh pathway activity.
Conclusion and perspective
Diverse PTMs confer functional diversity to proteins. Since the discovery of the first protein lipidation in the 1970s, five decades of continuous investigations have led to a greater understanding of protein lipidation. Accumulating evidence from genetic, structural, and biomedical studies undoubtedly shows that lipidations play an important role in a wide range of physiological and pathological processes. Genetic evidence for lipidation-related enzymes in diseases and the role of lipidation in modulating protein functions are summarised in Tables 3 and 4.
We now have a comprehensive understanding of regulatory enzymes and catalytic processes involved in protein lipidation, as well as their role in physiology and disease. Based on this, several drugs have been developed and tested in preclinical and clinical studies, some of which including asciminib and lonafarnib are FDA-approved for therapeutic use. Nevertheless, given that lipidation involves delicate and complex regulation, there is still a fair amount of challenges for researchers to overcome. For instance, although chemical biology tools such as click chemistry-based metabolic labelling have facilitated the elucidation of protein lipidation, revealing the dynamic modification processes remains challenging. More convenient and intuitive detection techniques need to be further developed. In addition, some proteins (e.g., Ras) are not only dependent on S-palmitoylation and S-prenylation for their functions but also undergo other PTMs. How the crosstalk of protein PTMs is precisely orchestrated during this process remains unknown. There are more palmitoylation catalytic enzymes than other lipidation catalytic enzymes, and the understanding of the potential functional redundancy of DHHC-PATs in the S-palmitoylation machinery remains rudimentary, thus hindering the development of specifically targeted agents. Targeting DHHC remains an urgent need. Only a few crystal structures of DHHCs have been determined, and structure identification is required for guided rational drug design. To date, most DHHC inhibitors used in preclinical studies are broad-spectrum inhibitors. The design of selective inhibitors to pharmacologically control different DHHCs is an unsolved challenge. Whether DHHCs can serve as pharmacological targets for the treatment of human diseases needs more research. In addition, lipidation proteome analyses have revealed a large number of lipid-modified protein substrates, but most of them remain unannotated. Therefore, the functional impact on other proteins that undergo the same lipidation when using inhibitors or agonists of lipid modification is unknown. Substantial additional work is needed to elucidate the functional impact on these modified proteins. Moreover, revealing the three-dimensional structure of specific lipid-modified proteins of pathological importance to obtain valuable insights into substrate–enzyme relationships for further specialised inhibitor optimisation design may avoid off-target effects; however, this still requires tremendous effort. Drug resistance in the application of lipidation catalytic enzyme inhibitors is common. In addition to high-throughput screening and the development of novel small-molecule inhibitors, exploring potential drug synergies, especially targeting proteins that undergo multiple lipidations such as Ras, may improve drug resistance and therapeutic efficacy.
In summary, targeting lipidation in various diseases provides an innovative approach to achieve therapeutic goals, but it is fraught with challenges.
References
Ambrogelly, A., Palioura, S. & Soll, D. Natural expansion of the genetic code. Nat. Chem. Biol. 3, 29–35 (2007).
Gupta, R. et al. Post-translational modifications: regulators of neurodegenerative proteinopathies. Ageing Res. Rev. 68, 101336 (2021).
Xu, H. et al. PTMD: a database of human disease-associated post-translational modifications. Genomics Proteom. Bioinforma. 16, 244–251 (2018).
Wang, R. & Chen, Y. Q. Protein lipidation types: current strategies for enrichment and characterization. Int. J. Mol. Sci. 23, 2365 (2022).
Chamberlain, L. H. & Shipston, M. J. The physiology of protein S-acylation. Physiol. Rev. 95, 341–376 (2015).
Schmidt, M. F. & Schlesinger, M. J. Fatty acid binding to vesicular stomatitis virus glycoprotein: a new type of post-translational modification of the viral glycoprotein. Cell 17, 813–819 (1979).
Schmidt, M. F., Bracha, M. & Schlesinger, M. J. Evidence for covalent attachment of fatty acids to Sindbis virus glycoproteins. Proc. Natl. Acad. Sci. USA 76, 1687–1691 (1979).
Mitchell, D. A., Vasudevan, A., Linder, M. E. & Deschenes, R. J. Protein palmitoylation by a family of DHHC protein S-acyltransferases. J. Lipid Res. 47, 1118–1127 (2006).
Won, S. J., Cheung See Kit, M. & Martin, B. R. Protein depalmitoylases. Crit. Rev. Biochem. Mol. Biol. 53, 83–98 (2018).
Qu, M., Zhou, X., Wang, X. & Li, H. Lipid-induced S-palmitoylation as a vital regulator of cell signaling and disease development. Int. J. Biol. Sci. 17, 4223–4237 (2021).
Li, X. et al. Protein palmitoylation modification during viral infection and detection methods of palmitoylated proteins. Front. Cell Infect. Microbiol. 12, 821596 (2022).
Fhu, C. W. & Ali, A. Protein lipidation by palmitoylation and myristoylation in cancer. Front. Cell Dev. Biol. 9, 673647 (2021).
Meinnel, T., Dian, C. & Giglione, C. Myristoylation, an ancient protein modification mirroring eukaryogenesis and evolution. Trends Biochem. Sci. 45, 619–632 (2020).
Jiang, H. et al. Protein lipidation: occurrence, mechanisms, biological functions, and enabling technologies. Chem. Rev. 118, 919–988 (2018).
Aitken, A. et al. Identification of the NH2-terminal blocking group of calcineurin B as myristic acid. FEBS Lett. 150, 314–318 (1982).
Stevenson, F. T., Bursten, S. L., Locksley, R. M. & Lovett, D. H. Myristyl acylation of the tumor necrosis factor alpha precursor on specific lysine residues. J. Exp. Med. 176, 1053–1062 (1992).
Feldman, J. L., Baeza, J. & Denu, J. M. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem. 288, 31350–31356 (2013).
Wang, M. & Casey, P. J. Protein prenylation: unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 17, 110–122 (2016).
Kamiya, Y., Sakurai, A., Tamura, S. & Takahashi, N. Structure of rhodotorucine A, a novel lipopeptide, inducing mating tube formation in Rhodosporidium toruloides. Biochem. Biophys. Res. Commun. 83, 1077–1083 (1978).
Wolda, S. L. & Glomset, J. A. Evidence for modification of lamin B by a product of mevalonic acid. J. Biol. Chem. 263, 5997–6000 (1988).
Marchwicka, A. et al. Protein prenyltransferases and their inhibitors: structural and functional characterization. Int. J. Mol. Sci. 23, 5424 (2022).
Winter-Vann, A. M. & Casey, P. J. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat. Rev. Cancer 5, 405–412 (2005).
Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biol. 10, 190290 (2020).
Kundu, S. et al. Profiling glycosylphosphatidylinositol (GPI)-interacting proteins in the cell membrane using a bifunctional GPI analogue as the probe. J. Proteome Res. 22, 919–930 (2023).
Poisson, G., Chauve, C., Chen, X. & Bergeron, A. FragAnchor: a large-scale predictor of glycosylphosphatidylinositol anchors in eukaryote protein sequences by qualitative scoring. Genomics Proteom. Bioinforma. 5, 121–130 (2007).
Porter, J. A., Young, K. E. & Beachy, P. A. Cholesterol modification of hedgehog signaling proteins in animal development. Science 274, 255–259 (1996).
Zhang, Y. & Beachy, P. A. Cellular and molecular mechanisms of Hedgehog signalling. Nat. Rev. Mol. Cell Biol. 24, 668–687 (2023).
Stix, R., Lee, C. J., Faraldo-Gomez, J. D. & Banerjee, A. Structure and mechanism of DHHC protein acyltransferases. J. Mol. Biol. 432, 4983–4998 (2020).
Ko, P. J. & Dixon, S. J. Protein palmitoylation and cancer. EMBO Rep. 19, e46666 (2018).
Ohno, Y., Kihara, A., Sano, T. & Igarashi, Y. Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochim Biophys. Acta 1761, 474–483 (2006).
Runkle, K. B. et al. Inhibition of DHHC20-mediated EGFR palmitoylation creates a dependence on EGFR signaling. Mol. Cell 62, 385–396 (2016).
Gorleku, O. A. et al. Endoplasmic reticulum localization of DHHC palmitoyltransferases mediated by lysine-based sorting signals. J. Biol. Chem. 286, 39573–39584 (2011).
Chen, J. J., Fan, Y. & Boehning, D. Regulation of dynamic protein S-acylation. Front. Mol. Biosci. 8, 656440 (2021).
Mesquita, F. S. et al. S-acylation controls SARS-CoV-2 membrane lipid organization and enhances infectivity. Dev. Cell 56, 2790–2807.e2798 (2021).
Rana, M. S. et al. Fatty acyl recognition and transfer by an integral membrane S-acyltransferase. Science 359, eaao6326 (2018).
Roth, A. F., Feng, Y., Chen, L. & Davis, N. G. The yeast DHHC cysteine-rich domain protein Akr1p is a palmitoyl transferase. J. Cell Biol. 159, 23–28 (2002).
Jennings, B. C. & Linder, M. E. DHHC protein S-acyltransferases use similar ping-pong kinetic mechanisms but display different acyl-CoA specificities. J. Biol. Chem. 287, 7236–7245 (2012).
Verardi, R., Kim, J. S., Ghirlando, R. & Banerjee, A. Structural basis for substrate recognition by the ankyrin repeat domain of human DHHC17 palmitoyltransferase. Structure 25, 1337–1347.e1336 (2017).
Ohno, Y. et al. Analysis of substrate specificity of human DHHC protein acyltransferases using a yeast expression system. Mol. Biol. Cell 23, 4543–4551 (2012).
Lemonidis, K. et al. The Golgi S-acylation machinery comprises zDHHC enzymes with major differences in substrate affinity and S-acylation activity. Mol. Biol. Cell 25, 3870–3883 (2014).
Swarthout, J. T. et al. DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H- and N-Ras. J. Biol. Chem. 280, 31141–31148 (2005).
Fredericks, G. J. et al. Stable expression and function of the inositol 1,4,5-triphosphate receptor requires palmitoylation by a DHHC6/selenoprotein K complex. Proc. Natl. Acad. Sci. USA 111, 16478–16483 (2014).
Abrami, L. et al. Identification and dynamics of the human ZDHHC16-ZDHHC6 palmitoylation cascade. eLife 6, e27826 (2017).
Plain, F. et al. Control of protein palmitoylation by regulating substrate recruitment to a zDHHC-protein acyltransferase. Commun. Biol. 3, 411 (2020).
Stix, R., Song, J., Banerjee, A. & Faraldo-Gomez, J. D. DHHC20 palmitoyl-transferase reshapes the membrane to foster catalysis. Biophys. J. 118, 980–988 (2020).
Ning, W. et al. GPS-Palm: a deep learning-based graphic presentation system for the prediction of S-palmitoylation sites in proteins. Brief. Bioinforma. 22, 1836–1847 (2021).
Greaves, J. & Chamberlain, L. H. DHHC palmitoyl transferases: substrate interactions and (patho)physiology. Trends Biochem. Sci. 36, 245–253 (2011).
Lanyon-Hogg, T., Faronato, M., Serwa, R. A. & Tate, E. W. Dynamic protein acylation: new substrates, mechanisms, and drug targets. Trends Biochem. Sci. 42, 566–581 (2017).
Thomas, G. M. et al. Palmitoylation by DHHC5/8 targets GRIP1 to dendritic endosomes to regulate AMPA-R trafficking. Neuron 73, 482–496 (2012).
Sanders, S. S. et al. The palmitoyl acyltransferase ZDHHC14 controls Kv1-family potassium channel clustering at the axon initial segment. eLife 9, e56058 (2020).
Huang, K. et al. Neuronal palmitoyl acyl transferases exhibit distinct substrate specificity. FASEB J. 23, 2605–2615 (2009).
Matt, L., Kim, K., Chowdhury, D. & Hell, J. W. Role of palmitoylation of postsynaptic proteins in promoting synaptic plasticity. Front. Mol. Neurosci. 12, 8 (2019).
Zheng, B. et al. 2-Bromopalmitate analogues as activity-based probes to explore palmitoyl acyltransferases. J. Am. Chem. Soc. 135, 7082–7085 (2013).
Abrami, L. et al. Palmitoylated acyl protein thioesterase APT2 deforms membranes to extract substrate acyl chains. Nat. Chem. Biol. 17, 438–447 (2021).
Long, J. Z. & Cravatt, B. F. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem. Rev. 111, 6022–6063 (2011).
Azizi, S. A., Kathayat, R. S. & Dickinson, B. C. Activity-based sensing of S-depalmitoylases: chemical technologies and biological discovery. Acc. Chem. Res. 52, 3029–3038 (2019).
Duncan, J. A. & Gilman, A. G. A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein alpha subunits and p21(RAS). J. Biol. Chem. 273, 15830–15837 (1998).
Hirano, T. et al. Thioesterase activity and subcellular localization of acylprotein thioesterase 1/lysophospholipase 1. Biochim. Biophys. Acta 1791, 797–805 (2009).
Kong, E. et al. Dynamic palmitoylation links cytosol-membrane shuttling of acyl-protein thioesterase-1 and acyl-protein thioesterase-2 with that of proto-oncogene H-ras product and growth-associated protein-43. J. Biol. Chem. 288, 9112–9125 (2013).
Kathayat, R. S. et al. Active and dynamic mitochondrial S-depalmitoylation revealed by targeted fluorescent probes. Nat. Commun. 9, 334 (2018).
Vartak, N. et al. The autodepalmitoylating activity of APT maintains the spatial organization of palmitoylated membrane proteins. Biophys. J. 106, 93–105 (2014).
Adachi, N., Hess, D. T., McLaughlin, P. & Stamler, J. S. S-palmitoylation of a novel site in the beta2-adrenergic receptor associated with a novel intracellular itinerary. J. Biol. Chem. 291, 20232–20246 (2016).
Tomatis, V. M., Trenchi, A., Gomez, G. A. & Daniotti, J. L. Acyl-protein thioesterase 2 catalyzes the deacylation of peripheral membrane-associated GAP-43. PLoS ONE 5, e15045 (2010).
Won, S. J. et al. Molecular mechanism for isoform-selective inhibition of acyl protein thioesterases 1 and 2 (APT1 and APT2). ACS Chem. Biol. 11, 3374–3382 (2016).
Camp, L. A. & Hofmann, S. L. Purification and properties of a palmitoyl-protein thioesterase that cleaves palmitate from H-Ras. J. Biol. Chem. 268, 22566–22574 (1993).
Hellsten, E. et al. Human palmitoyl protein thioesterase: evidence for lysosomal targeting of the enzyme and disturbed cellular routing in infantile neuronal ceroid lipofuscinosis. EMBO J. 15, 5240–5245 (1996).
Yuan, W. et al. GFAP hyperpalmitoylation exacerbates astrogliosis and neurodegenerative pathology in PPT1-deficient mice. Proc. Natl. Acad. Sci. USA 118, e2022261118 (2021).
Cao, M., Luo, X., Wu, K. & He, X. Targeting lysosomes in human disease: from basic research to clinical applications. Signal Transduct. Target Ther. 6, 379 (2021).
Lu, J. Y., Verkruyse, L. A. & Hofmann, S. L. Lipid thioesters derived from acylated proteins accumulate in infantile neuronal ceroid lipofuscinosis: correction of the defect in lymphoblasts by recombinant palmitoyl-protein thioesterase. Proc. Natl. Acad. Sci. USA 93, 10046–10050 (1996).
Gorenberg, E. L. et al. Identification of substrates of palmitoyl protein thioesterase 1 highlights roles of depalmitoylation in disulfide bond formation and synaptic function. PLoS Biol. 20, e3001590 (2022).
Calero, G. et al. The crystal structure of palmitoyl protein thioesterase-2 (PPT2) reveals the basis for divergent substrate specificities of the two lysosomal thioesterases, PPT1 and PPT2. J. Biol. Chem. 278, 37957–37964 (2003).
Gupta, P. et al. Disruption of PPT2 in mice causes an unusual lysosomal storage disorder with neurovisceral features. Proc. Natl. Acad. Sci. USA 100, 12325–12330 (2003).
Yuan, C. et al. Overexpression of PPT2 represses the clear cell renal cell carcinoma progression by reducing epithelial-to-mesenchymal transition. J. Cancer 11, 1151–1161 (2020).
Levin, S. W. et al. Oral cysteamine bitartrate and N-acetylcysteine for patients with infantile neuronal ceroid lipofuscinosis: a pilot study. Lancet Neurol. 13, 777–787 (2014).
Rebecca, V. W. et al. PPT1 promotes tumor growth and is the molecular target of chloroquine derivatives in cancer. Cancer Discov. 9, 220–229 (2019).
Lin, D. T. & Conibear, E. ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. eLife 4, e11306 (2015).
Cao, Y. et al. ABHD10 is an S-depalmitoylase affecting redox homeostasis through peroxiredoxin-5. Nat. Chem. Biol. 15, 1232–1240 (2019).
Remsberg, J. R. et al. ABHD17 regulation of plasma membrane palmitoylation and N-Ras-dependent cancer growth. Nat. Chem. Biol. 17, 856–864 (2021).
Yokoi, N. et al. Identification of PSD-95 depalmitoylating enzymes. J. Neurosci. 36, 6431–6444 (2016).
Dekker, F. J. et al. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat. Chem. Biol. 6, 449–456 (2010).
Rusch, M. et al. Identification of acyl protein thioesterases 1 and 2 as the cellular targets of the Ras-signaling modulators palmostatin B and M. Angew. Chem. Int. Ed. Engl. 50, 9838–9842 (2011).
Brun, S. et al. GNS561, a clinical-stage PPT1 inhibitor, is efficient against hepatocellular carcinoma via modulation of lysosomal functions. Autophagy 18, 678–694 (2022).
Zhou, B. et al. The palmitoylation of AEG-1 dynamically modulates the progression of hepatocellular carcinoma. Theranostics 12, 6898–6914 (2022).
Han, J., Wu, P., Wang, F. & Chen, J. S-palmitoylation regulates AMPA receptors trafficking and function: a novel insight into synaptic regulation and therapeutics. Acta Pharm. Sin. B 5, 1–7 (2015).
Glatz, J. F. C. & Luiken, J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 59, 1084–1093 (2018).
Zhao, L. et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J. Hepatol. 69, 705–717 (2018).
Wang, J. et al. DHHC4 and DHHC5 facilitate fatty acid uptake by palmitoylating and targeting CD36 to the plasma membrane. Cell Rep. 26, 209–221 e205 (2019).
Hao, J. W. et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat. Commun. 11, 4765 (2020).
Martin-Belmonte, F. & Perez-Moreno, M. Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer 12, 23–38 (2011).
Chen, B. et al. ZDHHC7-mediated S-palmitoylation of Scribble regulates cell polarity. Nat. Chem. Biol. 12, 686–693 (2016).
Patterson, S. I. Posttranslational protein S-palmitoylation and the compartmentalization of signaling molecules in neurons. Biol. Res. 35, 139–150 (2002).
Hayashi, T., Rumbaugh, G. & Huganir, R. L. Differential regulation of AMPA receptor subunit trafficking by palmitoylation of two distinct sites. Neuron 47, 709–723 (2005).
Lv, K. et al. Depalmitoylation rewires FLT3-ITD signaling and exacerbates leukemia progression. Blood 138, 2244–2255 (2021).
Tortosa, E. & Hoogenraad, C. C. Polarized trafficking: the palmitoylation cycle distributes cytoplasmic proteins to distinct neuronal compartments. Curr. Opin. Cell Biol. 50, 64–71 (2018).
Ernst, A. M. et al. S-palmitoylation sorts membrane cargo for anterograde transport in the Golgi. Dev. Cell 47, 479–493.e477 (2018).
Hancock, J. F., Magee, A. I., Childs, J. E. & Marshall, C. J. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 57, 1167–1177 (1989).
Misaki, R. et al. Palmitoylated Ras proteins traffic through recycling endosomes to the plasma membrane during exocytosis. J. Cell Biol. 191, 23–29 (2010).
Yao, H. et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat. Biomed. Eng. 3, 306–317 (2019).
Wu, M. et al. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J. Hematol. Oncol. 15, 24 (2022).
Chen, X. et al. Oct4A palmitoylation modulates tumorigenicity and stemness in human glioblastoma cells. Neuro Oncol. 25, 82–96 (2023).
Zhou, L., Zeng, H., Cui, J. & Jin, S. Palmitoylation facilitates inflammation through suppressing NOD2 degradation mediated by the selective autophagy receptor SQSTM1. Autophagy 18, 2254–2255 (2022).
Yuan, M. et al. ZDHHC12-mediated claudin-3 S-palmitoylation determines ovarian cancer progression. Acta Pharm. Sin. B 10, 1426–1439 (2020).
Rossin, A. et al. Fas palmitoylation by the palmitoyl acyltransferase DHHC7 regulates Fas stability. Cell Death Differ. 22, 643–653 (2015).
Wang, L. et al. Palmitoylation prevents sustained inflammation by limiting NLRP3 inflammasome activation through chaperone-mediated autophagy. Mol. Cell 83, 281–297.e210 (2023).
Du, W. et al. Loss of optineurin drives cancer immune evasion via palmitoylation-dependent IFNGR1 lysosomal sorting and degradation. Cancer Discov. 11, 1826–1843 (2021).
Zheng, M., Karki, R., Vogel, P. & Kanneganti, T. D. Caspase-6 is a key regulator of innate immunity, inflammasome activation, and host defense. Cell 181, 674–687.e613 (2020).
Wang, X. J. et al. Crystal structures of human caspase 6 reveal a new mechanism for intramolecular cleavage self-activation. EMBO Rep. 11, 841–847 (2010).
Skotte, N. H. et al. Palmitoylation of caspase-6 by HIP14 regulates its activation. Cell Death Differ. 24, 433–444 (2017).
Jeyifous, O. et al. Palmitoylation regulates glutamate receptor distributions in postsynaptic densities through control of PSD95 conformation and orientation. Proc. Natl. Acad. Sci. USA 113, E8482–E8491 (2016).
Mukai, K. et al. Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 7, 11932 (2016).
Hansen, A. L. et al. STING palmitoylation as a therapeutic target. Cell Mol. Immunol. 16, 236–241 (2019).
Zhu, Z. et al. STING suppresses mitochondrial VDAC2 to govern RCC growth independent of innate immunity. Adv. Sci. 10, e2203718 (2023).
Liu, Z. et al. Emerging roles of protein palmitoylation and its modifying enzymes in cancer cell signal transduction and cancer therapy. Int. J. Biol. Sci. 18, 3447–3457 (2022).
Fritsch, J., Sarchen, V. & Schneider-Brachert, W. Regulation of death receptor signaling by S-palmitoylation and detergent-resistant membrane micro domains-greasing the gears of extrinsic cell death induction, survival, and inflammation. Cancers 13, 2513 (2021).
Zhang, Y. et al. Function of protein S-palmitoylation in immunity and immune-related diseases. Front. Immunol. 12, 661202 (2021).
Shi, C. et al. ZDHHC18 negatively regulates cGAS‐mediated innate immunity through palmitoylation. EMBO J. 41, e109272 (2022).
Zhang, M. et al. A STAT3 palmitoylation cycle promotes T(H)17 differentiation and colitis. Nature 586, 434–439 (2020).
Tewari, D. et al. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: a novel therapeutic strategy. Semin. Cancer Biol. 80, 1–17 (2022).
Sun, Y. et al. ZDHHC2-mediated AGK palmitoylation activates AKT/mTOR signaling to reduce sunitinib sensitivity in renal cell carcinoma. Cancer Res. 83, 2034–2051 (2023).
Tao, L. et al. GRK6 palmitoylation increasing its membrance translocation promotes LPS-induced inflammation by PI3K/ AKT pathway in Kuppfer cells. Int. Immunopharmacol. 117, 109933 (2023).
Sun, Y. et al. S-palmitoylation of PCSK9 induces sorafenib resistance in liver cancer by activating the PI3K/AKT pathway. Cell Rep. 40, 111194 (2022).
Bailey, M. H. et al. Comprehensive characterization of cancer driver genes and mutations. Cell 174, 1034–1035 (2018).
Blanc, M., David, F. P. A. & van der Goot, F. G. SwissPalm 2: protein S-palmitoylation database. Methods Mol. Biol. 2009, 203–214 (2019).
Zhang, Q. et al. Reprogramming of palmitic acid induced by dephosphorylation of ACOX1 promotes beta-catenin palmitoylation to drive colorectal cancer progression. Cell Discov. 9, 26 (2023).
Pei, X. et al. Palmitoylation of MDH2 by ZDHHC18 activates mitochondrial respiration and accelerates ovarian cancer growth. Sci. China Life Sci. 65, 2017–2030 (2022).
Zhang, Z. et al. DHHC9-mediated GLUT1 S-palmitoylation promotes glioblastoma glycolysis and tumorigenesis. Nat. Commun. 12, 5872 (2021).
Chen, Q. T. et al. HK1 from hepatic stellate cell-derived extracellular vesicles promotes progression of hepatocellular carcinoma. Nat. Metab. 4, 1306–1321 (2022).
Chen, S. et al. Palmitoylation-dependent activation of MC1R prevents melanomagenesis. Nature 549, 399–403 (2017).
Chen, S. et al. Targeting MC1R depalmitoylation to prevent melanomagenesis in redheads. Nat. Commun. 10, 877 (2019).
Sun, Y. et al. AMPK phosphorylates ZDHHC13 to increase MC1R activity and suppress melanomagenesis. Cancer Res. 83, 1062–1073 (2023).
Huang, J. et al. ZDHHC22-mediated mTOR palmitoylation restrains breast cancer growth and endocrine therapy resistance. Int. J. Biol. Sci. 18, 2833–2850 (2022).
Cho, E. & Park, M. Palmitoylation in Alzheimer’s disease and other neurodegenerative diseases. Pharm. Res. 111, 133–151 (2016).
Cervilla-Martinez, J. F. et al. Altered cortical palmitoylation induces widespread molecular disturbances in Parkinson’s disease. Int. J. Mol. Sci. 23, 14018 (2022).
Ho, G. P. H. et al. Upregulation of cellular palmitoylation mitigates alpha-synuclein accumulation and neurotoxicity. Mov. Disord. 36, 348–359 (2021).
Ho, G. P. H., Wilkie, E. C., White, A. J. & Selkoe, D. J. Palmitoylation of the Parkinson’s disease-associated protein synaptotagmin-11 links its turnover to alpha-synuclein homeostasis. Sci. Signal. 16, eadd7220 (2023).
Cassinelli, S. et al. Palmitoylation of voltage-gated ion channels. Int. J. Mol. Sci. 23, 9357 (2022).
Essandoh, K., Philippe, J. M., Jenkins, P. M. & Brody, M. J. Palmitoylation: A fatty regulator of myocardial electrophysiology. Front. Physiol. 11, 108 (2020).
Pei, Z. et al. Cardiac sodium channel palmitoylation regulates channel availability and myocyte excitability with implications for arrhythmia generation. Nat. Commun. 7, 12035 (2016).
Cha, A. et al. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron 22, 73–87 (1999).
Zhou, B. et al. Zdhhc2 is essential for plasmacytoid dendritic cells mediated inflammatory response in psoriasis. Front. Immunol. 11, 607442 (2020).
Hansen, A. L. et al. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc. Natl. Acad. Sci. USA 115, E7768–E7775 (2018).
Schnell, A., Littman, D. R. & Kuchroo, V. K. T(H)17 cell heterogeneity and its role in tissue inflammation. Nat. Immunol. 24, 19–29 (2023).
Xiong, W. et al. Metformin alleviates inflammation through suppressing FASN-dependent palmitoylation of Akt. Cell Death Dis. 12, 934 (2021).
Lu, Y. et al. Palmitoylation of NOD1 and NOD2 is required for bacterial sensing. Science 366, 460–467 (2019).
Ramadan, A. A. et al. Identification of SARS-CoV-2 spike palmitoylation inhibitors that results in release of attenuated virus with reduced infectivity. Viruses 14, 531 (2022).
Li, D. et al. Palmitoylation of SARS-CoV-2 S protein is critical for S-mediated syncytia formation and virus entry. J. Med. Virol. 94, 342–348 (2022).
Xie, F. et al. Engineering extracellular vesicles enriched with palmitoylated ACE2 as COVID-19 therapy. Adv. Mater. 33, e2103471 (2021).
Qiu, N. et al. Artemisinin inhibits NRas palmitoylation by targeting the protein acyltransferase ZDHHC6. Cell Chem. Biol. 29, 530–537.e537 (2022).
Fan, X. et al. Local anesthetics impair the growth and self-renewal of glioblastoma stem cells by inhibiting ZDHHC15-mediated GP130 palmitoylation. Stem Cell Res. Ther. 12, 107 (2021).
Haag, S. M. et al. Targeting STING with covalent small-molecule inhibitors. Nature 559, 269–273 (2018).
Jia, M. et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 21, 727–735 (2020).
Vetting, M. W. et al. Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 433, 212–226 (2005).
Giang, D. K. & Cravatt, B. F. A second mammalian N-myristoyltransferase. J. Biol. Chem. 273, 6595–6598 (1998).
Ducker, C. E., Upson, J. J., French, K. J. & Smith, C. D. Two N-myristoyltransferase isozymes play unique roles in protein myristoylation, proliferation, and apoptosis. Mol. Cancer Res. 3, 463–476 (2005).
Yang, S. H. et al. N-myristoyltransferase 1 is essential in early mouse development. J. Biol. Chem. 280, 18990–18995 (2005).
Shrivastav, A. et al. Requirement of N-myristoyltransferase 1 in the development of monocytic lineage. J. Immunol. 180, 1019–1028 (2008).
Perinpanayagam, M. A. et al. Regulation of co- and post-translational myristoylation of proteins during apoptosis: interplay of N-myristoyltransferases and caspases. FASEB J. 27, 811–821 (2013).
Castrec, B. et al. Structural and genomic decoding of human and plant myristoylomes reveals a definitive recognition pattern. Nat. Chem. Biol. 14, 671–679 (2018).
Farazi, T. A., Waksman, G. & Gordon, J. I. The biology and enzymology of protein N-myristoylation. J. Biol. Chem. 276, 39501–39504 (2001).
Rudnick, D. A. et al. Kinetic and structural evidence for a sequential ordered Bi Bi mechanism of catalysis by Saccharomyces cerevisiae myristoyl-CoA:protein N-myristoyltransferase. J. Biol. Chem. 266, 9732–9739 (1991).
Dian, C. et al. High-resolution snapshots of human N-myristoyltransferase in action illuminate a mechanism promoting N-terminal Lys and Gly myristoylation. Nat. Commun. 11, 1132 (2020).
Takamune, N. et al. Suppression of human immunodeficiency virus type-1 production by coexpression of catalytic-region-deleted N-myristoyltransferase mutants. Biol. Pharm. Bull. 33, 2018–2023 (2010).
Glover, C. J., Hartman, K. D. & Felsted, R. L. Human N-myristoyltransferase amino-terminal domain involved in targeting the enzyme to the ribosomal subcellular fraction. J. Biol. Chem. 272, 28680–28689 (1997).
Thinon, E. et al. Global profiling of co- and post-translationally N-myristoylated proteomes in human cells. Nat. Commun. 5, 4919 (2014).
Kosciuk, T. et al. NMT1 and NMT2 are lysine myristoyltransferases regulating the ARF6 GTPase cycle. Nat. Commun. 11, 1067 (2020).
Jiang, H. et al. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496, 110–113 (2013).
Choudhary, C. et al. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550 (2014).
Bagchi, R. A. et al. Reversible lysine fatty acylation of an anchoring protein mediates adipocyte adrenergic signaling. Proc. Natl. Acad. Sci. USA 119, e2119678119 (2022).
Peitzsch, R. M. & McLaughlin, S. Binding of acylated peptides and fatty acids to phospholipid vesicles: pertinence to myristoylated proteins. Biochemistry 32, 10436–10443 (1993).
Shahinian, S. & Silvius, J. R. Doubly-lipid-modified protein sequence motifs exhibit long-lived anchorage to lipid bilayer membranes. Biochemistry 34, 3813–3822 (1995).
Resh, M. D. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Biophys. Acta 1451, 1–16 (1999).
Ames, J. B., Tanaka, T., Stryer, L. & Ikura, M. Portrait of a myristoyl switch protein. Curr. Opin. Struct. Biol. 6, 432–438 (1996).
Zhou, W., Parent, L. J., Wills, J. W. & Resh, M. D. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J. Virol. 68, 2556–2569 (1994).
McLaughlin, S. & Aderem, A. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem. Sci. 20, 272–276 (1995).
van den Bout, I. et al. Investigation into the mechanism regulating MRP localization. Exp. Cell Res. 314, 330–341 (2008).
John, K. & Bar, M. Travelling lipid domains in a dynamic model for protein-induced pattern formation in biomembranes. Phys. Biol. 2, 123–132 (2005).
Braun, T., McIlhinney, R. A. & Vergeres, G. Myristoylation-dependent N-terminal cleavage of the myristoylated alanine-rich C kinase substrate (MARCKS) by cellular extracts. Biochimie 82, 705–715 (2000).
Xiong, W. H., Qin, M. & Zhong, H. Myristoylation alone is sufficient for PKA catalytic subunits to associate with the plasma membrane to regulate neuronal functions. Proc. Natl. Acad. Sci. USA 118, e2021658118 (2021).
Tanaka, T. et al. Sequestration of the membrane-targeting myristoyl group of recoverin in the calcium-free state. Nature 376, 444–447 (1995).
Ames, J. B. et al. Molecular mechanics of calcium-myristoyl switches. Nature 389, 198–202 (1997).
Godsel, L. M. & Engman, D. M. Flagellar protein localization mediated by a calcium-myristoyl/palmitoyl switch mechanism. EMBO J. 18, 2057–2065 (1999).
Cadwallader, K. A., Paterson, H., Macdonald, S. G. & Hancock, J. F. N-terminally myristoylated Ras proteins require palmitoylation or a polybasic domain for plasma membrane localization. Mol. Cell Biol. 14, 4722–4730 (1994).
Barylko, B. et al. Myristoylation-dependent palmitoylation of the receptor tyrosine kinase adaptor FRS2alpha. Biochemistry 58, 2809–2813 (2019).
Santonico, E. et al. Multiple modification and protein interaction signals drive the Ring finger protein 11 (RNF11) E3 ligase to the endosomal compartment. Oncogene 29, 5604–5618 (2010).
Alvarez, R. et al. G protein-membrane interactions I: Galphai1 myristoyl and palmitoyl modifications in protein-lipid interactions and its implications in membrane microdomain localization. Biochim Biophys. Acta 1851, 1511–1520 (2015).
Zhang, Q. et al. ACSL1-induced ferroptosis and platinum resistance in ovarian cancer by increasing FSP1 N-myristylation and stability. Cell Death Discov. 9, 83 (2023).
Tan, X. P. et al. Blockade of NMT1 enzymatic activity inhibits N-myristoylation of VILIP3 protein and suppresses liver cancer progression. Signal Transduct. Target Ther. 8, 14 (2023).
Kennedy, M. T., Brockman, H. & Rusnak, F. Contributions of myristoylation to calcineurin structure/function. J. Biol. Chem. 271, 26517–26521 (1996).
Bachmair, A., Finley, D. & Varshavsky, A. In vivo half-life of a protein is a function of its amino-terminal residue. Science 234, 179–186 (1986).
Varshavsky, A. N-degron and C-degron pathways of protein degradation. Proc. Natl. Acad. Sci. USA 116, 358–366 (2019).
Timms, R. T. et al. A glycine-specific N-degron pathway mediates the quality control of protein N-myristoylation. Science 365, eaaw4912 (2019).
Patwardhan, P. & Resh, M. D. Myristoylation and membrane binding regulate c-Src stability and kinase activity. Mol. Cell Biol. 30, 4094–4107 (2010).
Resh, M. D. & Ling, H. P. Identification of a 32K plasma membrane protein that binds to the myristylated amino-terminal sequence of p60v-src. Nature 346, 84–86 (1990).
Sigal, C. T. & Resh, M. D. The ADP/ATP carrier is the 32-kilodalton receptor for an NH2-terminally myristylated src peptide but not for pp60src polypeptide. Mol. Cell Biol. 13, 3084–3092 (1993).
Bryant, M. & Ratner, L. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc. Natl. Acad. Sci. USA 87, 523–527 (1990).
Gottlinger, H. G., Sodroski, J. G. & Haseltine, W. A. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 86, 5781–5785 (1989).
Jouvenet, N., Simon, S. M. & Bieniasz, P. D. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. USA 106, 19114–19119 (2009).
Zhu, Y. et al. Heme oxygenase 2 binds myristate to regulate retrovirus assembly and TLR4 signaling. Cell Host Microbe 21, 220–230 (2017).
Kim, S. et al. Blocking myristoylation of Src inhibits its kinase activity and suppresses prostate cancer progression. Cancer Res. 77, 6950–6962 (2017).
Liang, J. et al. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat. Commun. 6, 7926 (2015).
Li, C. et al. Myristoylated Naked2 escorts transforming growth factor alpha to the basolateral plasma membrane of polarized epithelial cells. Proc. Natl. Acad. Sci. USA 101, 5571–5576 (2004).
Kharas, M. G. et al. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 115, 1406–1415 (2010).
Koutelou, E. et al. Neuralized-like 1 (Neurl1) targeted to the plasma membrane by N-myristoylation regulates the notch ligand Jagged1. J. Biol. Chem. 283, 3846–3853 (2008).
Wang, B. et al. Protein N-myristoylation: functions and mechanisms in control of innate immunity. Cell Mol. Immunol. 18, 878–888 (2021).
Jia, M. et al. Myristic acid as a checkpoint to regulate STING-dependent autophagy and interferon responses by promoting N-myristoylation. Nat. Commun. 14, 660 (2023).
Beauchamp, E. et al. Targeting N-myristoylation for therapy of B-cell lymphomas. Nat. Commun. 11, 5348 (2020).
Udenwobele, D. I. et al. Myristoylation: an important protein modification in the immune response. Front. Immunol. 8, 751 (2017).
Herzig, S. & Shaw, R. J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19, 121–135 (2018).
Wen, Z. et al. N-myristoyltransferase deficiency impairs activation of kinase AMPK and promotes synovial tissue inflammation. Nat. Immunol. 20, 313–325 (2019).
Zhang, J. et al. Myristoylation-mediated phase separation of EZH2 compartmentalizes STAT3 to promote lung cancer growth. Cancer Lett. 516, 84–98 (2021).
Sun, Y. et al. N-myristoyltransferase-1 deficiency blocks myristoylation of LAMTOR1 and inhibits bladder cancer progression. Cancer Lett. 529, 126–138 (2022).
Zhang, Q. et al. Metabolic reprogramming of ovarian cancer involves ACSL1-mediated metastasis stimulation through upregulated protein myristoylation. Oncogene 40, 97–111 (2021).
Guo, H. et al. Targeting EGFR-dependent tumors by disrupting an ARF6-mediated sorting system. Nat. Commun. 13, 6004 (2022).
King, M. J., Pugazhenthi, S., Khandelwal, R. L. & Sharma, R. K. In vivo modulation of N-myristoyltransferase activity by orthovanadate. Mol. Cell Biochem. 153, 151–155 (1995).
King, M. J., Pugazhenthi, S., Khandelwal, R. L. & Sharma, R. K. Membrane-associated N-myristoyltransferase activity is reduced in obese (fa/fa) Zucker rat liver. Biochem. Biophys. Res. Commun. 196, 665–670 (1993).
Holzer, R. G. et al. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 147, 173–184 (2011).
Barthel, A., Kohn, A. D., Luo, Y. & Roth, R. A. A constitutively active version of the Ser/Thr kinase Akt induces production of the ob gene product, leptin, in 3T3-L1 adipocytes. Endocrinology 138, 3559–3562 (1997).
Weyand, C. M. & Goronzy, J. J. Immunometabolism in the development of rheumatoid arthritis. Immunol. Rev. 294, 177–187 (2020).
Matsubara, M. et al. Myristoyl moiety of HIV Nef is involved in regulation of the interaction with calmodulin in vivo. Protein Sci. 14, 494–503 (2005).
Aiken, C. et al. Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell 76, 853–864 (1994).
Schwartz, O. et al. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 2, 338–342 (1996).
Mazzolini, J. et al. Inhibition of phagocytosis in HIV-1-infected macrophages relies on Nef-dependent alteration of focal delivery of recycling compartments. Blood 115, 4226–4236 (2010).
Paige, L. A. et al. Metabolic activation of 2-substituted derivatives of myristic acid to form potent inhibitors of myristoyl CoA:protein N-myristoyltransferase. Biochemistry 29, 10566–10573 (1990).
Kallemeijn, W. W. et al. Validation and invalidation of chemical probes for the human N-myristoyltransferases. Cell Chem. Biol. 26, 892–900 e894 (2019).
Bhandarkar, S. S. et al. Tris (dibenzylideneacetone) dipalladium, a N-myristoyltransferase-1 inhibitor, is effective against melanoma growth in vitro and in vivo. Clin. Cancer Res. 14, 5743–5748 (2008).
Rajala, R. V. et al. Expression of N-myristoyltransferase inhibitor protein and its relationship to c-Src levels in human colon cancer cell lines. Biochem. Biophys. Res. Commun. 273, 1116–1120 (2000).
Selvakumar, P. et al. Potential role of N-myristoyltransferase in cancer. Prog. Lipid Res. 46, 1–36 (2007).
Shrivastav, A. et al. Potent inhibitor of N-myristoylation: a novel molecular target for cancer. Cancer Res. 63, 7975–7978 (2003).
Hughes, T. P. et al. Asciminib in chronic myeloid leukemia after ABL kinase inhibitor failure. New Engl. J. Med. 381, 2315–2326 (2019).
Rea, D. et al. A phase 3, open-label, randomized study of asciminib, a STAMP inhibitor, vs bosutinib in CML after 2 or more prior TKIs. Blood 138, 2031–2041 (2021).
Hochhaus, A. et al. Asciminib vs bosutinib in chronic-phase chronic myeloid leukemia previously treated with at least two tyrosine kinase inhibitors: longer-term follow-up of ASCEMBL. Leukemia 37, 617–626 (2023).
Deeks, E. D. Asciminib: first approval. Drugs 82, 219–226 (2022).
Priyamvada, L. et al. Inhibition of vaccinia virus L1 N-myristoylation by the host N-myristoyltransferase inhibitor IMP-1088 generates non-infectious virions defective in cell entry. PLoS Pathog. 18, e1010662 (2022).
Mousnier, A. et al. Fragment-derived inhibitors of human N-myristoyltransferase block capsid assembly and replication of the common cold virus. Nat. Chem. 10, 599–606 (2018).
Georgopapadakou, N. H. Antifungals targeted to protein modification: focus on protein N-myristoyltransferase. Expert Opin. Investig. Drugs 11, 1117–1125 (2002).
Kawasaki, K. et al. Design and synthesis of novel benzofurans as a new class of antifungal agents targeting fungal N-myristoyltransferase. Part 3. Bioorg. Med Chem. Lett. 13, 87–91 (2003).
Yuan, M. et al. N-myristoylation: from cell biology to translational medicine. Acta Pharm. Sin. 41, 1005–1015 (2020).
Brand, S. et al. Discovery of a novel class of orally active trypanocidal N-myristoyltransferase inhibitors. J. Med. Chem. 55, 140–152 (2012).
Goncalves, V. et al. Discovery of Plasmodium vivax N-myristoyltransferase inhibitors: screening, synthesis, and structural characterization of their binding mode. J. Med Chem. 55, 3578–3582 (2012).
Rackham, M. D. et al. Discovery of novel and ligand-efficient inhibitors of Plasmodium falciparum and Plasmodium vivax N-myristoyltransferase. J. Med Chem. 56, 371–375 (2013).
Goldston, A. M., Sharma, A. I., Paul, K. S. & Engman, D. M. Acylation in trypanosomatids: an essential process and potential drug target. Trends Parasitol. 30, 350–360 (2014).
Cox, A. D. & Der, C. J. Protein prenylation: more than just glue? Curr. Opin. Cell Biol. 4, 1008–1016 (1992).
Seabra, M. C. et al. Protein farnesyltransferase and geranylgeranyltransferase share a common alpha subunit. Cell 65, 429–434 (1991).
Taylor, J. S. et al. Structure of mammalian protein geranylgeranyltransferase type-I. EMBO J. 22, 5963–5974 (2003).
Zhang, F. L. & Casey, P. J. Protein prenylation: molecular mechanisms and functional consequences. Annu. Rev. Biochem 65, 241–269 (1996).
Reid, T. S., Terry, K. L., Casey, P. J. & Beese, L. S. Crystallographic analysis of CaaX prenyltransferases complexed with substrates defines rules of protein substrate selectivity. J. Mol. Biol. 343, 417–433 (2004).
Roberts, P. J. et al. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 283, 25150–25163 (2008).
Amaya, M., Baranova, A. & van Hoek, M. L. Protein prenylation: a new mode of host-pathogen interaction. Biochem. Biophys. Res. Commun. 416, 1–6 (2011).
Trueblood, C. E., Ohya, Y. & Rine, J. Genetic evidence for in vivo cross-specificity of the CaaX-box protein prenyltransferases farnesyltransferase and geranylgeranyltransferase-I in Saccharomyces cerevisiae. Mol. Cell Biol. 13, 4260–4275 (1993).
Lobell, R. B. et al. Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 61, 8758–8768 (2001).
Liu, A. et al. RhoB alteration is necessary for apoptotic and antineoplastic responses to farnesyltransferase inhibitors. Mol. Cell Biol. 20, 6105–6113 (2000).
Calero, M. et al. Dual prenylation is required for Rab protein localization and function. Mol. Biol. Cell 14, 1852–1867 (2003).
Kuchay, S. et al. GGTase3 is a newly identified geranylgeranyltransferase targeting a ubiquitin ligase. Nat. Struct. Mol. Biol. 26, 628–636 (2019).
Shirakawa, R. et al. A SNARE geranylgeranyltransferase essential for the organization of the Golgi apparatus. EMBO J. 39, e104120 (2020).
Boyartchuk, V. L., Ashby, M. N. & Rine, J. Modulation of Ras and a-factor function by carboxyl-terminal proteolysis. Science 275, 1796–1800 (1997).
Ma, Y. T., Gilbert, B. A. & Rando, R. R. Inhibitors of the isoprenylated protein endoprotease. Biochemistry 32, 2386–2393 (1993).
Pei, J. & Grishin, N. V. Type II CAAX prenyl endopeptidases belong to a novel superfamily of putative membrane-bound metalloproteases. Trends Biochem. Sci. 26, 275–277 (2001).
Hampton, S. E., Dore, T. M. & Schmidt, W. K. Rce1: mechanism and inhibition. Crit. Rev. Biochem. Mol. Biol. 53, 157–174 (2018).
Wright, L. P. et al. Topology of mammalian isoprenylcysteine carboxyl methyltransferase determined in live cells with a fluorescent probe. Mol. Cell Biol. 29, 1826–1833 (2009).
Hrycyna, C. A., Sapperstein, S. K., Clarke, S. & Michaelis, S. The Saccharomyces cerevisiae STE14 gene encodes a methyltransferase that mediates C-terminal methylation of a-factor and RAS proteins. EMBO J. 10, 1699–1709 (1991).
Diver, M. M. & Long, S. B. Mutational analysis of the integral membrane methyltransferase isoprenylcysteine carboxyl methyltransferase (ICMT) reveals potential substrate binding sites. J. Biol. Chem. 289, 26007–26020 (2014).
Chelsky, D., Ruskin, B. & Koshland, D. E. Jr. Methyl-esterified proteins in a mammalian cell line. Biochemistry 24, 6651–6658 (1985).
Philips, M. R. et al. Carboxyl methylation of Ras-related proteins during signal transduction in neutrophils. Science 259, 977–980 (1993).
Lu, Q. et al. Isoprenylcysteine carboxyl methyltransferase modulates endothelial monolayer permeability: involvement of RhoA carboxyl methylation. Circ. Res. 94, 306–315 (2004).
Cushman, I., Cushman, S. M., Potter, P. M. & Casey, P. J. Control of RhoA methylation by carboxylesterase I. J. Biol. Chem. 288, 19177–19183 (2013).
Konstantinopoulos, P. A., Karamouzis, M. V. & Papavassiliou, A. G. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 6, 541–555 (2007).
Yang, S. et al. Control of antiviral innate immune response by protein geranylgeranylation. Sci. Adv. 5, eaav7999 (2019).
Scott-Solomon, E. & Kuruvilla, R. Prenylation of axonally translated Rac1 controls NGF-dependent axon growth. Dev. Cell. 53, 691–705.e697 (2020).
Hancock, J. F., Cadwallader, K. & Marshall, C. J. Methylation and proteolysis are essential for efficient membrane binding of prenylated p21K-ras(B). EMBO J. 10, 641–646 (1991).
Li, G. & Stahl, P. D. Post-translational processing and membrane association of the two early endosome-associated rab GTP-binding proteins (rab4 and rab5). Arch. Biochem. Biophys. 304, 471–478 (1993).
Porfiri, E., Evans, T., Chardin, P. & Hancock, J. F. Prenylation of Ras proteins is required for efficient hSOS1-promoted guanine nucleotide exchange. J. Biol. Chem. 269, 22672–22677 (1994).
Chandra, A. et al. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 14, 148–158 (2011).
Schmick, M. et al. KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell 157, 459–471 (2014).
Dharmaiah, S. et al. Structural basis of recognition of farnesylated and methylated KRAS4b by PDEdelta. Proc. Natl. Acad. Sci. USA 113, E6766–E6775 (2016).
Akula, M. K. et al. Protein prenylation restrains innate immunity by inhibiting Rac1 effector interactions. Nat. Commun. 10, 3975 (2019).
Pylypenko, O. et al. Farnesylation of the SNARE protein Ykt6 increases its stability and helical folding. J. Mol. Biol. 377, 1334–1345 (2008).
Hoffman, G. R., Nassar, N. & Cerione, R. A. Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell 100, 345–356 (2000).
Boulter, E. et al. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat. Cell Biol. 12, 477–483 (2010).
Backlund, P. S. Jr. Post-translational processing of RhoA. Carboxyl methylation of the carboxyl-terminal prenylcysteine increases the half-life of Rhoa. J. Biol. Chem. 272, 33175–33180 (1997).
Bergo, M. O. et al. Inactivation of Icmt inhibits transformation by oncogenic K-Ras and B-Raf. J. Clin. Investig. 113, 539–550 (2004).
Stubbs, E. B. Jr. & Von Zee, C. L. Prenylation of Rho G-proteins: a novel mechanism regulating gene expression and protein stability in human trabecular meshwork cells. Mol. Neurobiol. 46, 28–40 (2012).
Adjei, A. A. Blocking oncogenic Ras signaling for cancer therapy. J. Natl. Cancer Inst. 93, 1062–1074 (2001).
Goldstein, J. L. & Brown, M. S. Regulation of the mevalonate pathway. Nature 343, 425–430 (1990).
Liao, P., Hemmerlin, A., Bach, T. J. & Chye, M. L. The potential of the mevalonate pathway for enhanced isoprenoid production. Biotechnol. Adv. 34, 697–713 (2016).
Xu, N. et al. The alteration of protein prenylation induces cardiomyocyte hypertrophy through Rheb-mTORC1 signalling and leads to chronic heart failure. J. Pathol. 235, 672–685 (2015).
Su, W. et al. Protein prenylation drives discrete signaling programs for the differentiation and maintenance of effector T(reg) cells. Cell Metab. 32, 996–1011.e1017 (2020).
Du, X. et al. Mevalonate metabolism-dependent protein geranylgeranylation regulates thymocyte egress. J. Exp. Med. 217, e20190969 (2020).
Scott, M. P. et al. Drosophila tempura, a novel protein prenyltransferase α subunit, regulates notch signaling via Rab1 and Rab11. PLoS Biol. 12, e1001777 (2014).
Odeniyide, P. et al. Targeting farnesylation as a novel therapeutic approach in HRAS-mutant rhabdomyosarcoma. Oncogene 41, 2973–2983 (2022).
Adamson, P., Marshall, C. J., Hall, A. & Tilbrook, P. A. Post-translational modifications of p21rho proteins. J. Biol. Chem. 267, 20033–20038 (1992).
McTaggart, S. J. Isoprenylated proteins. Cell Mol. Life Sci. 63, 255–267 (2006).
Gilardi, M. et al. Tipifarnib as a precision therapy for HRAS-mutant head and neck squamous cell carcinomas. Mol. Cancer Ther. 19, 1784–1796 (2020).
Gao, S., Yu, R. & Zhou, X. The role of geranylgeranyltransferase I-mediated protein prenylation in the brain. Mol. Neurobiol. 53, 6925–6937 (2016).
Jeong, A. et al. Protein farnesylation is upregulated in Alzheimer’s human brains and neuron-specific suppression of farnesyltransferase mitigates pathogenic processes in Alzheimer’s model mice. Acta Neuropathol. Commun. 9, 129 (2021).
Gelb, M. H. et al. Therapeutic intervention based on protein prenylation and associated modifications. Nat. Chem. Biol. 2, 518–528 (2006).
Walters, C. E. et al. Inhibition of Rho GTPases with protein prenyltransferase inhibitors prevents leukocyte recruitment to the central nervous system and attenuates clinical signs of disease in an animal model of multiple sclerosis. J. Immunol. 168, 4087–4094 (2002).
Jo, A. et al. PARIS farnesylation prevents neurodegeneration in models of Parkinson’s disease. Sci. Transl. Med. 13, eaax8891 (2021).
Eriksson, M. et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423, 293–298 (2003).
Price, C. T., Jones, S. C., Amundson, K. E. & Kwaik, Y. A. Host-mediated post-translational prenylation of novel dot/icm-translocated effectors of legionella pneumophila. Front. Microbiol. 1, 131 (2010).
Pronin, A. V., Narovlyansky, A. N. & Sanin, A. V. New approaches to the prevention and treatment of viral diseases. Arch. Immunol. Ther. Exp. 69, 10 (2021).
Glenn, J. S., Watson, J. A., Havel, C. M. & White, J. M. Identification of a prenylation site in delta virus large antigen. Science 256, 1331–1333 (1992).
Bordier, B. B. et al. A prenylation inhibitor prevents production of infectious hepatitis delta virus particles. J. Virol. 76, 10465–10472 (2002).
Loureiro, D. et al. New therapies for hepatitis delta virus infection. Liver Int. 41(Suppl 1), 30–37 (2021).
Mathews, E. S., Jezewski, A. J. & Odom John, A. R. Protein prenylation and Hsp40 in thermotolerance of Plasmodium falciparum malaria parasites. mBio 12, e0076021 (2021).
Wu, X. et al. Targeting protein modifications in metabolic diseases: molecular mechanisms and targeted therapies. Signal Transduct. Target Ther. 8, 220 (2023).
Xu, N. et al. Protein prenylation and human diseases: a balance of protein farnesylation and geranylgeranylation. Sci. China Life Sci. 58, 328–335 (2015).
Lopez-Posadas, R. et al. Rho-A prenylation and signaling link epithelial homeostasis to intestinal inflammation. J. Clin. Investig. 126, 611–626 (2016).
Shen, N. et al. An early response transcription factor, Egr-1, enhances insulin resistance in type 2 diabetes with chronic hyperinsulinism. J. Biol. Chem. 286, 14508–14515 (2011).
Silva, T., Teixeira, J., Remiao, F. & Borges, F. Alzheimer’s disease, cholesterol, and statins: the junctions of important metabolic pathways. Angew. Chem. Int. Ed. Engl. 52, 1110–1121 (2013).
Jiang, W. et al. Statins: a repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 40, 241 (2021).
Zhong, W. B., Wang, C. Y., Chang, T. C. & Lee, W. S. Lovastatin induces apoptosis of anaplastic thyroid cancer cells via inhibition of protein geranylgeranylation and de novo protein synthesis. Endocrinology 144, 3852–3859 (2003).
Collisson, E. A. et al. Atorvastatin prevents RhoC isoprenylation, invasion, and metastasis in human melanoma cells. Mol. Cancer Ther. 2, 941–948 (2003).
Nam, G. H. et al. Statin-mediated inhibition of RAS prenylation activates ER stress to enhance the immunogenicity of KRAS mutant cancer. J. Immunother. Cancer 9, e002474 (2021).
Gordon, R. E. et al. Statins synergize with hedgehog pathway inhibitors for treatment of medulloblastoma. Clin. Cancer Res. 24, 1375–1388 (2018).
Misirkic, M. et al. Inhibition of AMPK-dependent autophagy enhances in vitro antiglioma effect of simvastatin. Pharm. Res. 65, 111–119 (2012).
Viswanathan, V. S. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017).
Zhou, W. et al. Targeting the mevalonate pathway suppresses ARID1A-inactivated cancers by promoting pyroptosis. Cancer Cell 41, 740–756.e710 (2023).
Ahern, T. P. et al. Statin prescriptions and breast cancer recurrence risk: a Danish nationwide prospective cohort study. J. Natl. Cancer Inst. 103, 1461–1468 (2011).
Kwan, M. L. et al. Post-diagnosis statin use and breast cancer recurrence in a prospective cohort study of early stage breast cancer survivors. Breast Cancer Res. Treat. 109, 573–579 (2008).
Ng, K. et al. Relationship between statin use and colon cancer recurrence and survival: results from CALGB 89803. J. Natl. Cancer Inst. 103, 1540–1551 (2011).
Kim, S. T. et al. Simvastatin plus capecitabine-cisplatin versus placebo plus capecitabine-cisplatin in patients with previously untreated advanced gastric cancer: a double-blind randomised phase 3 study. Eur. J. Cancer 50, 2822–2830 (2014).
Konings, I. R. et al. The addition of pravastatin to chemotherapy in advanced gastric carcinoma: a randomised phase II trial. Eur. J. Cancer 46, 3200–3204 (2010).
Green, J. R. Bisphosphonates: preclinical review. Oncologist 9(Suppl 4), 3–13 (2004).
Jeong, A. et al. Isoprenoids and protein prenylation: implications in the pathogenesis and therapeutic intervention of Alzheimer’s disease. Crit. Rev. Biochem. Mol. Biol. 53, 279–310 (2018).
Caraglia, M. et al. Emerging anti-cancer molecular mechanisms of aminobisphosphonates. Endocr. Relat. Cancer 13, 7–26 (2006).
Fisher, J. E. et al. Alendronate mechanism of action: geranylgeraniol, an intermediate in the mevalonate pathway, prevents inhibition of osteoclast formation, bone resorption, and kinase activation in vitro. Proc. Natl. Acad. Sci. USA 96, 133–138 (1999).
Coxon, F. P. et al. Phosphonocarboxylate inhibitors of Rab geranylgeranyl transferase disrupt the prenylation and membrane localization of Rab proteins in osteoclasts in vitro and in vivo. Bone 37, 349–358 (2005).
Duez, S. et al. Towards the synthesis of bisubstrate inhibitors of protein farnesyltransferase: Synthesis and biological evaluation of new farnesylpyrophosphate analogues. Bioorg. Med. Chem. 18, 543–556 (2010).
Manne, V. et al. Bisubstrate inhibitors of farnesyltransferase: a novel class of specific inhibitors of ras transformed cells. Oncogene 10, 1763–1779 (1995).
Karasic, T. B., Chiorean, E. G., Sebti, S. M. & O’Dwyer, P. J. A phase I study of GGTI-2418 (Geranylgeranyl transferase I inhibitor) in patients with advanced solid tumors. Target Oncol. 14, 613–618 (2019).
Venet, M., End, D. & Angibaud, P. Farnesyl protein transferase inhibitor ZARNESTRA R115777—history of a discovery. Curr. Top. Med. Chem. 3, 1095–1102 (2003).
Morgillo, F. & Lee, H. Y. Lonafarnib in cancer therapy. Expert Opin. Investig. Drugs 15, 709–719 (2006).
Berndt, N., Hamilton, A. D. & Sebti, S. M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 11, 775–791 (2011).
Whyte, D. B. et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 272, 14459–14464 (1997).
Ho, A. L. et al. Tipifarnib in head and neck squamous cell carcinoma with HRAS mutations. J. Clin. Oncol. 39, 1856–1864 (2021).
Lee, H. W. et al. A phase II trial of tipifarnib for patients with previously treated, metastatic urothelial carcinoma harboring HRAS mutations. Clin. Cancer Res. 26, 5113–5119 (2020).
Adjei, A. A. et al. A Phase I trial of the farnesyl protein transferase inhibitor R115777 in combination with gemcitabine and cisplatin in patients with advanced cancer. Clin. Cancer Res. 9, 2520–2526 (2003).
Borthakur, G. et al. Pilot study of lonafarnib, a farnesyl transferase inhibitor, in patients with chronic myeloid leukemia in the chronic or accelerated phase that is resistant or refractory to imatinib therapy. Cancer 106, 346–352 (2006).
Harousseau, J. L. et al. A phase 2 study of the oral farnesyltransferase inhibitor tipifarnib in patients with refractory or relapsed acute myeloid leukemia. Blood 109, 5151–5156 (2007).
Khuri, F. R. et al. Phase I study of the farnesyltransferase inhibitor lonafarnib with paclitaxel in solid tumors. Clin. Cancer Res. 10, 2968–2976 (2004).
Perez-Ruixo, J. J. et al. Exposure-toxicity relationships for tipifarnib in cancer patients. Br. J. Clin. Pharm. 64, 219–232 (2007).
Dhillon, S. Lonafarnib: first approval. Drugs 81, 283–289 (2021).
Sjogren, A. K. et al. GGTase-I deficiency reduces tumor formation and improves survival in mice with K-RAS-induced lung cancer. J. Clin. Investig. 117, 1294–1304 (2007).
Morgan, M. A. et al. Synergistic cytotoxic effects in myeloid leukemia cells upon cotreatment with farnesyltransferase and geranylgeranyl transferase-I inhibitors. Leukemia 17, 1508–1520 (2003).
Liu, M. et al. Targeting the protein prenyltransferases efficiently reduces tumor development in mice with K-RAS-induced lung cancer. Proc. Natl. Acad. Sci. USA 107, 6471–6476 (2010).
Shen, M. et al. Farnesyltransferase and geranylgeranyltransferase I: structures, mechanism, inhibitors and molecular modeling. Drug Discov. Today 20, 267–276 (2015).
Hasan, Z. et al. Geranylgeranyl transferase regulates CXC chemokine formation in alveolar macrophages and neutrophil recruitment in septic lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 304, L221–229, (2013).
Kazi, A. et al. Dual farnesyl and geranylgeranyl transferase inhibitor thwarts mutant KRAS-driven patient-derived pancreatic tumors. Clin. Cancer Res. 25, 5984–5996 (2019).
Dailey, J. M. et al. Inhibiting isoprenylation suppresses FcepsilonRI-mediated mast cell function and allergic inflammation. J. Immunol. 211, 527–538 (2023).
Martin, N. E. et al. A phase I trial of the dual farnesyltransferase and geranylgeranyltransferase inhibitor L-778,123 and radiotherapy for locally advanced pancreatic cancer. Clin. Cancer Res. 10, 5447–5454 (2004).
Hahn, S. M. et al. A Phase I trial of the farnesyltransferase inhibitor L-778,123 and radiotherapy for locally advanced lung and head and neck cancer. Clin. Cancer Res. 8, 1065–1072 (2002).
Bergo, M. O. et al. Absence of the CAAX endoprotease Rce1: effects on cell growth and transformation. Mol. Cell Biol. 22, 171–181 (2002).
Marin-Ramos, N. I. et al. A potent isoprenylcysteine carboxylmethyltransferase (ICMT) inhibitor improves survival in Ras-driven acute myeloid leukemia. J. Med Chem. 62, 6035–6046 (2019).
Hanker, A. B. et al. Differential requirement of CAAX-mediated posttranslational processing for Rheb localization and signaling. Oncogene 29, 380–391 (2010).
Yang, W. S. et al. Isoprenyl carboxyl methyltransferase inhibitors: a brief review including recent patents. Amino Acids 49, 1469–1485 (2017).
Winter-Vann, A. M. et al. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc. Natl. Acad. Sci. USA 102, 4336–4341 (2005).
Wang, M. et al. A small molecule inhibitor of isoprenylcysteine carboxymethyltransferase induces autophagic cell death in PC3 prostate cancer cells. J. Biol. Chem. 283, 18678–18684 (2008).
Lau, H. Y. et al. An improved isoprenylcysteine carboxylmethyltransferase inhibitor induces cancer cell death and attenuates tumor growth in vivo. Cancer Biol. Ther. 15, 1280–1291 (2014).
Ramanujulu, P. M. et al. Functionalized indoleamines as potent, drug-like inhibitors of isoprenylcysteine carboxyl methyltransferase (Icmt). Eur. J. Med. Chem. 63, 378–386 (2013).
Chen, X. et al. A small-molecule ICMT inhibitor delays senescence of Hutchinson-Gilford progeria syndrome cells. eLife 10, e63284 (2021).
Gerber, L. D., Kodukula, K. & Udenfriend, S. Phosphatidylinositol glycan (PI-G) anchored membrane proteins. Amino acid requirements adjacent to the site of cleavage and PI-G attachment in the COOH-terminal signal peptide. J. Biol. Chem. 267, 12168–12173 (1992).
Takeda, J. & Kinoshita, T. GPI-anchor biosynthesis. Trends Biochem. Sci. 20, 367–371 (1995).
Moran, P., Raab, H., Kohr, W. J. & Caras, I. W. Glycophospholipid membrane anchor attachment. Molecular analysis of the cleavage/attachment site. J. Biol. Chem. 266, 1250–1257 (1991).
Masuishi, Y., Kimura, Y., Arakawa, N. & Hirano, H. Identification of glycosylphosphatidylinositol-anchored proteins and omega-sites using TiO2-based affinity purification followed by hydrogen fluoride treatment. J. Proteom. 139, 77–83 (2016).
Gamage, D. G. & Hendrickson, T. L. GPI transamidase and GPI anchored proteins: oncogenes and biomarkers for cancer. Crit. Rev. Biochem Mol. Biol. 48, 446–464 (2013).
Benghezal, M. et al. Yeast Gpi8p is essential for GPI anchor attachment onto proteins. EMBO J. 15, 6575–6583 (1996).
Yu, J. et al. The affected gene underlying the class K glycosylphosphatidylinositol (GPI) surface protein defect codes for the GPI transamidase. Proc. Natl. Acad. Sci. USA 94, 12580–12585 (1997).
Zhang, H. et al. Structure of human glycosylphosphatidylinositol transamidase. Nat. Struct. Mol. Biol. 29, 203–209 (2022).
Sharma, D. K., Vidugiriene, J., Bangs, J. D. & Menon, A. K. A cell-free assay for glycosylphosphatidylinositol anchoring in African trypanosomes. Demonstration of a transamidation reaction mechanism. J. Biol. Chem. 274, 16479–16486 (1999).
Xu, Y. et al. Structures of liganded glycosylphosphatidylinositol transamidase illuminate GPI-AP biogenesis. Nat. Commun. 14, 5520 (2023).
Tanaka, S., Maeda, Y., Tashima, Y. & Kinoshita, T. Inositol deacylation of glycosylphosphatidylinositol-anchored proteins is mediated by mammalian PGAP1 and yeast Bst1p. J. Biol. Chem. 279, 14256–14263 (2004).
Fujita, M. et al. GPI glycan remodeling by PGAP5 regulates transport of GPI-anchored proteins from the ER to the Golgi. Cell 139, 352–365 (2009).
Fujita, M. et al. Sorting of GPI-anchored proteins into ER exit sites by p24 proteins is dependent on remodeled GPI. J. Cell Biol. 194, 61–75 (2011).
Maeda, Y. et al. Fatty acid remodeling of GPI-anchored proteins is required for their raft association. Mol. Biol. Cell 18, 1497–1506 (2007).
Tashima, Y. et al. PGAP2 is essential for correct processing and stable expression of GPI-anchored proteins. Mol. Biol. Cell 17, 1410–1420 (2006).
Kinoshita, T. & Fujita, M. Biosynthesis of GPI-anchored proteins: special emphasis on GPI lipid remodeling. J. Lipid Res. 57, 6–24 (2016).
Lukacs, M., Roberts, T., Chatuverdi, P. & Stottmann, R. W. Glycosylphosphatidylinositol biosynthesis and remodeling are required for neural tube closure, heart development, and cranial neural crest cell survival. eLife 8, e45248 (2019).
Itzhaky, D., Raz, N. & Hollander, N. The glycosylphosphatidylinositol-anchored form and the transmembrane form of CD58 associate with protein kinases. J. Immunol. 160, 4361–4366 (1998).
Harder, T. & Simons, K. Caveolae, DIGs, and the dynamics of sphingolipid-cholesterol microdomains. Curr. Opin. Cell Biol. 9, 534–542 (1997).
Maeda, Y. & Kinoshita, T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Prog. Lipid Res. 50, 411–424 (2011).
Sutherland, D. R. et al. Identification of a cell-surface antigen associated with activated T lymphoblasts and activated platelets. Blood 77, 84–93 (1991).
Murray, L. J. et al. CD109 is expressed on a subpopulation of CD34+ cells enriched in hematopoietic stem and progenitor cells. Exp. Hematol. 27, 1282–1294 (1999).
Hasegawa, M. et al. CD109, a new marker for myoepithelial cells of mammary, salivary, and lacrimal glands and prostate basal cells. Pathol. Int. 57, 245–250 (2007).
Emori, M. et al. High expression of CD109 antigen regulates the phenotype of cancer stem-like cells/cancer-initiating cells in the novel epithelioid sarcoma cell line ESX and is related to poor prognosis of soft tissue sarcoma. PLoS ONE 8, e84187 (2013).
Sakakura, H. et al. CD109 is a component of exosome secreted from cultured cells. Biochem. Biophys. Res. Commun. 469, 816–822 (2016).
Hagiwara, S. et al. Processing of CD109 by furin and its role in the regulation of TGF-beta signaling. Oncogene 29, 2181–2191 (2010).
Mii, S. et al. CD109: a multifunctional GPI-anchored protein with key roles in tumor progression and physiological homeostasis. Pathol. Int. 69, 249–259 (2019).
Chuang, C. H. et al. Molecular definition of a metastatic lung cancer state reveals a targetable CD109-Janus kinase-Stat axis. Nat. Med. 23, 291–300 (2017).
Minata, M. et al. Phenotypic plasticity of invasive edge glioma stem-like cells in response to ionizing radiation. Cell Rep. 26, 1893–1905.e1897 (2019).
Lee, K. Y. et al. Elevation of CD109 promotes metastasis and drug resistance in lung cancer via activation of EGFR-AKT-mTOR signaling. Cancer Sci. 111, 1652–1662 (2020).
Goyert, S. M. et al. The CD14 monocyte differentiation antigen maps to a region encoding growth factors and receptors. Science 239, 497–500 (1988).
Wu, Z., Zhang, Z., Lei, Z. & Lei, P. CD14: Biology and role in the pathogenesis of disease. Cytokine Growth Factor Rev. 48, 24–31 (2019).
Plociennikowska, A., Hromada-Judycka, A., Borzecka, K. & Kwiatkowska, K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol. Life Sci. 72, 557–581 (2015).
Wennerberg, E. et al. Expression of the mono-ADP-ribosyltransferase ART1 by tumor cells mediates immune resistance in non-small cell lung cancer. Sci. Transl. Med. 14, eabe8195 (2022).
Wennerberg, E., Mukherjee, S., Sainz, R. M. & Stiles, B. M. The ART of tumor immune escape. Oncoimmunology 11, 2076310 (2022).
Ronn, L. C., Hartz, B. P. & Bock, E. The neural cell adhesion molecule (NCAM) in development and plasticity of the nervous system. Exp. Gerontol. 33, 853–864 (1998).
Matsue, H. et al. Folate receptor allows cells to grow in low concentrations of 5-methyltetrahydrofolate. Proc. Natl. Acad. Sci. USA 89, 6006–6009 (1992).
Sabharanjak, S. & Mayor, S. Folate receptor endocytosis and trafficking. Adv. Drug Deliv. Rev. 56, 1099–1109 (2004).
Nawaz, F. Z. & Kipreos, E. T. Emerging roles for folate receptor FOLR1 in signaling and cancer. Trends Endocrinol. Metab. 33, 159–174 (2022).
Jiang, W. W. et al. Alterations of GPI transamidase subunits in head and neck squamous carcinoma. Mol. Cancer 6, 74 (2007).
Ho, J. C. et al. Increased expression of glycosyl-phosphatidylinositol anchor attachment protein 1 (GPAA1) is associated with gene amplification in hepatocellular carcinoma. Int. J. Cancer 119, 1330–1337 (2006).
Zhao, P. et al. Proteomic identification of glycosylphosphatidylinositol anchor-dependent membrane proteins elevated in breast carcinoma. J. Biol. Chem. 287, 25230–25240 (2012).
Zhang, X. X. et al. GPAA1 promotes gastric cancer progression via upregulation of GPI-anchored protein and enhancement of ERBB signalling pathway. J. Exp. Clin. Cancer Res. 38, 214 (2019).
Estreicher, A. et al. The receptor for urokinase type plasminogen activator polarizes expression of the protease to the leading edge of migrating monocytes and promotes degradation of enzyme inhibitor complexes. J. Cell Biol. 111, 783–792 (1990).
Smith, H. W. & Marshall, C. J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 11, 23–36 (2010).
Takeda, J. et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell 73, 703–711 (1993).
Ricklin, D., Hajishengallis, G., Yang, K. & Lambris, J. D. Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 11, 785–797 (2010).
Hill, A., DeZern, A. E., Kinoshita, T. & Brodsky, R. A. Paroxysmal nocturnal haemoglobinuria. Nat. Rev. Dis. Prim. 3, 17028 (2017).
Foliaki, S. T. et al. Temporary alteration of neuronal network communication is a protective response to redox imbalance that requires GPI-anchored prion protein. Redox Biol. 63, 102733 (2023).
Howard, M. F. et al. Mutations in PGAP3 impair GPI-anchor maturation, causing a subtype of hyperphosphatasia with mental retardation. Am. J. Hum. Genet. 94, 278–287 (2014).
Murakami, Y. et al. Null mutation in PGAP1 impairing Gpi-anchor maturation in patients with intellectual disability and encephalopathy. PLoS Genet. 10, e1004320 (2014).
Davids, M. et al. Homozygous splice-variants in human ARV1 cause GPI-anchor synthesis deficiency. Mol. Genet. Metab. 130, 49–57 (2020).
Nguyen, T. T. M. et al. Bi-allelic variants in the GPI transamidase subunit PIGK cause a neurodevelopmental syndrome with hypotonia, cerebellar atrophy, and epilepsy. Am. J. Hum. Genet. 106, 484–495 (2020).
Krawitz, P. M. et al. PGAP2 mutations, affecting the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation syndrome. Am. J. Hum. Genet. 92, 584–589 (2013).
Murakami, Y. et al. Mutations in PIGB cause an inherited GPI biosynthesis defect with an axonal neuropathy and metabolic abnormality in severe cases. Am. J. Hum. Genet. 105, 384–394 (2019).
Salian, S. et al. C18orf32 loss-of-function is associated with a neurodevelopmental disorder with hypotonia and contractures. Hum. Genet. 141, 1423–1429 (2022).
Mugnier, M. R., Stebbins, C. E. & Papavasiliou, F. N. Masters of disguise: antigenic variation and the VSG coat in Trypanosoma brucei. PLoS Pathog. 12, e1005784 (2016).
Borges, A. R., Link, F., Engstler, M. & Jones, N. G. The glycosylphosphatidylinositol anchor: a linchpin for cell surface versatility of trypanosomatids. Front. Cell Dev. Biol. 9, 720536 (2021).
Cowman, A. F. & Crabb, B. S. Invasion of red blood cells by malaria parasites. Cell 124, 755–766 (2006).
Grimme, S. J., Colussi, P. A., Taron, C. H. & Orlean, P. Deficiencies in the essential Smp3 mannosyltransferase block glycosylphosphatidylinositol assembly and lead to defects in growth and cell wall biogenesis in Candida albicans. Microbiol. 150, 3115–3128 (2004).
Essen, L. O., Vogt, M. S. & Mosch, H. U. Diversity of GPI-anchored fungal adhesins. Biol. Chem. 401, 1389–1405 (2020).
Klis, F. M., Sosinska, G. J., de Groot, P. W. & Brul, S. Covalently linked cell wall proteins of Candida albicans and their role in fitness and virulence. FEMS Yeast Res. 9, 1013–1028 (2009).
Hagel, K. R. et al. Systematic interrogation of tumor cell resistance to chimeric antigen receptor T-cell therapy in pancreatic cancer. Cancer Res. 83, 613–625 (2023).
Guo, Z. et al. CDC91L1 (PIG-U) is a newly discovered oncogene in human bladder cancer. Nat. Med. 10, 374–381 (2004).
Nagpal, J. K. et al. Profiling the expression pattern of GPI transamidase complex subunits in human cancer. Mod. Pathol. 21, 979–991 (2008).
Urbaniak, M. D. et al. Fragment screening reveals salicylic hydroxamic acid as an inhibitor of Trypanosoma brucei GPI GlcNAc-PI de-N-acetylase. Carbohydr. Res. 387, 54–58 (2014).
Tu, J. et al. Discovery of a new chemical scaffold for the treatment of superbug Candida auris infections. Emerg. Microbes Infect. 12, 2208687 (2023).
Yadav, U. & Khan, M. A. Targeting the GPI biosynthetic pathway. Pathog. Glob. Health 112, 115–122 (2018).
Hodges, M. R. et al. Safety and pharmacokinetics of intravenous and oral fosmanogepix, a first-in-class antifungal agent, in healthy volunteers. Antimicrob. Agents Chemother. 67, e0162322 (2023).
Vazquez, J. A. et al. Clinical efficacy and safety of a novel antifungal, fosmanogepix, in patients with candidemia caused by Candida auris: results from a phase 2 trial. Antimicrob. Agents Chemother. 67, e0141922 (2023).
Chen, X. et al. Processing and turnover of the Hedgehog protein in the endoplasmic reticulum. J. Cell Biol. 192, 825–838 (2011).
Hall, T. M. et al. Crystal structure of a Hedgehog autoprocessing domain: homology between Hedgehog and self-splicing proteins. Cell 91, 85–97 (1997).
Ciepla, P., Magee, A. I. & Tate, E. W. Cholesterylation: a tail of hedgehog. Biochem Soc. Trans. 43, 262–267 (2015).
Cooper, M. K., Porter, J. A., Young, K. E. & Beachy, P. A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 280, 1603–1607 (1998).
Xiao, X. et al. Cholesterol modification of smoothened is required for hedgehog signaling. Mol. Cell 66, 154–162.e110 (2017).
Hu, A. et al. Cholesterylation of Smoothened is a calcium-accelerated autoreaction involving an intramolecular ester intermediate. Cell Res. 32, 288–301 (2022).
Kong, Z. et al. The cation-pi interaction in cysteine-rich domain of Smoothened is critical for its cholesterylation and function. Acta Biochim. Biophys. Sin. 54, 1171–1179 (2022).
Qi, X. & Li, X. Mechanistic insights into the generation and transduction of hedgehog signaling. Trends Biochem. Sci. 45, 397–410 (2020).
Tukachinsky, H. et al. Dispatched and scube mediate the efficient secretion of the cholesterol-modified hedgehog ligand. Cell Rep. 2, 308–320 (2012).
Goetz, S. C. & Anderson, K. V. The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 11, 331–344 (2010).
Briscoe, J. & Therond, P. P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 14, 416–429 (2013).
Kaushal, J. B., Batra, S. K. & Rachagani, S. Hedgehog signaling and its molecular perspective with cholesterol: a comprehensive review. Cell Mol. Life Sci. 79, 266 (2022).
Huang, P. et al. Cellular cholesterol directly activates smoothened in hedgehog signaling. Cell 166, 1176–1187.e1114 (2016).
Das, A. et al. Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. eLife 3, e02882 (2014).
Bidet, M. et al. The hedgehog receptor patched is involved in cholesterol transport. PLoS ONE 6, e23834 (2011).
Huang, X., Litingtung, Y. & Chiang, C. Region-specific requirement for cholesterol modification of sonic hedgehog in patterning the telencephalon and spinal cord. Development 134, 2095–2105 (2007).
Li, Y., Zhang, H., Litingtung, Y. & Chiang, C. Cholesterol modification restricts the spread of Shh gradient in the limb bud. Proc. Natl. Acad. Sci. USA 103, 6548–6553 (2006).
Beachy, P. A., Karhadkar, S. S. & Berman, D. M. Tissue repair and stem cell renewal in carcinogenesis. Nature 432, 324–331 (2004).
Jiang, S. Q. & Paulus, H. A high-throughput, homogeneous, fluorescence polarization assay for inhibitors of hedgehog protein autoprocessing. J. Biomol. Screen 15, 1082–1087 (2010).
Qiu, Z. P., Hu, A. & Song, B. L. The 3-beta-hydroxysteroid-Delta(8), Delta(7)-isomerase EBP inhibits cholesterylation of Smoothened. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1866, 159041 (2021).
Vujic, I. et al. Acyl protein thioesterase 1 and 2 (APT-1, APT-2) inhibitors palmostatin B, ML348 and ML349 have different effects on NRAS mutant melanoma cells. Oncotarget 7, 7297–7306 (2016).
Harrison, C. F. et al. Exploring anti-bacterial compounds against intracellular Legionella. PLoS ONE 8, e74813 (2013).
Virlogeux, A. et al. Increasing brain palmitoylation rescues behavior and neuropathology in Huntington disease mice. Sci. Adv. 7, eabb0799 (2021).
Zhuang, Z., Gu, J., Li, B. & Yang, L. Inhibition of gasdermin D palmitoylation by disulfiram is crucial for the treatment of myocardial infarction. Transl. Res. S1931-5244, 00158–00155 (2023).
Cordo, S. M., Candurra, N. A. & Damonte, E. B. Myristic acid analogs are inhibitors of Junin virus replication. Microbes Infect. 1, 609–614 (1999).
Harper, D. R., Gilbert, R. L., Blunt, C. & McIlhinney, R. A. Inhibition of varicella-zoster virus replication by an inhibitor of protein myristoylation. J. Gen. Virol. 74, 1181–1184 (1993).
Frearson, J. A. et al. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature 464, 728–732 (2010).
Schlott, A. C. et al. Inhibition of protein N-myristoylation blocks Plasmodium falciparum intraerythrocytic development, egress and invasion. PLoS Biol. 19, e3001408 (2021).
Bell, A. S. et al. Novel thienopyrimidine inhibitors of Leishmania N-myristoyltransferase with on-target activity in intracellular amastigotes. J. Med. Chem. 63, 7740–7765 (2020).
Masubuchi, M. et al. Synthesis and biological activities of benzofuran antifungal agents targeting fungal N-myristoyltransferase. Bioorg. Med. Chem. 11, 4463–4478 (2003).
Sangha, R. et al. Novel, first-in-human, oral PCLX-001 treatment in a patient with relapsed diffuse large B-cell lymphoma. Curr. Oncol. 29, 1939–1946 (2022).
Tabernero, J. et al. Phase I pharmacokinetic and pharmacodynamic study of weekly 1-hour and 24-hour infusion BMS-214662, a farnesyltransferase inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 23, 2521–2533 (2005).
Cortes, J. et al. Phase I study of BMS-214662, a farnesyl transferase inhibitor in patients with acute leukemias and high-risk myelodysplastic syndromes. J. Clin. Oncol. 23, 2805–2812 (2005).
Van Cutsem, E. et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J. Clin. Oncol. 22, 1430–1438 (2004).
Harousseau, J. L. et al. A randomized phase 3 study of tipifarnib compared with best supportive care, including hydroxyurea, in the treatment of newly diagnosed acute myeloid leukemia in patients 70 years or older. Blood 114, 1166–1173 (2009).
Suzuki, M. et al. FDA approval summary for lonafarnib (Zokinvy) for the treatment of Hutchinson-Gilford progeria syndrome and processing-deficient progeroid laminopathies. Genet Med. 25, 100335 (2023).
Sun, J. et al. Geranylgeranyltransferase I inhibitor GGTI-2154 induces breast carcinoma apoptosis and tumor regression in H-Ras transgenic mice. Cancer Res. 63, 8922–8929 (2003).
Mohammed, I. et al. 8-Hydroxyquinoline-based inhibitors of the Rce1 protease disrupt Ras membrane localization in human cells. Bioorg. Med. Chem. 24, 160–178 (2016).
Wan, W. et al. Isoprenylcysteine carboxyl methyltransferase is critical for glioblastoma growth and survival by activating Ras/Raf/Mek/Erk. Cancer Chemother. Pharm. 89, 401–411 (2022).
Wang, M. et al. Inhibition of isoprenylcysteine carboxylmethyltransferase induces autophagic-dependent apoptosis and impairs tumor growth. Oncogene 29, 4959–4970 (2010).
Fenollar, A. et al. Compounds targeting GPI biosynthesis or N-glycosylation are active against Plasmodium falciparum. Comput Struct. Biotechnol. J. 20, 850–863 (2022).
Tsukahara, K. et al. Medicinal genetics approach towards identifying the molecular target of a novel inhibitor of fungal cell wall assembly. Mol. Microbiol. 48, 1029–1042 (2003).
McLellan, C. A. et al. Inhibiting GPI anchor biosynthesis in fungi stresses the endoplasmic reticulum and enhances immunogenicity. ACS Chem. Biol. 7, 1520–1528 (2012).
Mann, P. A. et al. Chemical genomics-based antifungal drug discovery: targeting glycosylphosphatidylinositol (GPI) precursor biosynthesis. ACS Infect. Dis. 1, 59–72 (2015).
Zhou, Q. et al. The ER-associated protein ZDHHC1 is a positive regulator of DNA virus-triggered, MITA/STING-dependent innate immune signaling. Cell Host Microbe 16, 450–461 (2014).
Wang, S. et al. The absence of DHHC3 affects primary and latent herpes simplex virus 1 infection. J. Virol. 92, e01599–01517 (2018).
Baldwin, T. A. et al. Palmitoylation-dependent regulation of cardiomyocyte Rac1 signaling activity and minor effects on cardiac hypertrophy. J. Biol. Chem. 299, 105426 (2023).
Kerkenberg, N. et al. Acute stress reveals different impacts in male and female Zdhhc7-deficient mice. Brain Struct. Funct. 226, 1613–1626 (2021).
Mukai, J. et al. Molecular substrates of altered axonal growth and brain connectivity in a mouse model of schizophrenia. Neuron 86, 680–695 (2015).
Shimell, J. J. et al. The X-linked intellectual disability gene Zdhhc9 is essential for dendrite outgrowth and inhibitory synapse formation. Cell Rep. 29, 2422–2437.e2428 (2019).
Liu, Y. et al. ZDHHC11 modulates innate immune response to DNA virus by mediating MITA-IRF3 association. Cell Mol. Immunol. 15, 907–916 (2018).
Liu, K. M. et al. Cyclic alopecia and abnormal epidermal cornification in Zdhhc13-deficient mice reveal the importance of palmitoylation in hair and skin differentiation. J. Investig. Dermatol. 135, 2603–2610 (2015).
Mejias, R. et al. Increased novelty-induced locomotion, sensitivity to amphetamine, and extracellular dopamine in striatum of Zdhhc15-deficient mice. Transl. Psychiatry 11, 65 (2021).
Singaraja, R. R. et al. Altered palmitoylation and neuropathological deficits in mice lacking HIP14. Hum. Mol. Genet. 20, 3899–3909 (2011).
Sanders, S. S. et al. Sudden death due to paralysis and synaptic and behavioral deficits when Hip14/Zdhhc17 is deleted in adult mice. BMC Biol. 14, 108 (2016).
Wu, Y. et al. ZDHHC19 localizes to the cell membrane of spermatids and is involved in spermatogenesisdagger. Biol. Reprod. 106, 477–486 (2022).
Wang, S. et al. ZDHHC19 is dispensable for spermatogenesis, but is essential for sperm functions in mice. Int. J. Mol. Sci. 22, 8894 (2021).
Haines, R. J. et al. Targeting palmitoyl acyltransferase ZDHHC21 improves gut epithelial barrier dysfunction resulting from burn-induced systemic inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 313, G549–G557 (2017).
Dong, G. et al. Palmitoylation couples insulin hypersecretion with beta cell failure in diabetes. Cell Metab. 35, 332–344.e337 (2023).
Mijimolle, N. et al. Protein farnesyltransferase in embryogenesis, adult homeostasis, and tumor development. Cancer Cell 7, 313–324 (2005).
Lee, R. et al. Genetic studies on the functional relevance of the protein prenyltransferases in skin keratinocytes. Hum. Mol. Genet. 19, 1603–1617 (2010).
Boi, R. et al. Podocyte geranylgeranyl transferase type-I is essential for maintenance of the glomerular filtration barrier. J. Am. Soc. Nephrol. 34, 641–655 (2023).
Lopez-Posadas, R. et al. Inhibiting PGGT1B disrupts function of RHOA, resulting in T-cell expression of integrin alpha4beta7 and development of colitis in mice. Gastroenterology 157, 1293–1309 (2019).
Hirata, T. et al. Loss of the N-acetylgalactosamine side chain of the GPI-anchor impairs bone formation and brain functions and accelerates the prion disease pathology. J. Biol. Chem. 298, 101720 (2022).
Lee, G. H. et al. A GPI processing phospholipase A2, PGAP6, modulates Nodal signaling in embryos by shedding CRIPTO. J. Cell Biol. 215, 705–718 (2016).
Rampoldi, F. et al. Immunosuppression and aberrant T cell development in the absence of N-myristoylation. J. Immunol. 195, 4228–4243 (2015).
Hu, T. et al. Myristoylated Naked2 antagonizes Wnt-beta-catenin activity by degrading Dishevelled-1 at the plasma membrane. J. Biol. Chem. 285, 13561–13568 (2010).
Kinoshita-Kikuta, E. et al. Protein-N-myristoylation-dependent phosphorylation of serine 13 of tyrosine kinase Lyn by casein kinase 1gamma at the Golgi during intracellular protein traffic. Sci. Rep. 10, 16273 (2020).
Sonowal, H., Rice, W. G. & Howell, S. B. Luxeptinib interferes with LYN-mediated activation of SYK and modulates BCR signaling in lymphoma. PLoS ONE 18, e0277003 (2023).
Lu, X. et al. HGAL localization to cell membrane regulates B-cell receptor signaling. Blood 125, 649–657 (2015).
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No. 82073095), and the medical Sci-Tech innovation platform Foundation of Zhongnan Hospital, Wuhan University (No. PTXM2023005).
Author information
Authors and Affiliations
Contributions
X.X.H., Y.C., Q.Z. and Y.Y. designed this study. Y.Y. drafted the manuscript and drew the figures. Y.Y., P.Y.L., J.H.L., X.X.H., Y.C. and Q.Z. revised the manuscript. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yuan, Y., Li, P., Li, J. et al. Protein lipidation in health and disease: molecular basis, physiological function and pathological implication. Sig Transduct Target Ther 9, 60 (2024). https://doi.org/10.1038/s41392-024-01759-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01759-7
