
Abstract
Ferroptosis is an iron-dependent form of regulated cell death with distinct characteristics, including altered iron homeostasis, reduced defense against oxidative stress, and abnormal lipid peroxidation. Recent studies have provided compelling evidence supporting the notion that ferroptosis plays a key pathogenic role in many diseases such as various cancer types, neurodegenerative disease, diseases involving tissue and/or organ injury, and inflammatory and infectious diseases. Although the precise regulatory networks that underlie ferroptosis are largely unknown, particularly with respect to the initiation and progression of various diseases, ferroptosis is recognized as a bona fide target for the further development of treatment and prevention strategies. Over the past decade, considerable progress has been made in developing pharmacological agonists and antagonists for the treatment of these ferroptosis-related conditions. Here, we provide a detailed overview of our current knowledge regarding ferroptosis, its pathological roles, and its regulation during disease progression. Focusing on the use of chemical tools that target ferroptosis in preclinical studies, we also summarize recent advances in targeting ferroptosis across the growing spectrum of ferroptosis-associated pathogenic conditions. Finally, we discuss new challenges and opportunities for targeting ferroptosis as a potential strategy for treating ferroptosis-related diseases.
Similar content being viewed by others

Introduction
Ferroptosis is a unique form of iron-dependent regulated cell death originally identified by screening RSL (RAS-selective lethal) compounds.1 The key characteristic of ferroptosis is an extensive accumulation of lipid peroxides,1 making ferroptosis genetically, morphologically, and biochemically distinct from other types of cell death. In the decade since ferroptosis was first reported, increasing evidence has emerged suggesting that ferroptosis may play a pivotal role in many biological processes such as tumor suppression and immunity, indicating that ferroptosis is important for maintaining health by regulating metabolism and redox homeostasis. Recently, a plethora of studies have shown that ferroptosis also plays a critical role in a variety of pathophysiological processes such as ischemic organ injury, stroke, cardiac myopathy, and neurodegenerative diseases; ferroptosis has also been implicated in many oncogenic pathways, suggesting it might serve as a target for novel cancer therapeutics.2
In mammalian cells, ferroptosis is regulated mainly by iron homeostasis, lipid metabolism, and glutathione-dependent redox balance. Thanks to rapid progress in the study of ferroptosis, numerous efforts have made to identify potent, druggable ferroptosis modulators for use in clinical applications, opening new avenues for developing novel treatment strategies to target many ferroptosis-related diseases, including cancer and heart injury. With respect to cancer, novel ferroptosis agonists have shown promise in several cancer types. On the other hand, antagonists of ferroptosis have been shown to help alleviate ferroptosis-related diseases such as ischemia/reperfusion (I/R)-induced damage, neurodegenerative diseases, and inflammatory diseases.3,4,5,6,7
Here, we provide a comprehensive update of recent efforts to target ferroptosis for treating a variety of relevant diseases, and we discuss the safety and efficacy of ferroptosis agonists and antagonists, as well as their pharmacokinetics profiles, their experimental stage in the drug discovery process, and opportunities for further clinical development. We also review the biological roles, molecular mechanisms, and clinical implications of ferroptosis, and we discuss insights into ferroptosis-focused translational research, as well as current knowledge gaps and opportunities. Finally, we discuss future research orientations that will lead to the clinical implementation of these novel ferroptosis modulators for treating a diverse range of diseases.
Discovery and characteristics of ferroptosis
Although the term “ferroptosis” was first coined by Dixon et al. in 2012 (Fig. 1a), a similar form of neuronal cell death triggered by cysteine depletion called “oxytosis” was reported by Murphy et al. back in 1989;8 this neuronal cell death was induced by the excitotoxin glutamate by inhibiting SLC7A11 (solute carrier family 7 member 11), a component of the cystine-glutamate antiporter system Xc− known to play a role in ferroptosis. Therefore, oxytosis and ferroptosis have been suggested to share several key characteristics, including their gene expression patterns, high activity of lipoxygenases, and high accumulation of reactive oxygen species (ROS).9 By 2003, a distinct form of erastin-induced cell death in RAS-expressing cancer cells was receiving considerable attention;10 this form of cell death was not inhibited by caspase inhibitors but could be suppressed by treating the cells with iron-chelating agents.11 Yang et al. subsequently found that RAS-selective lethal small molecule 3 (RSL3) could also trigger this iron-dependent form of cell death.12 Based on these characteristics, Dixon et al. named this type of cell death ferroptosis.1 Morphologically, cells undergoing ferroptosis generally have shrunken mitochondria with increased membrane density and reduced—or absent—mitochondrial cristae. Biochemically, ferroptosis is characterized by increased oxidative stress and depleted antioxidative defense. Although ferroptosis is generally well-accepted as a tightly regulated cellular metabolic process, its precise mechanisms such as how excessive phospholipid (PL) peroxidation drives ferroptosis and the tissue- and disease-specific epilipidome signatures remain unknown.

Timeline of the identification and characterization of ferroptosis, and the underlying mechanisms. a, Timeline depicting the past, present, and future of ferroptosis. The first time period (1980–2012) ended with the term “ferroptosis” being coined by Dixon et al. in 2012. The present period (2012–2023) developed rapidly, with important details emerging such as the GCH1-BH4 and DHODH-CoQ10 pathways. The future (2023-?) is expected to bring numerous new applications for targeting ferroptosis. b, Three pathways mediate ferroptosis, including iron metabolism, redox, and lipid metabolism. Dysregulation of oxidative-reductive systems, iron metabolism, and/or peroxidation of PUFAs can induce ferroptosis. ACSL4, acyl-CoA synthetase long-chain family member 4; BH4, tetrahydrobiopterin; CBS, cystathionine beta-synthase; CD36, cluster differentiation 36; CoQ10, coenzyme Q10; CTH, cystathionine gamma-lyase; DHODH, dihydroorotate dehydrogenase; DMT1, proton-coupled divalent metal ion transporter 1; FPN, ferroportin; FSP1, ferroptosis suppressor protein 1; GCH1, GTP cyclohydrolase 1; GPX4, glutathione peroxidase 4; GSH, glutathione; GSSG, glutathione disulfide; HO-1, heme oxygenase 1; Keap1, Kelch-like ECH-associated protein 1; LOX, lipoxygenase; LPCAT, lysophosphatidylcholine acyltransferase; MUFA, monounsaturated fatty acid; NCOA4, nuclear receptor coactivator 4; Nrf2, nuclear factor erythroid 2-related factor 2; PL-PUFA, phospholipid-containing polyunsaturated fatty acid; PUFA, polyunsaturated fatty acid; RNF217, E3 ubiquitin protein ligase RNF217; SCD1, stearoyl-coenzyme A desaturase 1; SLC, solute carrier family; SQS, squalene synthase; STEAP, 6-transmembrane epithelial antigen of the prostate metalloreductase family; TF, transferrin; TFR1, transferrin receptor protein 1; TRPML, lysosomal cation channel mucolipin. Created with BioRender.com
The mechanisms that regulate ferroptosis have begun to emerge (Fig. 1a). For example, Yang et al. found that the glutathione/glutathione peroxidase 4 (GSH/GPX4) pathway serves as an important regulator of ferroptosis.13 Soon after, other groups identified ACSL4 (acyl-CoA synthetase long-chain family member 4) as a predominant mediator of ferroptosis.14,15 In addition, selenium was shown to protect against ferroptosis.16 In 2019, two groups independently reported that the FSP1 (ferroptosis suppressor protein 1)-CoQ10 (coenzyme Q10)-NAD(P)H pathway inhibits ferroptosis via a GPX4-independent mechanism.17,18 In addition, the GCH1 (GTP cyclohydrolase 1)-BH4 (tetrahydrobiopterin) pathway was reported as an additional GPX4-independent modulator of ferroptosis.19 Recently, Mao et al. identified the DHODH (dihydroorotate dehydrogenase)-CoQ10 axis, which is located at the mitochondrial inner membrane, as an important pathway that protects against ferroptosis.20 Even more recently, Mishima et al. reported that vitamin K is a potent FSP1-dependent inhibitor of ferroptosis by functionally screening vitamin compounds in Gpx4-deficient mouse embryonic fibroblasts.21
Pathways that regulate ferroptosis
A growing body of evidence indicates that ferroptosis is regulated by a complex network involving iron homeostasis, lipid metabolism, and the oxidative-reductive system (Fig. 1b). For example, cellular ferroptosis is driven directly by an accumulation of toxic PL peroxides in membranes, which are produced primarily by the free iron‒induced Fenton reaction and oxidized PL-containing polyunsaturated fatty acids (PL-PUFAs).
Because iron chelators such as deferoxamine (DFO) can block ferroptosis, ferroptosis was originally defined as iron-dependent.1 In contrast, iron overload has been shown to sensitize numerous cells types to ferroptosis.3,22,23,24 Therefore, maintaining iron homeostasis is critical in order to protect cells from ferroptosis. The intracellular labile iron pool (LIP) is regulated by the uptake, export, storage, and utilization of iron (Fig. 1b). Cellular iron uptake is regulated primarily by the transferrin/transferrin receptor (TF/TFR) system, which has been shown to play a role in ferroptosis.25 Recently, we reported that TF protects against ferroptosis-induced liver damage via SLC39A14 (solute carrier family 39, member 4)-mediated non-TF-bound iron (NTBI).23 Excess intracellular iron is sequestered largely by the principal iron-storage protein ferritin (FTH), which protects cells from ferroptosis. Consistent with this protective role, we found that mice lacking Fth in cardiomyocytes develop ferroptosis-induced cardiomyopathy.24 Notably, ferritinophagy—an NCOA4 (nuclear receptor coactivator 4)-mediated autophagic degradation of ferritin in the lysosome—has been shown to induce ferroptosis by releasing free iron from ferritin.26,27 Moreover, the iron exporter ferroportin (FPN) has also been shown to regulate cellular sensitivity to ferroptosis in vitro.28
The metabolism of lipids—particularly PL-PUFAs—critically regulates ferroptosis (Fig. 1b). Using a combination of genome-wide CRISPR screening and microarray analysis of ferroptosis-resistant cell lines, Doll et al. found that the enzyme ACSL4 is a major mediator of ferroptosis.15 ACSL4 catalyzes the conversion of free PUFAs to acyl-CoA derivatives (PUFA-CoAs), which can be further catalyzed by lysophosphatidylcholine acyltransferase 3 (LPCAT3) to produce PL-PUFAs. PL-PUFAs participate in the biosynthesis of cellular membranes and are highly susceptible to peroxidation due to their bis-allylic hydrogen atoms. Interestingly, loss of either ACSL4 or LPCAT3 increases cellular insusceptibility to ferroptosis by reducing the production of substrates for PL peroxidation.15,29,30 In addition, inhibiting lipoxygenases (LOXs), which are iron-containing enzymes, suppresses erastin-induced ferroptosis via iron- and oxygen-triggered free radical chain reactions, independent of their enzymatic activity.31
With respect to oxidative and reductive reactions, several major pathways are involved in protecting against ferroptosis (Fig. 1b). First, the well-characterized system Xc–-GSH-GPX4 axis serves as a GPX4-dependent mechanism for scavenging PL peroxides via system Xc–-mediated GSH synthesis.1,13 Moreover, inhibiting either component of system Xc– (i.e., SLC7A11 or SLC3A2) induces ferroptosis by disrupting cystine uptake, thereby limiting GSH synthesis.1 In this pathway, GPX4 serves as an essential regulator of ferroptosis.13 Second, in 2019 an FSP1-dependent metabolic pathway was shown to protect against ferroptosis via an GPX4-independent process.17,18 Specifically, two groups simultaneously reported that FSP1 traps lipid peroxyl radicals to suppress ferroptosis by reducing CoQ10 levels at the plasma membrane,17,18 a process that is independent of the system Xc–-GSH-GPX4 pathway. Third, Mao et al. showed that mitochondrial inner membrane-located DHODH can protect against ferroptosis by reducing CoQ to form ubiquinol, an antioxidant that inhibits ferroptosis; thus, inhibiting DHODH in cancer cells potently activates ferroptosis in parallel with mitochondrial GPX4, independent of cytosolic GPX4 and FSP1.20 Finally, Liang et al. recently identified the PL-modifying enzymes MBOAT1 and MBOAT2 as novel sex hormone‒dependent ferroptosis inhibitors.32 Mechanistically, the authors showed that MBOAT1/2 suppress ferroptosis by remodeling cellular PLs to protect cells from ferroptosis independent of GPX4, providing novel ferroptosis-targeted therapeutic strategies to sensitize either estrogen receptor (ER) antagonists in ER-positive breast cancer and androgen receptor (AR) antagonists in AR-positive prostate cancer, respectively.
The pathological role of ferroptosis in disease
Intracellular accumulation of iron and PL peroxides are two central biochemical events that occur during ferroptosis, and these processes have also been implicated in a wide range of diseases (Tables 1 and 2). Indeed, a plethora of studies suggest a putative causal connection between ferroptosis and the pathophysiological processes underlying many conditions and diseases, including cancer, neurodegeneration, I/R-induced tissue injury, acute kidney damage, and hematologic diseases (Fig. 2).

Ferroptosis-related diseases that can present throughout the human lifespan. As we age, iron levels in the body accumulate, inducing ferroptosis and increasing our susceptibility to hypoxic-ischemic brain damage, organ injury‒related diseases, and neurodegenerative diseases. Reduced ferroptosis can cause various forms of cancers in all stages of life. NAFLD non-alcoholic fatty liver disease, NASH non-alcoholic steatohepatitis. Created with BioRender.com
Cancer
Apoptosis was long considered the principal form of cell death in various cancer types; however, apoptosis-based cancer therapies have limited clinical benefit.33 Therefore, identifying novel cancer therapeutics that target pathways other than apoptosis is now an urgent unmet clinical need. Indeed, the notion of ferroptosis originated from lethal screening of compounds in RAS-mutated cancer cells.1 In support of this notion, ferroptosis has been functionally validated in cancer types with increased PL peroxidation, including hepatocellular carcinoma (HCC),34 pancreatic ductal adenocarcinoma,35 triple-negative breast cancer (TNBC),36,37 and renal cell carcinoma.38,39 Thus, these specific cancer types may be more sensitive to ferroptosis-inducing compounds.
It is important to note that ferroptosis has also been closely linked to resistance to various cancer treatments. For example, several studies found that cancer cells with a high-mesenchymal state are more resistant to a variety of cancer treatments, but are particularly susceptible to ferroptosis-inducing compounds.15,40 In addition, priming cancer cells with ferroptosis-inducing compounds can sensitize the cancer cells to subsequent immunotherapies.41 Taken together, these findings highlight the promise of targeting ferroptosis as a novel strategy for treating various forms of cancer.
Neurodegenerative diseases
Iron deposition and lipid peroxidation are common pathophysiological features in various neurological diseases. Notably, the disease group known as neurodegeneration with brain iron accumulation (NBIA) refers to neurodegenerative disorders associated with altered iron metabolism in the lesion area and includes Alzheimer’s disease (AD),42 Parkinson’s disease (PD),43 and Huntington’s disease (HD).44 In addition, ablating or inactivating GPX4 promotes neuronal damage and neurodegeneration.6,7 In experimental models of degenerative brain conditions, both ferroptosis inhibitors and iron chelators were shown to improve outcome and prognosis.45,46,47,48
With respect to AD, several studies have shown that dysregulated iron metabolism is linked to ROS production, mitochondrial dysfunction, and neurodegeneration.42 Iron deposition in the brain, accompanied by a decrease in endogenous antioxidant capacity, has been associated with the pathogenesis of AD, and iron levels in the brain have been correlated with disease progression.49 Furthermore, accumulated iron interacts with the Aβ peptide and tau protein via the formation of a peptide-hemin complex, possibly implicating ferroptosis.50
PD is characterized by reduced dopaminergic neurons selectively in the substantia nigra and the presence of Lewy bodies.51 In the substantia nigra pars compacta of affected patients, iron was shown to accumulate in glia cells and dopaminergic neurons, with the level of iron correlated with disease severity.52 Moreover, reports suggest that the increased oxidative stress present in PD may be attributed to decreased GSH levels in the substantia nigra.53 In addition, the brain consumes high amounts of oxygen, making this organ more sensitive to lipid peroxidation.54
HD, a hereditary neurodegenerative disease due to an abnormally high number of CAG repeats in the huntingtin (HTT) gene,55 has also been linked to ferroptosis in animal models.56 Excess iron remains a major cause of oxidative stress in neurons, which directly induces ferroptosis during the pathogenesis of HD.57 Interestingly, similar to PD, reduced levels of GSH have also been observed in HD.58 Together, these findings suggest that ferroptosis may be involved in the pathogenesis of HD.
Tissue and organ injury
Ferroptosis was originally identified as the major pathogenic driver in several conditions related to organ injury. In 2014, mice lacking Gpx4 were shown to develop spontaneous acute renal failure and hepatic I/R-induced damage via ferroptosis.5 Subsequently, the pathogenic role of ferroptosis in the progression of I/R-induced acute damage in the lungs, heart, and intestinal tract has been validated in animal models.3,4,59 We previously systematically reviewed the varied role of ferroptosis in liver disease, and we refer the reader to this review.60 Notably, tissue-specific Gpx4 knockout mice present with tissue damage accompanied by massive death of photoreceptor cells61 and endothelial cells.62
Acute kidney injury (AKI) refers to sudden-onset kidney failure and/or kidney damage characterized by a rapid loss of the kidney’s excretory function caused by massive levels of cell death and inflammation.63 In mouse kidney tubular cells, Gpx4 has been reported to prevent AKI by blocking ferroptosis.5 Around the same time, Linkermann et al. independently found that ferroptosis plays a critical role in synchronizing kidney tubular cell death in both severe I/R-induced injury and oxalate crystal‒induced models of AKI; moreover, the authors showed that SRS 16–86—a novel third-generation ferroptosis-specific inhibitor—potently protected against AKI.64 Recently, Li et al. reported that ferroptosis is the primary form of cell death causing folic acid‒induced AKI in mice.65
In the heart, we first identified ferroptosis as the major pathogenic mechanism in both doxorubicin- and I/R-induced cardiomyopathy, and we showed that targeting ferroptosis significantly alleviated heart injury in mouse models.3 In addition, inhibiting glutaminolysis protected against I/R-induced ferroptotic heart damage in vitro, indicating that glutaminolysis plays an essential role in regulating ferroptosis during heart injury.25 Recently, Jacobs et al. suggested the possible presence of ferroptosis in the myocardium of a patient with COVID-19‒induced myocarditis.66 Using the antibody E06 to stain oxidized phosphatidylcholine, they found that a ferroptosis “signature” may be specific to injured cardiomyocytes in COVID-19‒induced myocarditis, as this signature was not present in either myocarditis of unknown etiology or in COVID-19 patients who presented without myocarditis.66 Recently, we summarized the role of ferroptosis in regulating cardiovascular disease, and we refer to the reader to this review.67
Previous studies using animal models of brain injury showed features resembling ferroptosis, including increased lipid peroxidation, increased intracellular iron levels, and decreased GSH levels.68,69 A subsequent study also showed that inhibiting ferroptosis prevented the death of primary oligodendrocytes.70 Notably, organotypic hippocampal slice cultures with ferroptosis-specific inhibitors were shown to prevent neuronal death and decrease hemoglobin-induced iron accumulation, suggesting a pathogenic role of ferroptosis in intracerebral hemorrhage.71 In addition, neurons obtained from an ischemic stroke mouse model were shown to have significantly decreased levels of GSH72 and increased lipid peroxidation,73 suggesting neuronal ferroptosis.74 In addition, carvacrol (the major monoterpenic phenol found in some essential oils and known to reduce oxidative stress and apoptosis) has been shown to help protect hippocampal neurons from ferroptosis by upregulating Gpx4 expression in a gerbil model of cerebral ischemia.75 In spinal cord injury, recent studies suggest that ferroptosis contributes to secondary injury, and blocking ferroptosis may help repair traumatic spinal cord injury.76
The liver serves as a central metabolic organ and is highly susceptible to various toxic metabolites. We previously showed that high dietary iron directly leads to ferroptosis-induced liver injury, and this damage was aggravated by either knocking out Slc7a11 expression77 or increasing non-TF-bound iron (NTBI).23 Ferroptosis was recently linked to various chemical-induced forms of liver injury such as alcoholic liver disease,78 methionine/choline-deficient‒induced non-alcoholic steatohepatitis (NASH),79 and arsenic-induced NASH.80 In addition, both ferroptosis and a concomitant accumulation of lipid ROS were also observed in CCl4-induced liver fibrosis,23 acetaminophen-induced liver damage,81 and I/R-induced liver injury.82
Critically ill patients with multiorgan dysfunction syndrome (MODS) were shown to present with a ferroptosis signature that includes increased plasma levels of MDA (malondialdehyde, a lipid peroxidation degradation product) and catalytic iron.83 In addition, a ferroptosis signature was shown in tissue samples obtained from a patient with COVID-19‒induced MODS (including cardiac and renal failure).66 Using an experimental model of MODS, Van Coillie et al. showed that excess iron can trigger ferroptosis in multiple organs; moreover, they showed that the novel ferroptosis inhibitor UAMC-3203 may be a viable therapeutic agent for the clinical treatment of MODS.83
Inflammatory and infectious diseases
Ferroptosis has also been reported to directly modulate the host’s immune response and inflammation during inflammatory and infectious diseases.84 Moreover, the functions of various immune cell types such as T cells, B cells, and macrophages can be affected by ferroptosis. For example, Gpx4-deficient CD4+ and CD8+ T cells undergo ferroptosis due to accumulated lipid peroxides.85 Thus, these ferroptotic T cells are unable to proliferate and have reduced function against the acute lymphoblastic choriomeningitis virus and Leishmania major parasites, suggesting that Gpx4 is required for maintaining T cell‒mediated immunity by protecting the cells against ferroptosis.85 With respect to B cells, various B cell populations have been shown to respond differently to ferroptosis induction.86 Similarly, in bone marrow cells inducible nitric oxide synthase (iNOS) is more abundant in M1 macrophages than in M2 macrophages, and M1 macrophages tend to be more resistant to iron deposits and ferroptosis.87 Interestingly, a recent study found that Gpx4-dependent ferroptosis in neutrophils drives the pathogenesis of systemic lupus erythematosus (SLE),88 suggesting that targeting ferroptosis-induced neutropenia may be a potential strategy for treating autoimmune diseases such as SLE.
The aerobic gram-negative bacterium Pseudomonas aeruginosa can infect both immunocompetent and immunocompromised hosts. Interestingly, this pathogen expresses lipoxygenase (which oxidizes AA-PE to 15-hydroperoxide AA-PE in host cells) and drives ferroptosis in human bronchial epithelial cells, as well as in clinically isolated cells from patients with persistent lower respiratory tract infection.89 Thus, targeting ferroptosis may be a viable strategy for managing P. aeruginosa infection.
The bacterium Mycobacterium tuberculosis is one of the major pathogens that causes tuberculosis, an infectious disease that affects the lungs, bone tissue, brain, and spine. Interestingly, M. tuberculosis has been shown to trigger ferroptosis in macrophages, accompanied by decreased GPX4 expression, increased iron content, and elevated levels of membrane lipid peroxidation.90 Similar results were also obtained in mice acutely infected with M. tuberculosis.90 Given these findings, ferroptosis may also be a promising therapeutic target in patients with tuberculosis and other infectious diseases.
Notably, ferroptosis has also been closely linked to sterile inflammation, a process that occurs in the absence of pathogens and is commonly triggered by release of the intracellular contents from damaged and/or necrotic cells. Sterile inflammation is often triggered by acute conditions such as I/R-induced injury, trauma, oxalate crystal‒induced inflammation, and chronic inflammatory diseases such as atherosclerosis. For instance, ferroptosis increases the recruitment of neutrophils to damaged heart tissue under ischemic conditions, and ferroptosis-specific inhibitors can alleviate this inflammatory damage.91 In addition, blocking ferroptosis was shown to significantly reduce atherosclerosis in ApoE−/− mice fed a high-fat diet.92
Pharmacological agents that target ferroptosis-regulating pathways
Given that iron metabolism, oxidative-reductive pathways, and lipid metabolism coordinately control ferroptosis, altering these three pathways using genetic and pharmacological approaches has been shown to affect ferroptosis (Table 3). Below, we summarize the bioactive pharmacological agents that affect ferroptosis by targeting core components in these three ferroptosis-regulating processes (see also Table 4).
Targeting dysregulated iron metabolism
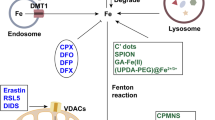
Iron is essential to many physiological processes, including electron delivery, oxygen transport, and DNA biosynthesis. Iron is stored and transported as Fe3+, while Fe2+ serves as an electron donor to catalyze the Fenton reaction. During iron overload conditions, excess iron is involved in the generation of free radicals, lipid peroxidation, and DNA damage, which in turn promotes ferroptosis. At the cellular level, transferrin (TF) mediates the cellular uptake of Fe3+ via endocytosis of iron-loaded TF-bound transferrin receptor 1 (TFR1), followed by release from the endosome,93,94 in which Fe3+ is then converted to Fe2+ via the metalloreductase STEAP3.95 When the binding capacity of TF-2Fe3+ is saturated, ferrireductases reduce Fe3+ to Fe2+, which can be transported to the LIP via NTBI transporters such as DMT1 (divalent metal transporter 1)96 and SLC39A14.97 The LIP can also increase due to the degradation of either heme or hemoglobin, which are captured by hemopexin and haptoglobin, respectively. Upon binding to CD163 (a monocyte/macrophage-specific scavenger receptor)98 and LRP1 (LDL receptor related protein 1),99 heme-hemopexin and hemoglobin-haptoglobin complexes are internalized via endosomes, after which the rate-limiting enzyme heme oxygenase-1 (HO-1) catabolizes heme to produce biliverdin, releasing iron into the LIP. Iron is reserved primarily in ferritin, a complex consisting of 24 light chain (FTL) and heavy chain (FTH1) subunits.100 FTH also has ferroxidase activity, converting Fe2+ to Fe3+ to prevent iron-induced toxicity. Excess iron is exported by FPN, the body’s sole iron exporter,101 which is regulated primarily by the hepatic peptide hepcidin,102 the master regulator of iron homeostasis. Thus, together with our recent finding that the newly identified E3 ubiquitin ligase RNF217 mediates the degradation of FPN,103 these results indicate that systemic iron homeostasis is tightly controlled by the hepcidin-FPN axis, and targeting dysregulated iron metabolism may directly affect ferroptosis (Fig. 3).

Targeting ferroptosis in iron metabolism. a Overview of systemic iron homeostasis. Labile iron binds to transferrin (TF) in the blood, and senescent erythrocytes are phagocytized by macrophages, releasing iron ions back into the circulation. The primary regulatory mechanism of iron homeostasis involves liver-derived hepcidin, which controls the cellular export of iron via ferroportin (FPN). b Overview of the various processes that involve ferritin under iron-deficient and iron-sufficient conditions. When cellular iron is sufficient, ferritin stores iron. Under iron-deficient conditions, ferritin undergoes NCOA4-mediated ferritinophagy and releases iron. c The active labile iron pool can be used either directly for incorporation into iron-containing proteins or transported into the mitochondria. d Overview of iron transporters in the plasma membrane and in lysosomes. Molecules in the pink and green text boxes are inhibitors or activators, respectively, of the pathways that regulate iron metabolism and suppress or trigger, respectively, ferroptosis. ALAS 5-amibolevulinic acid synthase, DMT1 proton-coupled divalent metal ion transporter 1, EPO erythropoietin; FLVCR1b, FLVCR heme transporter 1b, HERC2 HECT and RLD domain containing E3 ubiquitin protein ligase 2, HO-1 heme oxygenase 1, LIP labile iron pool, MFRN mitoferrin, NCOA4 nuclear receptor coactivator 4, PCBP poly(rC)-binding protein, RNF217 E3 ubiquitin protein ligase RNF217, SLC solute carrier family, STEAP 6-transmembrane epithelial antigen of the prostate metalloreductase family, TFR1 transferrin receptor protein 1, TRPML lysosomal cation channel mucolipin. Created with BioRender.com
Using iron chelators to inhibit ferroptosis
The term “ferroptosis” originated from the rescue effect of the iron chelator DFO. In the clinic, DFO, deferiprone (DFP), and deferasirox (DFX) are commonly used iron chelators.104 DFO is derived from Streptomyces pilus and is approved for managing iron overload‒related diseases such as hemochromatosis, thalassemia, and sickle cell anemia, as well as other chronic iron overload‒induced conditions such as blood transfusion.105,106,107 Although DFO has been reported to prevent ferroptosis in several animal disease models, including neurodegeneration, I/R-induced injury, NASH, and hemorrhagic stroke,3,46,82,108,109 DFO can cause toxic side effects such as anemia and edema.110 DFP, an effective, orally administered hydrophilic iron chelator, was developed as an alternative to DFO.111 Compared to DFO, DFP is relatively inexpensive and more effective, with lower toxicity, making DFP more suitable for use in iron-overloaded transfusion patients who currently cannot receive any form of chelation therapy.112 In addition, DFP can cross the blood-brain barrier (BBB) and can transfer iron in DFP-Fe to transferrin.113,114 DFP has been shown to improve motor function by decreasing iron content in the brain in patients with Friedreich’s ataxia.115
Similar to DFP, DFX is a highly selective iron chelator showing a high affinity for Fe3+ and a relatively long biological half-life.116 Studies have shown that DFX treatment can prevent the accumulation of hemosiderin (a complex composed of partially digested ferritin and lysosomes for iron storage) and can ameliorate ferroptosis-related kidney and neuronal damage.117,118
Currently, dexrazoxane (DXZ) is the only iron chelator approved by the FDA for protecting against doxorubicin (DOX)-induced cardiotoxicity by chelating DOX-induced mitochondrial iron.119 Compared to DXZ, the novel orally administered iron chelator CN128 is more potent and has fewer side effects and is currently being studied in a phase II clinical trial for treating β-thalassemia following regular blood transfusion.120 Finally, ciclopirox olamine, which is currently approved for treating cutaneous fungal infections, has also been used as an iron chelator in various model systems due to its inhibitory activity on iron-dependent ribonucleotide reductase.121,122
Iron receptors and transporters
The TF receptor TFR1 is a glycoprotein that interacts with iron-bound TF in order to mediate cellular iron uptake. Gao et al. reported that TFR1-mediated cellular uptake of TF-bound iron is required for ferroptosis, showing that inhibiting TFR1 using RNA interference (RNAi) can efficiently block ferroptosis.25 Moreover, Wu et al. showed that knocking down TFR1 inhibits ferroptosis under cystine starvation conditions, whereas upregulating TFR1 significantly activates ferroptosis via the NF2-YAP signaling pathway.123 Using the TFR1-specific antibody 3F3-FMA, Feng and colleagues showed that TFR1 can serve as a marker for cells undergoing ferroptosis.124 Given that various cancer cells express relatively high levels of TFR1, antibodies that either neutralize of block TFR1 have been investigated as potential cancer therapies. For example, Horonchik and Wessling-Resnick found that the small-molecule TFR1 inhibitor ferristatin (also known as NSC306711) can inhibit iron uptake by mediating the degradation of TFR1(ref. 125). Based on these results, it is reasonable to speculate that TFR1 antibodies may be suitable for treating ferroptosis-induced diseases, warranting further study.
DMT1 (also known as SLC11A2, DCT1, and NRAMP2) is known for its ability to transport Fe2+ into the duodenum and out of the endosome during the TF cycle, but it can also transport NTBI.96 DMT1 inhibitors such as ebselen,126 pyrrolidine dithiocarbamate (PDTC),126 and benzylisothiourea127 have been shown to reduce iron-induced damage by potently reducing the DMT1-mediated cellular uptake of NTBI, indicating that DMT1 may be a promising target for regulating ferroptosis to manage ferroptosis-related diseases.
Two strategies have been suggested to modulate ferroptosis by targeting iron export, namely hepcidin agonists such as minihepcidins128 and FPN inhibitors such as VIT-2763 (ref. 128). Similarly, small molecules that directly upregulate hepcidin expression may affect ferroptosis by promoting intracellular iron accumulation. In addition, endogenous inducers of hepcidin such as the cytokine oncostatin M (encoded by the OSM gene)129 may promote ferroptosis. Conversely, clinical trials involving hepcidin antagonists such as PRS-080,128 NOX-H94,130 and LY2787106131 may suppress ferroptosis by reducing intracellular iron content. Thus, further studies are needed in order to ascertain the functional role of these FPN/hepcidin regulators in modulating ferroptosis under pathological conditions.
Ferritinophagy
Under iron-deficient conditions, NCOA4 binds to iron-loaded ferritin, thus promoting lysosomal ferritin degradation and releasing iron into the LIP, a process known as ferritinophagy.132 Ferritinophagy has been shown to initiate ferroptosis via iron overload and lipid peroxidation.26,27 JQ1, a thienotriazolodiazepine that inhibits bromodomain proteins such as BRD4, has been reported to potentiate ferroptosis via ferritinophagy in breast cancer cells.133 Additionally, DpdtC (2,2′-di-pyridylketone dithiocarbamate) mobilizes iron by inducing ferritinophagy and can increase ferritinophagy-induced ROS production in MGC-803 cells, a human gastric carcinoma cell line.134 MMRi62, a small molecule compound initially identified as an inducer of apoptosis, is shown to potently trigger ferroptotic cell death in pancreatic ductal adenocarcinoma cells by increasing lysosomal ferritinophagy.135 On the other hand, Fang et al. recently found that a novel compound called 9a potently suppresses ferritinophagy-induced ferroptosis by competitively binding to NCOA4 and perturbing the interaction between NCOA4 and FTH1.136
Iron recycling by macrophages
Macrophages in the spleen and Kupffer cells (also known as stellate macrophages) in the liver maintain iron homeostasis by recycling iron obtained from senescent erythrocytes and damaged cells.137 Macrophage-mediated iron recycling is processed primarily in the spleen, and recycling iron is returned to the storage pool for utilization in various processes such as heme biosynthesis and Fe/S cluster formations in the mitochondria. The enzyme HO-1, encoded by the HMOX1 gene, catalyzes heme to produce carbon monoxide, biliverdin, and free iron; biliverdin and free iron can be used to generate bilirubin and sequestered by ferritin, respectively. Upregulation of HMOX1 expression has been shown to play a cytoprotective role138 and to increase the resistance of HCC cells to ferroptosis mediated by the p62-KEAP1 (Kelch-like ECH-associated protein 1)-NRF2 (nuclear factor erythroid-2-related factor 2) pathway.34 On the other hand, high HMOX1 expression can also be toxic due to high levels of ferrous iron,139 which in turn accelerates the Fenton reaction, particularly in the context of insufficient levels of free radical scavengers. Together, these reports support the notion that HMOX1 expression has a dose-dependent differential role.140 Targeting HO-1 has been proposed as a viable strategy for treating many diseases and conditions, including cardiovascular disease3 and inflammation.141 Notably, we previously showed that inhibiting HO-1 prevents ferroptosis-induced cardiomyopathy in mice.3 To date, many HO-1 agonists and antagonists have been proposed for use in various disease models. For example, Vreman et al. found that metalloporphyrins—synthetic heme analogs used to treat jaundice in newborn infants—can inhibit HO-1.142 However, several metalloporphyrins can induce phototoxicity and/or off-target adverse effects; given this poor safety profile, azalanstat was subsequently developed as a safer HO-1 inhibitor;143 however, studies are needed to test its efficacy in ferroptosis-related diseases.
Targeting reductive-oxidative pathways
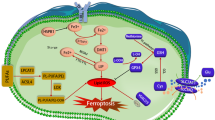
Under physiological conditions, ROS production is controlled by a coordinated network of antioxidative pathways. Among the various cellular antioxidative defense systems, the system Xc–-GSH-GPX4 metabolic pathway plays a key role in regulating ferroptosis. Intracellular cysteine is taken up primarily in the form of cystine via system Xc–144 or converted from methionine via the transsulfuration pathway,145 or is transported directly by alanine/serine/cysteine transporters (known as system ASC).146,147 In addition, the KEAP1/NRF2 antioxidative signaling pathway, the glutaminolysis pathway, the FSP1-CoQ10-NAD(P)H pathway, and the recently identified DHODH-mediated pathway have all been recognized as central mediators of ferroptosis by directly regulating reductive-oxidative pathways. Below, we summarize these ferroptosis modulators that target the reductive-oxidative pathways (see Fig. 4).

Targeting ferroptosis in reductive-oxidative pathways. Ferroptosis is tightly associated with levels of reactive oxygen species (ROS); therefore, homeostasis of the cellular reductive-oxidative response is important for regulating ferroptosis. The system Xc–-GSH-GPX4 pathway is a major ROS scavenger, and numerous molecules are designed to target the components involved in this pathway in order to modulate ferroptosis. The recently identified FSP1-CoQ10-NAD(P)H pathway and mitochondrial DHODH-mediated pathway are also potential targets for modulating ferroptosis. Moreover, NRF2 respond to cellular oxidative status by activating the transcription of genes involved in reductive-oxidative responses. Thus, targeting the KEAP1-NRF2 axis may be a viable strategy for modulating ferroptosis. Molecules listed in pink and green text boxes inhibit or induce, respectively, the indicated reductive-oxidative regulatory pathways, thereby suppressing or triggering, respectively, ferroptosis. CBS cystathionine beta-synthase, CoQ10 coenzyme Q10, CTH cystathionine gamma-lyase, DHODH dihydroorotate dehydrogenase, DPP4 dipeptidyl peptidase 4, FMN flavin mononucleotide, FMNH2 reduced flavin mononucleotide, FSP1 ferroptosis suppressor protein 1, GCL glutamate-cysteine ligase, GLS glutaminase, GLUD1 glutamate dehydrogenase 1, GPX4 glutathione peroxidase 4, GSH glutathione, GSR glutathione disulfide reductase, GSS glutathione synthetase, GSSG glutathione disulfide, KEAP1 Kelch-like ECH-associated protein 1, NOX1 NADPH oxidase 1, NRF2 nuclear factor erythroid 2-related factor 2, SLC solute carrier family, TCA tricarboxylic acid, TXNRD thioredoxin reductase. Created with BioRender.com
Pharmacological perturbation of the system Xc–-GSH-GPX4 pathway
Extracellular cystine imported by system Xc– is subsequently reduced to cysteine, the rate-limiting precursor of GSH,148,149 an essential cofactor for the antioxidant enzyme GPX4, which quenches PL hydroperoxides. GSH is produced from cysteine, glutamate, and glycine exclusively in the cytosol via glutamate-cysteine ligase and glutathione synthetase. Newly synthesized cytosolic GSH is then actively transported into the mitochondria via the recently identified GSH importer SLC25A39.150 Moreover, studies have shown that inhibiting either system Xc– or GPX4 potently induces ferroptosis via cysteine deprivation,1 driving GSH depletion and ultimately impairing the cell’s antioxidant defense mechanism.151
Triggering ferroptosis using system Xc– inhibitors
Erastin is a first-generation ferroptosis inducer that directly suppresses system Xc–1. Although the pro-ferroptotic effects of erastin have been reported in many types of malignant cancer cells in vitro, its further development has been limited due to its low efficacy in vivo. To optimize the chemical properties of erastin, Yang et al. developed a more effective erastin derivative, piperazine erastin, by inserting a piperazine group in the aniline ring of erastin, yielding improved drug-like properties such as increased water solubility and metabolic stability.13 Piperazine erastin was subsequently shown to reduce tumor growth in an HT-1080 xenograft mouse model, with no apparent toxicity.13 However, piperazine erastin showed limited activity against established tumors, perhaps due to its moderate potency.152 To overcome this limitation, imidazole ketone erastin (IKE) was developed and shown to have high (nanomolar) potency and high metabolic stability.152 The tumor-suppressing efficacy of IKE was then demonstrated in SUDHL6 (diffuse large B cell lymphoma) cell xenografts in mice.153 Capitalizing on the fact that IKE is soluble under acidic aqueous conditions, nanoparticles were used as an IKE delivery system to further improve its therapeutic index, with reduced toxicity compared to free IKE.153 Previous studies found that erastin was able to increase the sensitivity of various cancer cell lines to chemotherapy drugs, including doxorubicin, actinomycin D, cisplatin, temozolomide, and cytarabine,154,155,156,157 providing new motivation for exploring the feasibility of combination therapies. Recently, we solved the high-resolution structure of erastin-bound human system Xc– and showed that IKE is a highly potent inducer of ferroptosis, providing a structural basis for designing more effective ferroptosis modulators.158
The anti-rheumatic drug sulfasalazine (SAS) is clinically approved for treating inflammatory arthritis and inflammatory bowel disease. SAS has been reported to suppress the growth of lymphoma cells both in vitro and in vivo through the activation of ferroptosis by suppressing system Xc–(ref. 159). Moreover, a lot of studies have shown that SAS has anti-tumor activity in various xenograft tumor models, including glioblastoma,160 prostate cancer,161 small cell lung cancer,162 pancreatic cancer,163 and TNBC.164 Based on its excellent safety profile in animal studies, several phase I and phase II clinical studies have been initiated. However, various doses of SAS failed to produce a clinical response in malignant glioma, and side effects were reported, including anorexia, gastrointestinal toxicity, and hematological toxicity.165 In addition, long-term high-dose (8–12 g/day) treatment with SAS can induce several adverse side effects and should be avoided; therefore, combination therapy using SAS together with conventional radiotherapy and/or chemotherapy has been proposed as a promising therapeutic strategy.166,167,168,169,170,171,172,173 To mitigate its toxicity, studies involving SAS focus primarily on sensitizing agents and applications using nanoparticles, a novel strategy designed to increase the effectiveness of low-dose SAS.174
Sorafenib (SRF), a small molecular kinase inhibitor, is approved for treating various solid tumors, has well-characterized clinical efficacy and tolerability, and remains the only drug approved to treat advanced HCC. SRF has been shown to trigger ferroptosis in cancer cells by suppressing system Xc–.175,176 However, the relatively limited clinical benefits of SRF and the emergence of drug resistance are major hurdles hampering its further development. On the other hand, several studies found that combination therapies can increase SRF’s anti-tumor activity,177,178 as well as GSH starvation‒based nanoscience for cancer therapy.179,180 Notably, however, Zheng et al. recently reported that SRF failed to increase ferroptosis in various cancer cell lines.181 Thus, the precise role of SRF in ferroptosis, and its clinical value in the context of cancer, remains controversial and poorly understood.
Interestingly, a subset of FDA-approved drugs such as the muscle relaxant lanperisone and the analgesic acetaminophen (N-acetyl-p-aminophenol, or APAP) have been shown to activate ferroptosis by functionally suppressing system Xc–. For example, lanperisone has been shown to effectively target RAS-mutated cancers in vivo without causing overt toxicity.182 APAP is widely used as an analgesic and antipyretic, but can cause dose-dependent hepatotoxicity. Interestingly, Yamada et al. recently showed that APAP-induced hepatotoxicity is attributed to GSH depletion‒induced ferroptosis and proposed this as the predominant mechanism,183 providing new insights into the potential use of APAP in triggering ferroptosis in order to inhibit tumor growth. Indeed, several studies have shown a synergistic therapeutic effect of combining erastin and APAP to treat melanoma and lung cancer xenografts.184,185
GSH synthesis regulators
GSH has a central role in the protection of cells from oxidative damage and toxic reactive species. Therefore, targeting pathways involved in GSH synthesis has been studied extensively as a major strategy for treating ferroptosis-related diseases.
Auranofin (AUR) is a gold (I)‒containing compound used to treat rheumatic arthritis. Recent studies have shown that AUR has therapeutic potential for other diseases and conditions such as cancer, metabolic disease, and infectious and inflammatory diseases.186 Indeed, we provided the first report that AUR can be used in vivo as to induce ferroptosis in mice by suppressing the activity of thioredoxin reductase,22 providing compelling evidence supporting its further clinical testing—either alone or combined with other therapies—for treating ferroptosis-resistant disease conditions such as cancer. To expand the potential clinical applications of AUR, several studies have investigated repurposing AUR; for instance, AUR has been repurposed as an anticancer drug in TNBC cells by inhibiting GSH biosynthesis.187 Recently, we found that malic enzyme 1 (Me1) regulates ferroptosis in hepatic I/R-induced injury, and its inactivation further decreased the production of NADPH and failed to restore GSH synthesis,188 suggesting that targeting Me1 may be a viable strategy for treating ferroptosis-related diseases by altering GSH synthesis.
Buthionine sulfoximine (BSO) activates ferroptosis by inhibiting glutamate-cysteine ligase (GCL).189 Similar to erastin, BSO has also been used to deplete GSH and induce ferroptosis in several cancer cell lines.13,41 Based on early clinical trials, BSO is considered safe but has limited therapeutic benefit for treating refractory malignancies.190 To improve its clinical efficacy, researchers attempted to identify BSO-sensitive cancer types and patients, revealing that low GSH levels can serve as a marker for BSO susceptibility.191
1,3-bis-(2-chloroethyl)-1-nitrosourea (BCNU, also known as carmustine) is a selective glutathione disulfide reductase (GSR) inhibitor192 and is used as a chemotherapy drug for treating brain cancer and lymphoma. Based in its mode of action, BCNU is believed to induce ferroptosis by directly inhibiting GSH synthesis. Recently, a combination of BCNU and sorafenib was shown to significantly suppress the in vivo growth of liver cancer by promoting ferroptosis.193 In addition, both BCNU and AUR have been shown to cause cell death in oxidant-enriched tumorigenic endothelial (EOMA) cells,194 possibly by activating ferroptosis. These findings may therefore increase the clinical application of BCNU for the treatment of HCC and endothelial cell tumors.
N-acetylcysteine (NAC)195 is clinically approved to treat APAP overdose. As an antioxidant, NAC has been shown to inhibit ferroptosis by targeting cysteine metabolism. In addition to protecting the liver from APAP-induced ferroptosis, NAC has also been clinically shown to improve neurodegeneration-related symptoms by increasing cysteine levels and facilitating the synthesis of γ-glutamyl-cysteine and GSH.196 Due to its poor bioavailability, however, NAC must be administered in fairly high doses and requires a long treatment time in patients with severe APAP overdose, thereby increasing the risk of an anaphylactoid reaction (i.e., nonimmunologic anaphylaxis), fluid overload, and high fluid osmolarity.197 To overcome these issues, N-acetylcysteine amide (NACA), a modified form of NAC with increased membrane permeability198 and bioavailability (67% compared to only 15% for NAC),199 has been developed. To date, NACA has been shown to have antioxidant activity in several preclinical models, but is yet to be approved for clinical use.200
Promoting ferroptosis using GPX4 inhibitors
The essential antioxidant enzyme GPX4 is not only responsible for maintaining redox homeostasis, but is also recognized as the “ferroptosis gatekeeper” by transforming lipid ROS into lipid alcohols. Direct inactivation of GPX4 has been shown to drive ferroptosis independent of intracellular cysteine and GSH levels.13 Thus, numerous studies have focused on identifying novel activators of ferroptosis by targeting GPX4 in order to develop new cancer therapeutics.
As a covalent inhibitor of GPX4, RSL3 was originally identified as a pro-ferroptosis compound through chemical screening.1 Although sharing the more common features of ferroptosis, RSL3-mediated ferroptosis does not include decreased levels of cellular GSH. GPX4 was subsequently identified as the primary target of RSL3 using an unbiased, affinity-based chemoproteomics approach.13 Moreover, genetically knocking down and overexpressing GPX4 were shown to cause sensitization and resistance to RSL3, respectively, providing further evidence that GPX4 is the target of RSL3.13 Since these previous studies, RSL3 has been widely used as a ferroptosis agonist in vitro in a wide range of cancer cell types, including pancreatic cancer, adrenocortical carcinoma, fibrosarcoma,201 and breast cancer15 cells. In addition, RSL3 may also serve as a chemosensitizer for cisplatin, doxorubicin, and actinomycin D by activating ferroptosis in various cancer cells, including neuroblastoma,202 osteosarcoma,155 lung cancer,203 and rhabdomyosarcoma cell lines.154 In mouse xenograft models, RSL3 is well-tolerated, with no overt toxicity or body weight loss observed even at the relatively high dose of 400 mg/kg.13,204,205 As an experimental tool, RSL3 has been used extensively to study the role of ferroptosis in various disease models, including AKI, I/R-induced injury, and neurodegenerative disease.206,207,208
Similar to RSL3, ML162 was identified as a GPX4 inhibitor,209 and subsequent studies showed that ML162 can directly bind GPX4 and inhibit its activity.13 However, both RSL3 and ML162 have relatively poor selectivity and pharmacokinetics, as they covalently bind to GPX4 via a reactive alkyl chloride moiety.210 To develop a new GPX4 inhibitor containing a different moiety, Eaton et al. used masked nitrile-oxide electrophiles to create ML210, a more selective covalent suppressor of GPX4 with improved pharmacokinetics and comparable activity.211 However, given that the in vivo efficacy of all GPX4 inhibitors developed to date remains limited, efforts have focused on developing more efficacious GPX4-selective inhibitors.
Based on the structural optimization of CIL56 (caspase-independent lethal 56), FIN56 was developed as a highly selective inducer of ferroptosis that promotes GPX4 degradation as well as the GPX4-independent activation of squalene synthase (SQS).212 FIN56 potently induces ferroptosis in all TNBC cell lines tested to date, suggesting that FIN56 may be an effective additional pro-ferroptosis anticancer drug.213 In contrast, the endoperoxide-containing 1,2-dioxolane FINO2 was shown to potently and selectively induce ferroptosis in engineered cancer cells by directly oxidizing iron and indirectly inactivating GPX4.214 In addition, the naturally occurring C28 steroidal lactone withaferin A (WA), one of the best-studied withanolides, has a diverse range of pharmacological activities, including anti-inflammatory, anti-tumor, and antioxidant properties. In both high-risk neuroblastoma cell lines and neuroblastoma xenografts, WA has been shown to dose-dependently induce ferroptosis via the dual mechanisms of inactivating GPX4 and activating the NRF2 pathway.202 Thus, natural products may serve as an additional resource for the discovery of new GPX4 inhibitors.
Recently, Wu et al. identified creatine kinase B (CKB)-enhanced GPX4 as a novel mechanism for regulating ferroptosis, accounting for the resistance of HCC cells to ferroptosis by stabilized GPX4 via CKB phosphorylated GPX4 at residue S104.215 Mutations in either CKB or GPX4 significantly increase the ferroptosis-mediated cell death of HCC by inhibiting this phosphorylation.215 Importantly, the authors showed a positive correlation between phosphorylated GPX4 and HCC aggressiveness in clinical samples,215 suggesting a potential new strategy for treating HCC by blocking CKB/GPX4-mediated ferroptosis.
Although GPX4 inhibitors potently inhibit cells growth in vitro,216 all currently identified GPX4 inhibitors have limited prospects for further clinical development due to their poor pharmacokinetics and specificity. Therefore, more studies are needed in order to develop GPX4-specific inhibitors with improved pharmacological properties.
Suppressing ferroptosis using GPX4 activators
The essential micronutrient selenium (Se) is required for the production of selenocysteine,217 which serves as the active site for GPX4.16 Ionic selenite (SeO32-) is commonly used to deliver Se to cultured cells. For example, treating cultured neurons with SeO32- was shown to increase GPX4 transcription and inhibit ferroptosis induced by either hemin or homocysteic acid.74 In addition, systemically treating mice with Tat SelPep, a selenocysteine-containing peptide that can cross the BBB, was shown to improve functional recovery following hemorrhagic and ischemic stroke by blocking ferroptosis.74 Together, these studies suggest that Se supplementation may help protect against ferroptosis-related tissue damage and disease.
The neurotransmitter dopamine has many physiological roles, particularly in controlling various functions in the central nervous system, including movement, memory, motivation, mood, and attention. Insufficient levels of dopamine production are known to contribute to the progression of PD, and dopamine-based therapies such as levodopa and dopamine receptor agonists have been used clinically to treat PD and various cardiovascular conditions. Interestingly, non-oxidative dopamine was reported to potently inhibit erastin-induced ferroptosis in both cancerous and non-cancerous cells by stabilizing GPX4,218 suggesting that dopamine may be a promising candidate drug for alleviating ferroptosis-related tissue injury and disease, as well as certain neurodegenerative diseases.
Using a combination of computational prediction and experimental validation, Li et al. discovered eight new potential GPX4 activators and found that compound 1d4 was the most potent allosteric activator of GPX4, significantly increasing GPX4 activity by 50% when applied at 20 μM in a cell-free assay and 61 μM in cell extracts; moreover, they found that compound 1d4 potently inhibited ferroptosis when applied to HT-1080 fibrosarcoma cells.219
The diterpenoid triepoxide curculigoside is one of the primary bioactive phenolic compounds isolated from Curculigo orchioides Gaertn (commonly known as “Kali Musli”), an ancient medicinal plant well known for its immunomodulatory and rejuvenating effects. Recently, Wang et al. reported that curculigoside inhibits ferroptosis in IEC-6 cells (a cell line used to model intestinal iron transport) by upregulating GPX4 expression.220 In mice with dextran sulfate sodium (DSS)‒induced ulcerative colitis, curculigoside was shown to protect against disease progression by reducing ferroptosis via GPX4 induction,220 suggesting that activating GPX4 is a viable therapeutic option for ulcerative colitis. Another plant-derived bioactive compound, puerarin, is an isoflavone glycoside isolated from Pueraria lobata (also known by its Chinese name, Gegen) with various pharmacological effects,221 including antioxidant, anticancer, and anti-inflammation properties, in addition to alleviating pain, promoting bone formation, attenuating insulin resistance, and exerting cardiac and neuronal protective effects. However, its molecular target and mechanism remain unknown. Interestingly, the anti-ferroptosis activity of puerarin in heart disease has been linked to the induction of FTH1 and GPX4 in rats.222 Given that the majority of bioactive natural compounds have relatively low bioavailability, future studies should focus on developing rationally designed combination therapies and/or their incorporation in nanoparticles.
Targeting the p62-KEAP1-NRF2 signaling pathway
The p62-KEAP1-NRF2 signaling pathway plays a regulatory role in response to oxidative stress, environmental insult, and toxic chemicals. In response to oxidative stress, p62 activates NRF2 by directly binding to its ubiquitin ligase adapter KEAP1; as a result, NRF2 translocates to the nucleus and regulates cellular redox homeostasis by modulating the expression of target genes, including several genes encoding enzymes involved in both GSH synthesis and iron homeostasis.223 Thus, NRF2 signaling is important for regulating ferroptosis.
The polar hydrophilic alkaloid trigonelline can be extracted from many plant species, including coffee beans and fenugreek seeds, and has been shown to significantly sensitize cancer cells to ferroptosis inducers by inhibiting NRF2.34,204,224 In addition, sitagliptin, a selective inhibitor of dipeptidyl peptidase 4 (DPP4) used to treat type 2 diabetes, was shown recently to inhibit ROS production, inflammation, and excessive autophagy by promoting Nrf2 translocation to the nucleus in mouse models of acute lung injury induced by severe pancreatitis.225 In addition, a modest dose of WA was shown to induce ferroptosis and increase the LIP by directly targeting KEAP1, mediating the NRF2-mediated upregulation of HO-1.202
Additional reductive-oxidative pathways involved in ferroptosis
Other reductive-oxidative pathways, including the transsulfuration, FSP1-CoQ10-NAD(P)H, glutaminolysis, and DHODH-mediated pathways, have also been linked to ferroptosis and are discussed in detail below.
The transsulfuration pathway
When cysteine availability is restricted, the transsulfuration pathway is activated in order to increase the synthesis of cysteine from methionine. The enzyme cystathionine beta-synthase (CBS) activates the first step in this pathway, followed by the formation of cystathionine into cysteine by the enzyme cystathionine gamma-lyase (CTH). Sustained activation of the reverse transsulfuration pathway has been implicated in the resistance of ovarian cancer cells to erastin-induced ferroptosis.226 CH004, a pharmacological inhibitor of CBS, has been identified as a novel stimulator of ferroptosis in liver cancer both in vitro and in vivo,227 providing experimental evidence to support the therapeutic potential of targeting the transsulfuration pathway in liver cancer. Similarly, the CTH inhibitor propargylglycine has been shown to sensitize NSC-34 cells to both erastin- and RSL3-induced ferroptosis.228 In mice, propargylglycine was also shown to increase susceptibility to APAP-induced liver damage and mortality,229 possibly by inducing ferroptosis.
The FSP1-CoQ10-NAD(P)H pathway
The FSP1 gene was initially identified as a pro-apoptotic gene,230 but has also been identified as an anti-ferroptosis gene via FSP1’s oxidoreductase activity, which reduces CoQ10 to ubiquinol (CoQ10H2).17,18 Moreover, the FSP1 inhibitor iFSP1 was shown to selectively activate ferroptosis in GPX4-knockout FSP1-overexpressing cells and was identified by screening 10,000 drug-like compounds;17 iFSP1 was also shown to potently increase the sensitivity of cancer cells to RSL3-induced ferroptosis.17 Recently, Yoshioka et al. identified the novel compound called NPD4928 as an FSP1 inhibitor;231 the authors then showed that NPD4928 increased the cytotoxicity of various cell types in response to GPX4 antagonists, indicating that NPD4928 and GPX4 inhibitors may work synergistically to treat cancer by inducing ferroptosis.231 Most recently, Hendricks et al. developed another FSP1 inhibitor called FSEN1, which they found sensitized various cancer cells to ferroptosis.232 Together, these findings suggest that pharmacologically inhibiting the FSP1-CoQ10-NAD(P)H pathway may provide a plausible strategy for sensitizing cancer cells to ferroptosis-resistant therapeutic agents. On the other hand, Fang et al. recently identified the diphenylbutene derivative compound 3f as a ferroptosis inhibitor, and they showed that this compound can protect against ischemic stroke in rats by increasing FSP1 protein levels,233 suggesting that novel agents that upregulate FSP1 may be used to treat ferroptosis-related diseases.
The glutaminolysis pathway
Cellular uptake of the amino acid glutamine (Gln) is mediated primarily by specific transporters such as SLC38A1 and SLC1A5. Following its cellular uptake, intracellular Gln is converted by glutaminase (GLS) to produce glutamate, which can be further catalyzed to α-ketoglutarate either by glutamate dehydrogenase (GLUD1)‒mediated deamination or by transaminase-mediated transamination.234 Given that this reaction promotes ferroptosis by causing an accumulation of lipid peroxidation,25 blocking the glutaminolysis pathway has been proposed as a potential therapeutic approach for treating ferroptosis-induced tissue damage.
Inhibiting glutaminolysis using L-g-glutamyl-p-nitroanilide (GPNA, an inhibitor of the SLC38A1/SLC1A5 complex), compound 968 (an inhibitor of GLS), or amino-oxyacetic acid (AOA, an inhibitor of pan-transaminases) has been shown to block cystine deprivation‒induced ferroptosis in mouse embryonic fibroblasts, melanoma cells, as well as head and neck cancer cells.25,235,236 In addition, compound 968 was shown to potently prevent I/R-induced heart damage in an ex vivo model.25
Recent studies showed that low-concentration paclitaxel (PTX, a mitotic chemotherapeutic drug) suppressed cancer cell proliferation by promoting the production of lactate and changing the pH of the tumor microenvironment by downregulating glutaminolysis-related genes such as GLS, SLC7A11, and SLC1A5.237,238 In addition, low-dose PTX was shown to trigger ferroptosis by decreasing SLC7A11 expression by upregulating the well-characterized transcription factor p53(ref. 238). Moreover, a growing body of evidence suggests that p53 regulates ferroptosis either by transcriptional or posttranslational mechanisms such as modulating the downstream expression of SLC7A11,239 SAT1 (encoding spermidine/spermine N1-acetyltransferase 1),240 GLS2 (encoding glutaminase 2),25 and CDKN1A/p21 (encoding cyclin dependent kinase inhibitor 1A),241 or by directly inhibiting DPP4 activity in colorectal cancer cells.242
The DHODH-mediated pathway
The flavin-dependent mitochondrial enzyme DHODH catalyzes the de novo synthesis of pyrimidine.243 Originally, targeting DHODH was shown clinically to improve autoimmune diseases such as multiple sclerosis and rheumatoid arthritis. Although functional studies characterized DHODH as a potential target for treating cancer, several potent DHODH suppressors such as brequinar, leflunomide, and teriflunomide were clinically assessed but failed to receive FDA approval. Importantly, however, brequinar was recently shown to act synergistically with the FDA-approved drug SAS to suppress tumor growth by potently inducing ferroptosis,20 suggesting that DHODH may indeed serve as a viable target for treating cancer. Interestingly, DHODH inhibitors have also been developed as antiviral agents to act against cytomegalovirus, Ebola, influenza, and SARS-CoV-2(refs. 244,245). Whether ferroptosis is involved in the multifaceted activities of DHODH inhibitors remains an open question; nevertheless, targeting DHODH may have promise as a novel therapeutic approach for cancer and certain viral infections.
Targeting lipid metabolic pathways
Lipid metabolism has an important regulatory role in the balance between cell survival and cell death. In ferroptosis, the majority of oxygenated PL species are upregulated, resulting in damage to PL-containing cell membranes. Four types of PLs—namely, the double and triple oxygenated arachidonic acid (AA)- and adrenic acid (AdA)-containing phosphatidylethanolamine (PE) species (C18:0/C20:4 and C18:0/C22:4)—were previously identified as the most susceptible substrates for lipid peroxidation.30 In addition to PE, other PLs can also be oxidized upon the induction of ferroptosis. Because lipid metabolic pathways are important for regulating lipid peroxidation, targeting these pathways may provide a novel strategy for treating ferroptosis-related diseases (Fig. 5).

Targeting ferroptosis in lipid metabolism pathways. Cellular fatty acids are taken up by CD36 and FABPs, and stored in the free fatty acid (FFA) pool. Cells can also take up cholesterol via the LDLR or produce it from acetyl-CoA. These fatty acids can then be elongated to form long fatty acids and can be unsaturated to form monounsaturated fatty acids (MUFAs) or polyunsaturated fatty acids (PUFAs). MUFAs can be converted to PUFAs via the activity of FADS. ACSL4 and LPCAT3 are key enzymes that promote the incorporation of PUFAs into phospholipids (PLs) to form PL-PUFAs and induce ferroptosis. Molecules in the pink and green text boxes inhibit or induce, respectively, the indicated components in the lipid metabolism regulatory pathways, thereby suppressing or triggering, respectively, ferroptosis. ACC acetyl-CoA carboxylase, ACSL4 acyl-CoA synthetase long-chain family member 4, ALOX arachidonate lipoxygenase, CD36 cluster differentiation 36, FABP fatty acid binding protein, FADS fatty acid desaturase, FASN fatty acid synthase, HMG-CoA 3-hydroxy-3-methylglutaryl-CoA, HMGCR 3-hydroxy-3-methylglutaryl-CoA reductase, LDLR low density lipoprotein receptor, LPCAT3 lysophosphatidylcholine acyltransferase 3, PEBP1 phosphatidylethanolamine binding protein 1, SCD1 stearoyl-coenzyme A desaturase 1, SQLE squalene epoxidase, SQS squalene synthase. Created with BioRender.com
A haploid genetic screen in KBM7 cells (a chronic myeloid leukemia cell line) revealed that the enzymes ACSL4 and LPCAT3 serve as ferroptosis regulators.29 ACSL4 and LPCAT3 catalyze the addition of CoA to the long-chain polyunsaturated bonds of AA and AdA, respectively, thereby promoting the esterification of PUFAs to membrane PLs.29,30 PEs containing acylated AA or AdA (AA/AdA-CoA) can be subsequently oxidized by LOXs to form PL hydroperoxides (PE-AA/AdA-OOH), eventually leading to the membrane deposition of lipid ROS and driving ferroptosis.246 A number of inhibitors that target lipid metabolism have been shown to suppress ferroptosis, including LOX inhibitors, radical-trapping antioxidants (RTAs, which scavenge lipid hydroperoxyl radicals), ACSL4 inhibitors, and deuterated PUFAs/MUFAs (by decreasing PUFA-containing PLs).
Inhibiting ferroptosis using LOX inhibitors
LOXs are a class of non-heme iron-containing dioxygenases247 that can regulate ferroptosis by mediating PL oxidation.248 A total of 6 LOX isoforms (ALOX5, ALOX12, ALOX12B, ALOX15, ALOX15B, and ALOXE3) are expressed in humans, and a total of 7 LOX isoforms (Alox5, Alox12, Alox12b, Alox15, Alox15b, Aloxe3, and Alox12e) are expressed in mice.249,250 ALOX5/12 inhibitors, including AA-861,31 cinnamyl-3,4-dihydroxy-alpha-cyanocinnamate (CDC),251,252 the pan-LOX inhibitor nordihydroguaiaretic acid (NDGA),253 and BWA4C,248 have been either shown or suggested to suppress ferroptosis. In an effort to re-evaluate the selectivity profile of CDC for LOXs, Pergola et al. identified CDC as an ALOX5-specific antagonist with an IC50 value in the low nanomolar range (9–25 nM) measured in cell-free assays.251 Zileuton, another widely used ALOX5-selective inhibitor, was the first FDA-approved orally administered drug for treating asthma. Zileuton was also shown to exert a neuroprotective effect by blocking ALOX5-mediated glutamate toxicity and ferroptosis in HT22 cells (a mouse hippocampal cell line).254 With respect to ALOX12, ML355 is a potent and selective inhibitor255 that has been shown to reduce ALOX12-induced thrombosis and protect human pancreatic islets from ALOX12-triggered inflammatory injury.256,257 Recently, Zhang et al. reported that the compound IMA-1, which disrupts the interaction between ALOX12 and ACC1 (acetyl-CoA carboxylase 1), can reduce NASH progression in mice and Cynomolgus macaques,258 suggesting that the ALOX12-ACC1 complex may be a clinically viable target for the management of NASH.
ALOX15 was recently reported to play an essential role in exacerbating I/R-induced cerebral injury259 and ischemia-induced myocardial injury260 by driving the peroxidation of PL-PUFAs and inducing ferroptosis, providing compelling evidence supporting the viability of targeting ALOX15 to treat I/R-induced injury in these tissues. Interestingly, Walters et al. functionally confirmed that ALOX15 is involved in inducing oxidative stress in human spermatozoa, supporting the notion of increased ALOX15 expression in sperm cells261 and suggesting that ALOX15 may be a potential target for the treatment of oxidative stress‒induced male infertility.261 Indeed, the ALOX15-selective inhibitor PD146176 was shown to protect against oxidative damage by decreasing the production of membrane PL peroxidation and 4-hydroxynonenal (4-HNE) in spermatozoa.261 Moreover, the ALOX15-specific inhibitor ML351 was recently shown to reduce ferroptosis in cardiac I/R-induced injury.262
Taken together, various LOX inhibitors have been shown to effectively inhibit ferroptosis. In addition, the enzyme POR (cytochrome P450 oxidoreductase) was recently shown to promote PL peroxidation in a LOX-independent manner,263 suggesting that the regulation of enzymes other than LOXs may also contribute to lipid peroxidation by mediating the oxygenation of membrane PLs.
Suppressing ferroptosis using radical-trapping antioxidants (RTAs)
One of the principal features of ferroptosis, namely PL peroxidation initiated by the formation and propagation of lipid radicals, can be blocked by RTAs, a class of lipid chain‒breaking antioxidants. Among the various RTAs identified to date, the now-widely used ferrostatin-1 (Fer-1) was originally identified as a potent ferroptosis-specific inhibitor in erastin- and RSL3-treated HT-1080 cells.1 By scavenging free radicals, Fer-1 reduces ferroptosis-induced tissue injury and prolongs survival in mouse models of disease.3,47,64 However, Fer-1 has not been pursued in drug development, given its metabolic instability and poor pharmacokinetics. Currently, Fer-1 is used as a research tool for studying various ferroptosis-related processes. Compared to Fer-1, second-generation (SRS11-92) and third-generation (SRS16-86) RTAs have significantly higher activity but still contain an ester moiety.47,64 To develop an optimized set of RTAs, Devisscher et al. developed a series of compounds in which the ester was replaced with a sulfonamide, providing improved water solubility and better metabolic and kinetic properties.264 For example, they found that compound UAMC-3203 was more stable and more potent than Fer-1 (with a significantly lower IC50 compared to Fer-1).264 After intravenous injection, the terminal plasma half-life of UAMC-3203 is ~3 and 5 h in mice and rats, respectively,83 suggesting that this compound has relatively good pharmacokinetics.
Liproxstatin-1 (Lip-1), a spiroquinoxalinamine derivative, was identified by high-throughput screening and shown to inhibit ferroptosis in vivo with better pharmacological properties than Fer-1.5 However, Lip-1 also potently inhibits the enzymatic activity of CYP2D6 (with an IC50 of 4.1 μM), a member of the cytochrome P450 superfamily of drug-oxidizing enzymes, indicating that Lip-1 may not be suitable for use in clinical trials.5
In addition to inhibiting ferroptosis, potent RTAs such as Fer-1 and Lip-1 can insert into membrane PLs via arylamines. Notably, the catalytic activity of both Fer-1 and Lip-1 requires a relatively high temperature.265 Using three different cellular models of ferroptosis, Zilka et al. found that tetrahydronapthyridinols (THNs) inhibit ferroptosis induced by RSL3, cystine deprivation, and GPX4 inactivation.265 Compared to both Fer-1 and Lip-1, lipophilic THNs have similar potency, while hydrophilic THNs are ineffective at inhibiting ferroptosis.265
Diarylamines are RTAs commonly used to reduce the autoxidation of petroleum-derived products. The efficacy of this class of RTAs depends largely on the kinetics and stability of the rapid hydrogen atom transfer to one-electron oxidation by peroxidic species.266 Two diarylamine derivatives—phenothiazine and phenoxazine—have been shown to suppress ferroptosis in mouse embryonic fibroblasts.267 Interestingly, each phenoxazine molecule can trap >2 peroxyl radicals at moderate temperatures, suggesting new strategies for the development of more potent RTA-based ferroptosis inhibitors. Indeed, Mishima et al. showed that promethazine is more potent at protecting kidney function compared to Fer-1(ref. 268), suggesting that promethazine may be a viable tool for studying the role of ferroptosis in various disease models. Notably, edaravone—an RTA clinically approved for treating acute ischemic stroke and amyotrophic lateral sclerosis135—has been shown to protect against ferroptosis under various pathological conditions.269 In addition, Zilka et al. found that copper(II)-diacetyl-bis(N4-methylthiosemicarbazone) (CuATSM), a candidate drug for treating ALS and PD, suppresses ferroptosis via its RTA activity.270 Recently, a group of reduced forms of vitamin K (VKH2), including menaquinone and phylloquinone, was shown to have potent anti-ferroptosis properties,21 providing new mechanistic insights into the anti-ferroptotic activity of alternative RTAs.
Recently, Wu et al.271 and Barayeu et al.272 independently reported that endogenous sulfane sulfur (S0) species/hydropersulfides (H2S or RSSH) are potent RTAs. By supplying sulfur for S0 biosynthesis, cysteine can inhibit ferroptosis via a GPX4-independent mechanism.272 Genetic manipulation of enzymes involved in S0 biosynthesis clearly implicate S0 as playing a role in regulating ferroptosis by scavenging ROS and suppressing lipid peroxides.272 Both endogenous and exogenous S0 have been shown to provide cellular protection against ferroptosis.271,272 Together, these results suggest that regulating ferroptosis by targeting S0 warrants further clinical study.
Reducing ferroptosis using ACSL4 inhibitors
ACSL4 is a unique and important isozyme involved in the metabolism of PL-PUFAs such as AA, and deleting or inhibiting ACSL4 blocks ferroptosis by preventing the incorporation of PL-PUFAs into cell membranes.15,30 Thiazolidinediones (TZDs) such as rosiglitazone, pioglitazone, and troglitazone were originally identified as agonists of peroxisome proliferator-activated receptor γ (PPARγ) and are approved to treat adult type 2 diabetes. Interestingly, TZDs showed effects on suppressing ferroptosis by selectively inhibiting ACSL415,273 in breast cancer cell lines and reduce mortality due to acute renal failure in kidney-specific Gpx4 knockout mice.15
Modulators of lipid composition
Similar to ACSL4 inhibitors, deuterated PUFAs and MUFAs can also inhibit ACSL4 by decreasing PL-PUFAs. PUFAs deuterated at the bis-allylic position (D-PUFAs) have been shown to inhibit ferroptosis in cells and animal models of PD and Friedreich’s ataxia.31,274,275,276 Notably, exogenous lipid supplementation can modulate both apoptosis and ferroptosis.277 Magtanong et al. showed that exogenous MUFAs can also specifically reduce the accumulation of lipid ROS in the plasma membrane and can displace PUFAs from their cellular location, suppressing ferroptosis in an ACSL3-dependent manner.277
Molecules that affect the composition of intracellular lipids may also regulate ferroptosis. For example, sulfosuccinimidyl oleate (SSO), which irreversibly inhibits the fatty acid receptor CD36, has been shown to inhibit ferroptosis in CD8+ T cells by blocking the uptake of oxidized lipids.278 Moreover, inhibiting stearoyl-CoA desaturase-1 (SCD1) with CVT-11127 or inhibiting acetyl-CoA carboxylase (ACC) with CP-640186 can affect the production of MUFAs, reducing the growth of lung cancer cells.279 Thus, Batchuluun et al. proposed that other potent ACC inhibitors should be examined for their ability to affect ferroptosis.280
Recently, Minami et al. found that loss of CDKN2A expression remodels the lipidome of glioblastoma cells, and patient-derived CDKN2A-deficient glioblastoma cells have higher levels of lipid peroxidation, thereby sensitizing the cells to ferroptosis in response to GPX4 inhibition.281 These results suggest potential new therapeutic strategies in which targeting cellular lipidome remodeling can induce ferroptosis in glioblastoma cells, particularly in cells lacking CDKN2A expression.
The mevalonate pathway
The mevalonate pathway regulates ferroptosis by altering GPX4 production by affecting the maturation of selenocysteine tRNA.108 The small molecule FIN56 potently induces ferroptosis via two mechanisms, namely by reducing GPX4 levels and by decreasing CoQ10 activity by targeting the enzyme SQS, which acts downstream of HMG-CoA (3-hydroxy-3-methylglutaryl-CoA) reductase in the mevalonate pathway.212,213
Statins are well-known inhibitors of HMG-CoA reductase (HMGCR) and are commonly used as cholesterol-lowering medications to prevent and/or treat atherosclerosis. Statins have also been shown to increase FIN56-induced cellular ferroptosis212 and selectively kill cancer cells in a high-mesenchymal state.40 However, the concentration of statins needed to increase ferroptosis in cancer cells is 100–500 times the recommended dose for decreasing cholesterol, and high-dose statins often cause severe side effects such as myalgia, rhabdomyolysis, and hepatotoxicity.282,283 To mitigate these side effects, it may be possible to deliver the statins in nanoparticles, leading to a high concentration specifically in tumor cells.284
Additional strategies for targeting ferroptosis
Although numerous ferroptosis-targeting agents have been studied, their properties often make them poorly suited for use as therapeutic compounds; indeed, their low solubility, high metabolic clearance, low cellular permeability, and systemic toxicity limit their clinical development. Therefore, multi-target ferroptosis modulators and nanomaterial technologies have been investigated in an attempt to target ferroptosis more effectively in vivo.
Multi-target ferroptosis modulators
Artemisinins, which are derived from sweet wormwood (Artemisia annua) extracts, are commonly used as effective antimalarial drugs. The semi-synthetic derivative and major active metabolite of artemisinin, dihydroartemisinin (DHA), has been shown to increase ferroptosis in lung cancer cells by inactivating the PRIM2/SLC7A11 axis,285 as well as in head and neck cancers286 and glioblastoma cell lines287 by inhibiting GPX4. In addition, DHA plays a role in ferritinophagy, thereby inducing free iron‒induced ferroptosis.288
Glycyrrhizin, a natural antioxidant extracted from the glycyrrhiza root,289 was shown to have protective effects on the liver via its antioxidative, anti-inflammatory, antifibrotic, and antiviral properties.195,290,291 Wang et al. showed that glycyrrhizin can prevent ferroptosis in cells and in an animal model of acute liver failure by regulating iron levels, the GSH/GPX4 pathway, and the NRF2/HO-1/HMGB1 pathway.292
The flavonoid baicalein (in Chinese: Huangqin) has long been used as a popular antibacterial and antiviral agent.293 Near the end of the last century, Afanas’ev et al. reported that baicalein exerts its protective role against oxidative damage by inhibiting the iron-catalyzed Fenton reaction.294 In addition, baicalein is a specific antagonist of ALOX12 and was shown to protect against I/R-induced heart injury in mice.295 Subsequently, Leyen et al. proposed that baicalein may also protect against I/R-induced brain injury by inhibiting ALOX12/15 (ref. 296). Recently, baicalein was shown to prevent ferroptosis in erastin-treated PANC1 cells (a human pancreatic cancer cell line) by reducing ferrous iron, inhibiting GSH consumption, and inhibiting GPX4 degradation, thereby suppressing lipid peroxidation.297 These results suggest that baicalein may prevent ferroptosis-associated tissue damage.
Targeting ferroptosis using nanoparticles
Compared to small-molecule compounds, ferroptosis-inducing nanoparticles have been suggested to have a better preclinical profile. Importantly, nanoparticles can be loaded with chemotherapeutic drugs or tumor-selective molecules, making them ideally suited for developing nanoparticle-based cancer therapies as well as combination therapies.
Many nanoparticles have been designed to increase ferroptosis by triggering the Fenton reaction in targeted tumor cells (Fig. 6). For instance, the magnetic nanoparticle FeGd-HN@Pt@LF/RGD2,298 the PEGylated single-atom Fe-containing nanocatalyst PSAF,299 the nanoprobes UCNP@GA-FeIII300 and UCNP@LP(Azo-CA4),301 and the nano-platforms HA@MOF302 and PB@FePt-Ha-g-PEG303 can effectively deliver Fe2+ to cancer cells in order to accelerate the Fenton reaction and produce lethal amounts of ROS, specifically triggering ferroptosis. Interestingly, Zhang et al. found that the FePt@MoS2 nanocomposite efficiently induces ferroptosis by releasing >30% of loaded Fe2+ into the tumor microenvironment within 72 h.304 In addition, GOD-Fe3O4@DMSNs, in which the nanoparticle is loaded with a natural glucose oxidase, has been shown to exhaust glucose in tumor cells and generate excessive levels of H2O2, thereby accelerating the Fenton reaction catalyzed by Fe3O4(ref. 305). In the relatively acidic tumor microenvironment, several nanoparticles can be reduced in order to release two molecules such as iron and doxorubicin.306,307 For example, FePt-NP2 can deliver iron oxide in order to enhance the chemotherapeutic effects of cisplatin specifically in cancer cells.308 Moreover, Shen et al. designed FeGd-HN@Pt@LF/RGD2, a cisplatin-loaded Fe3O4/Gd2O3 hybrid nanoparticle conjugated to lactoferrin (LF) and an RGD (Arg-Gly-Asp) dimer in order to potently induce ferroptosis specifically in brain tumor cells.298 Thanks to its small size (6.6 nm) and LF receptor‒mediated transcytosis, FeGd-HN@Pt@LF/RGD2 nanoparticles can cross the BBB and deliver cisplatin together with reactants (e.g., Fe2+, Fe3+, and H2O2) to brain tumor cells, significantly slowing the growth of orthotopic brain tumors.298

Five strategies for developing nanoparticles to target ferroptosis. Five principal strategies have been used to design nanoparticles to modulate ferroptosis. In all five strategies, nanoparticles contain compounds that either directly modulate ferroptosis or affect signaling pathways that modulate ferroptosis. GSH, glutathione; GPX4, glutathione peroxidase 4; ROS, reactive oxygen species. Created with BioRender.com
An alternative nanoparticle-based strategy for triggering ferroptosis is to deplete GSH and/or inhibit GPX4 (Fig. 6). For example, arginine-rich manganese silicate nanobubbles have been shown to activate ferroptosis by depleting GSH and subsequently inactivating GPX4.309 SRF@FeIIITA is a rationally designed nanoparticle spontaneously formed by a Fe3+ and tannic acid (TA) network‒like corona onto a nanocore containing the kinase inhibitor sorafenib (SRF). In the acidic lysosomal environment, SRF@FeIIITA releases SRF, thereby inhibiting the production of GSH and inducing ferroptosis.310 Another SRF-based nanoparticle, FaPEG-MnMSN@SFB, has been validated to effectively activate ferroptosis and suppress tumor growth via the dual GSH-depleting effects of MnMSN (manganese-loaded mesoporous silica nanozyme) and SRF.179 Moreover, SRF@MPDA-SPIO nanoparticles were generated using SRF and ultrasmall superparamagnetic iron oxide (SPIO) nanoparticles, making the particles visible on MRI and potently inducing ferroptosis by supplying iron.311 In addition, the biomimetic magnetic nanoparticle Fe3O4-SAS@PLT was generated using SAS-loaded mesoporous magnetic nanoparticles (Fe3O4) and a platelet (PLT) membrane camouflage, thereby activating ferroptosis by blocking the system Xc− pathway.174 Notably, Fe3O4-SAS@PLT‒induced ferroptosis significantly increased PD-1 (programmed cell death 1)‒based cancer immunotherapy and long-term tumor elimination in mice transplanted with 4T1 metastatic breast cancer cells.174 Recently, Li et al. reported the development of the multifunctional nanosheet system Au/Cu-TCPP(Fe)@RSL3-PEG-iRGD, which triggers ferroptosis in tumor cells by simultaneously blocking the GSH/GPX4 and FSP1/CoQ10H2 pathways via its nanocatalytic activity, as well as synergizing with RSL3 to further inactivate GPX4 in tumor cells.312
Interestingly, nanoparticles can also be designed to directly deliver molecules that trigger ferroptosis (Fig. 6). For example, αMSH-PEG-C’ are ultrasmall (<10 nm in diameter) PEG-coated silica nanoparticles conjugated to MC1-R (melanocortin-1 receptor) in order to target alpha melanocyte‒stimulating hormone (αMSH) peptides; this nanoparticle was then shown to activate ferroptosis in a variety of starved cancer cells, as well as in mice xenografted with 786-O and HT-1080 cells.313 In addition, MON-p53, an encapsulated metal-organic network (MON) containing a plasmid expressing the tumor suppressor p53, is designed to treat cancer by switching apoptosis to ferroptosis, and long-term treatment (i.e., for 75 days) was shown to have potent anticancer activity in tumor-bearing mice.314 Similarly, RSL3 micelles—nanoparticles composed of an AA-conjugated amphiphilic copolymer encapsuling RSL3—have been shown to increase the efficacy of RSL3 in vivo, providing a novel approach to overcome multidrug resistance and improve cancer treatment.315 Moreover, a nanodrug consisting of nanoparticle ferritin‒bound erastin and rapamycin (NFER) with significantly improved drug-like properties, was proposed to increase ferroptosis by decreasing GPX4 activity as well as reducing lipid peroxidation levels.316 Interestingly, Sal-AuNP, a gold nanoparticle loaded with salinomycin, has been shown to induce ferroptosis in breast cancer stem cells by causing oxidative stress, mitochondrial dysfunction, and lipid oxidation.317 Recently, a hypercarbon-centered gold(I) cluster prodrug was engineered and then shown to accelerate ferroptosis in EJ cells (a human bladder cancer cell line) by inhibiting thioredoxin reductase (TrxR) activity.318
To maximize the efficacy of cancer therapeutics, nanoparticle-based combination therapies have been widely studied in various preclinical models. For example, CSO-SS-Cy7-Hex/SPION/Srfn319 and SPFeN320 have been studied as photothermal ferrotherapies for use in cancer, combining photosensitizers with ferroptosis-inducing molecules that can exert synergistic anticancer effects. Another photosensitizer—chlorin e6 (Ce6)—has been used in several delivery systems, including Ce6-erastin nanoparticles,321 SRF@Hb-Ce6,322 Ce6@MOF,323 and HAS-Ce6-IrO2.324 Liu et al. reported excellent chemodynamic/photodynamic synergy using mCMSN, a biodegradable cancer cell membrane‒coated mesoporous copper/manganese silicate nanosphere that increases ferroptosis via laser irradiation‒triggered ROS production and the GSH-activated Fenton reaction.325 To achieve a synergistic ferroptosis and immunomodulatory effect in treating cancer, Zhang et al. engineered a biomimetic magnetosome (Pa-M/Ti-NC) consisting of an Fe3O4 magnetic nanocluster (NC) as the core, pre-engineered leukocyte membranes loaded with TGF-β inhibitor (Ti), and a PD-1 antibody (Pa) anchored to the membrane surface;326 they found that this magnetosome potently promoted ferroptosis in cancer cells via cooperative activity between the Pa and Ti components, causing reprogramming of the immunogenic microenvironment. Interestingly, NMIL-100@Gox@C was designed to activate ferroptosis and induce cancer starvation;327 specifically, the glucose oxidase (Gox) enzyme catalyzes glucose to generate sufficient levels of H2O2 in order to induce ferroptosis, simultaneously causing starvation of the cancer cells via the Gox-mediated consumption of glucose.327 Yang et al. developed an HLCaP nanoreactor that encapsulates lipoxidase and hemin in biodegradable PLGA (poly(lactic-co-glycolic acid)), which releases its contents in the acidic tumor microenvironment, synergistically inducing ferroptosis using the PUFA substrates produced in the tumor debris using radiofrequency ablation;328 importantly, these so-called tumor debris‒fueled nanoreactors have been shown to prevent tumor recurrence and metastasis.328 Similarly, the mesoporous carbon nanoparticle FeCO-Dox@MCN is designed to exploit the combined effects of chemotherapy, photothermal therapy, and gas therapy in order to increase carbon monoxide‒induced ferroptosis.329
By contrast, relatively few studies have focused on ferroptosis-suppressing nanoparticles (Fig. 6). Here, we discuss two nanoparticles that have been reported to inhibit ferroptosis. The first is a carboxyl-modified polystyrene nanoparticle (CPS) that effectively inhibits ferroptosis by reducing cellular ROS mediated by TFEB (transcription factor EB).330 The second is DEF-HCC-PEG, a deferoxamine-binding nanoparticle that protects cells against both ferroptosis and senescence.331 Importantly, ferroptosis is a rapidly developing field, with new mechanisms underlying ferroptosis being identified; thus, additional studies are clearly helpful for accelerating the development of clinical applications using nanoparticles to target ferroptosis.
Conclusions and future perspectives
Over the past decade, a rapidly growing body of evidence has shown that ferroptosis plays a key role in a wide range of diseases and conditions, including cancer, neurodegenerative disease, tissue/organ injury, inflammation, and inflammatory diseases. Despite many obstacles along the way, it is now widely accepted that targeting ferroptosis holds great promise for promoting the development and clinical translation of ferroptosis-based therapeutic strategies.
Challenges and opportunities
To date, nearly all in vivo ferroptosis-related studies were based on preclinical animal models, and several challenges limit their translation to clinical practice.
As discussed in this review, numerous molecules—including experimental agents, naturally derived compounds, nanoparticles, and clinically approved drugs—have been shown to modulate ferroptosis, revealing promising new strategies for the development of ferroptosis-based therapies. However, with respect to small molecule compounds, only ~15% of proteins in the human proteome are estimated to be druggable.332 In addition, poor pharmacokinetics remains a major bottleneck for the further development of both ferroptosis antagonists such as Fer-1 and Lip-1 and ferroptosis agonists such as RSL3 and ML210. To develop novel bioactive ferroptosis-targeted drug candidates, researchers have studied natural products derived from microorganisms and plants, particularly traditional herbs. Furthermore, high-throughput screening, artificial intelligence, and other new technologies may also be used to identify more effective drug candidates to target ferroptosis.
Another challenge to ferroptosis-based drug discovery is the rational design of combination strategies to treat various ferroptosis-related diseases. It is generally well accepted that ferroptosis plays a pathogenic role in various diseases. In addition, many existing therapies can trigger apoptosis or other types of cell death. For conditions such as cancer, tissue injury, and neurodegenerative disease, various forms of cell death can occur simultaneously. Therefore, developing ferroptosis modulators that can act synergistically with existing therapies may serve as a promising strategy for treating these diseases, particularly drug-resistant cancer. Combining ferroptosis agonists with other anticancer therapies may also increase ferroptosis, as well as other types of cell death, thereby eradicating cancer more effectively.
Novel technologies for developing ferroptosis-targeting agents
In addition to conventional ferroptosis inhibitors and inducers, new technologies such as proteolysis-targeting chimeras (PROTACs), RNA-based therapies, gene editing, and peptide and protein drugs have been applied to develop ferroptosis-targeting drugs.
Targeted protein degradation
Targeted protein degradation (TPD) is a new and challenging therapeutic modality that can degrade “undrugged” targets and other difficult protein targets such as proteins with a broad and shallow active pocket and/or “smooth” surfaces. Three major classifications of protein degraders have been developed, including PROTACs, monomeric targeted protein degraders, and molecular glues (MGs). PROTACs are heterobifunctional small molecules consisting of two ligands connected via a linker. The ligands are designed to bind to an E3 ubiquitin ligase and a target of interest. This chemically induced close proximity between the protein and E3 ligase results in the target protein’s ubiquitylation and subsequent degradation via the ubiquitin-proteasome system (UPS). Recently, TPD has gained considerable attention due to encouraging results obtained from clinical trials involving ARV-110 and ARV-471, two PROTACs that target and degrade the androgen receptor (AR) and estrogen receptor (ER), respectively. Recently, several groups have attempted to develop PROTAC-based ferroptosis modulators. For example, Luo et al. developed the PROTAC dGPX4 to target GPX4 and showed in vivo that it potently induces ferroptosis selectively in cancer cells with no apparent side effects,333 providing a promising novel strategy for the development of clinically applicable GPX4-targeted drugs. Similar to PROTACs, photodegradation-targeting chimeras (PDTACs),334 lysosome-targeting chimeras (LYTACs),335 and autophagy-targeting chimeras (AUTACs)336 have also been developed for use in TPD-based therapies. For example, Liu et al. reported the targeted photodegradation of GPX4 using a PDTAC conjugated with the photosensitizer verteporfin upon red-light irradiation, and showed that this PDTAC can induce immunogenic ferroptosis and may improve the efficacy of cancer immunotherapy.334
Unlike PROTACs, monomeric targeted protein degraders are designed to either directly bind to their target protein or induce their target protein’s degradation. Therefore, monomeric degraders have a comparatively small molecular weight and can easily cross the BBB.337 Fulvestrant, a selective ER degrader, has been approved for treating ER-positive breast cancer, and some monomeric degraders have been tested in clinical trials.338 MGs are relatively simple small molecules that increase the interaction between E3 ligase and the protein substrate in order to induce the UPS-induced degradation of target proteins.339 Thanks to their low molecular weight, MGs are believed to have more favorable pharmacokinetics than other compounds. Thalidomide was developed as a MG to bring various targets in proximity to the E3 ubiquitin ligase CUL4-RBX1-DDB1-CRBN for degradation.340 Lenalidomide, a thalidomide derivative, has been approved to treat multiple myeloma by inducing the degradation of oncoproteins. Thus, monomeric degraders and MGs represent promising new strategies for targeted protein degradation. However, to the best of our knowledge no reports have yet been published regarding the use of monomeric targeted protein degraders or MG-based degraders to target ferroptosis-related proteins.
Artificial intelligence (AI)‒based drug discovery strategies
AI, which was first defined by John McCarthy back in 1956 as the “science and engineering of making intelligent machines”, primarily involves computer science, mathematics, and neuroscience, but can also be used for drug discovery.341 Given that the traditional process of drug discovery is time-consuming and labor-intensive, it is now widely accepted that AI—particularly machine learning—can significantly increase prediction accuracy and accelerate the drug discovery process. Deep learning, which combines machine learning and AI, can serve as a particularly cost-effective platform for discovering new drugs, as it directly enables rapid machine-based decision-making using an artificial neural network. Today, AI-based technologies have been applied to virtually every step in the drug discovery process, including target identification, drug design, drug screening, chemical synthesis, and prediction of the drug’s properties and mode of action.341,342 In addition, AI can be used to quickly identify lead compounds extracted from plants and microorganisms.343 Recently, AI was successfully used in cancer research and precision medicine.344 With respect to ferroptosis, Wu et al. used a combination of bioinformatics, network pharmacology, and AI to show that ferroptosis and the TGF-β signaling pathway may contribute to the protective effects of celastrol, a plant-derived triterpene, against type 2 diabetes.345 It is therefore reasonable to speculate that AI will significantly accelerate the discovery of new ferroptosis-targeted drugs.
RNA-based therapies
In addition to protein targets, a new line in the field of drug discovery focuses on RNA-based therapeutics, particularly for the management of genetic diseases. RNA-based therapeutics now include the use of messenger RNA (mRNA), RNAi, single-stranded antisense oligonucleotides, aptamers, ribozymes, and CRISPR-Cas endonuclease‒mediated gene editing. For example, mRNA-based therapies have been successfully used to develop personalized cancer vaccines,346 as well as vaccines against infectious diseases.347 With respect to RNAi, four RNAi-based therapies have been approved for use in patients, including patisiran for hereditary transthyretin amyloidosis, givosiran for acute hepatic porphyria, lumasiran for primary hyperoxaluria type 1, and inclisiran for heterozygous familial hypercholesterolemia and atherosclerotic cardiovascular disease. These four approved RNAi-based compounds are based on small-interfering RNA (siRNA) technology, which decrease the level of specific mRNAs in order to reduce the corresponding protein levels. Currently, >50 clinical trials are investigating siRNA-based compounds. Unfortunately, however, no clinical trials are investigating ferroptosis-targeting agents using RNA-based technologies. Nevertheless, although the inability to precisely deliver RNA-based tools remains a major barrier to developing RNA therapies, new RNA-based modalities—particularly CRISPR/Cas9-based gene editing—will likely gain considerable traction in the near future.
Peptide-based drugs and biologicals
Peptide-based drugs represent a unique pharmaceutical niche between small-molecule compounds and biologicals. Compared to chemical compounds, peptide-based drugs are more efficient, safer, and better tolerated. This category of drugs has several other advantages as well, including high selectivity and reduced tissue accumulation. Biological drugs such as monoclonal antibodies bind with high specificity to their target proteins. Recently, bispecific antibodies—which simultaneously target multiple antigens and/or epitopes using two specific antibodies—have attracted considerable attention and may represent the next generation of antibody-targeted T cell‒based cancer immunotherapies. Furthermore, antibody-drug conjugates have high specificity and are highly efficient at delivering toxic small molecules in order to specifically target cancer cells.
Conclusions
Ferroptosis is now generally accepted as playing a key pathogenic role in a wide range of diseases and conditions. As summarized in this review, a large amount of compelling evidence supports the notion that targeting ferroptosis can provide promising new options for the treatment of many ferroptosis-related diseases. As our understanding of ferroptosis-regulating pathways—including iron metabolism, lipid metabolism, and oxidative-reductive pathways—increases to reveal new druggable targets, ferroptosis modulators are likely to provide new opportunities to develop treatments for many currently incurable diseases. Although further studies are clearly needed, the novel technologies summarized in this review will facilitate the development of safer, more effective ferroptosis-targeted drugs.
References
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Friedmann Angeli, J. P., Krysko, D. V. & Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 19, 405–414 (2019).
Fang, X. et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl Acad. Sci. USA 116, 2672–2680 (2019).
Li, Y. et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 26, 2284–2299 (2019).
Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191 (2014).
Chen, L., Hambright, W. S., Na, R. & Ran, Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J. Biol. Chem. 290, 28097–28106 (2015).
Hambright, W. S., Fonseca, R. S., Chen, L., Na, R. & Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 12, 8–17 (2017).
Murphy, T. H., Miyamoto, M., Sastre, A., Schnaar, R. L. & Coyle, J. T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2, 1547–1558 (1989).
Lewerenz, J., Ates, G., Methner, A., Conrad, M. & Maher, P. Oxytosis/ferroptosis-(re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front Neurosci. 12, 214 (2018).
Dolma, S., Lessnick, S. L., Hahn, W. C. & Stockwell, B. R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296 (2003).
Yagoda, N. et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868 (2007).
Yang, W. S. & Stockwell, B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245 (2008).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014).
Yuan, H., Li, X., Zhang, X., Kang, R. & Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys. Res Commun. 478, 1338–1343 (2016).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2017).
Ingold, I. et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–422.e421 (2018).
Doll, S. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019).
Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019).
Kraft, V. A. N. et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent. Sci. 6, 41–53 (2020).
Mao, C. et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590 (2021).
Mishima, E. et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 608, 778–783 (2022).
Yang, L. et al. Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduct. Target Ther. 5, 138 (2020).
Yu, Y. et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood 136, 726–739 (2020).
Fang, X. et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ. Res 127, 486–501 (2020).
Gao, M., Monian, P., Quadri, N., Ramasamy, R. & Jiang, X. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59, 298–308 (2015).
Hou, W. et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428 (2016).
Gao, M. et al. Ferroptosis is an autophagic cell death process. Cell Res 26, 1021–1032 (2016).
Geng, N. et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur. Rev. Med Pharm. Sci. 22, 3826–3836 (2018).
Dixon, S. J. et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609 (2015).
Kagan, V. E. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90 (2017).
Yang, W. S. et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl Acad. Sci. USA 113, E4966–E4975 (2016).
Liang, D. et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell, (2023).
Liang, C., Zhang, X., Yang, M. & Dong, X. Recent progress in ferroptosis inducers for cancer therapy. Adv. Mater. 31, e1904197 (2019).
Sun, X. et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184 (2016).
Badgley, M. A. et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89 (2020).
Chen, M. S. et al. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2alpha-ATF4 pathway. Oncotarget 8, 114588–114602 (2017).
Yu, M. et al. Targeted exosome-encapsulated erastin induced ferroptosis in triple negative breast cancer cells. Cancer Sci. 110, 3173–3182 (2019).
Yang, W. H. et al. The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 28, 2501–2508.e2504 (2019).
Markowitsch, S. D. et al. Artesunate inhibits growth of sunitinib-resistant renal cell carcinoma cells through cell cycle arrest and induction of ferroptosis. Cancers (Basel) 12, 3150 (2020).
Viswanathan, V. S. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017).
Wang, W. et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 (2019).
Querfurth, H. W. & LaFerla, F. M. Alzheimer’s disease. N. Engl. J. Med. 362, 329–344 (2010).
Schneider, S. A. Neurodegenerations with Brain Iron Accumulation. Parkinsonism Relat. Disord. 22, S21–S25 (2016).
Bartzokis, G., Cummings, J., Perlman, S., Hance, D. B. & Mintz, J. Increased basal ganglia iron levels in Huntington disease. Arch. Neurol. 56, 569–574 (1999).
Guiney, S. J., Adlard, P. A., Bush, A. I., Finkelstein, D. I. & Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem Int 104, 34–48 (2017).
Zille, M. et al. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke 48, 1033–1043 (2017).
Skouta, R. et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 136, 4551–4556 (2014).
Zhang, Z. et al. Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res. 1701, 112–125 (2018).
Derry, P. J. & Kent, T. A. Correlating quantitative susceptibility mapping with cognitive decline in Alzheimer’s disease. Brain 140, 2069–2072 (2017).
Derry, P. J. et al. Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer’s disease from a ferroptosis perspective. Prog. Neurobiol. 184, 101716 (2020).
Hornykiewicz, O. Basic research on dopamine in Parkinson’s disease and the discovery of the nigrostriatal dopamine pathway: the view of an eyewitness. Neurodegener. Dis. 5, 114–117 (2008).
Pyatigorskaya, N. et al. High nigral iron deposition in LRRK2 and Parkin mutation carriers using R2* relaxometry. Mov. Disord. 30, 1077–1084 (2015).
Pearce, R. K., Owen, A., Daniel, S., Jenner, P. & Marsden, C. D. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J. Neural Transm. (Vienna) 104, 661–677 (1997).
Dexter, D. et al. Lipid peroxidation as cause of nigral cell death in Parkinson’s disease. Lancet 2, 639–640 (1986).
MacDonald, M. E. et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s disease collaborative research group. Cell 72, 971–983 (1993).
Lee, J. et al. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol. 121, 487–498 (2011).
Muller, M. & Leavitt, B. R. Iron dysregulation in Huntington’s disease. J. Neurochem 130, 328–350 (2014).
Klepac, N. et al. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects : a cross-sectional study. J. Neurol. 254, 1676–1683 (2007).
Li, Y. et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 27, 2635–2650 (2020).
Chen, J., Li, X., Ge, C., Min, J. & Wang, F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 29, 467–480 (2022).
Ueta, T. et al. Glutathione peroxidase 4 is required for maturation of photoreceptor cells. J. Biol. Chem. 287, 7675–7682 (2012).
Wortmann, M. et al. Combined deficiency in glutathione peroxidase 4 and vitamin E causes multiorgan thrombus formation and early death in mice. Circ. Res. 113, 408–417 (2013).
Kellum, J. A. et al. Acute kidney injury. Nat. Rev. Dis. Prim. 7, 52 (2021).
Linkermann, A. et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl Acad. Sci. USA 111, 16836–16841 (2014).
Li, X. et al. Pretreatment with roxadustat (FG-4592) attenuates folic acid-induced kidney injury through antiferroptosis via Akt/GSK-3beta/Nrf2 pathway. Oxid. Med. Cell Longev. 2020, 6286984 (2020).
Jacobs, W. et al. Fatal lymphocytic cardiac damage in coronavirus disease 2019 (COVID-19): autopsy reveals a ferroptosis signature. ESC Heart Fail. 7, 3772–3781 (2020).
Fang, X., Ardehali, H., Min, J. & Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 20, 7–23 (2023).
Dommergues, M. A., Gallego, J., Evrard, P. & Gressens, P. Iron supplementation aggravates periventricular cystic white matter lesions in newborn mice. Eur. J. Paediatr. Neurol. 2, 313–318 (1998).
Brault, S. et al. Cytotoxicity of the E(2)-isoprostane 15-E(2t)-IsoP on oligodendrocyte progenitors. Free Radic. Biol. Med. 37, 358–366 (2004).
Novgorodov, S. A. et al. Acid sphingomyelinase promotes mitochondrial dysfunction due to glutamate-induced regulated necrosis. J. Lipid Res. 59, 312–329 (2018).
Li, Q. et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2, e90777 (2017).
Jin, Z. L. et al. Paeonol inhibits the progression of intracerebral haemorrhage by mediating the HOTAIR/UPF1/ACSL4 axis. ASN Neuro 13, 17590914211010647 (2021).
Karuppagounder, S. S. et al. N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E(2) to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann. Neurol. 84, 854–872 (2018).
Alim, I. et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177, 1262–1279.e1225 (2019).
Guan, X. et al. The neuroprotective effects of carvacrol on ischemia/reperfusion-induced hippocampal neuronal impairment by ferroptosis mitigation. Life Sci. 235, 116795 (2019).
Zhou, H. et al. Proanthocyanidin promotes functional recovery of spinal cord injury via inhibiting ferroptosis. J. Chem. Neuroanat. 107, 101807 (2020).
Wang, H. et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 66, 449–465 (2017).
Liu, C. Y. et al. Ferroptosis is involved in alcohol-induced cell death in vivo and in vitro. Biosci. Biotechnol. Biochem. 84, 1621–1628 (2020).
Li, X. et al. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int. 40, 1378–1394 (2020).
Wei, S. et al. Ferroptosis mediated by the interaction between Mfn2 and IREalpha promotes arsenic-induced nonalcoholic steatohepatitis. Environ. Res. 188, 109824 (2020).
Lorincz, T., Jemnitz, K., Kardon, T., Mandl, J. & Szarka, A. Ferroptosis is involved in acetaminophen induced cell death. Pathol. Oncol. Res. 21, 1115–1121 (2015).
Yamada, N. et al. Iron overload as a risk factor for hepatic ischemia-reperfusion injury in liver transplantation: Potential role of ferroptosis. Am. J. Transpl. 20, 1606–1618 (2020).
Van Coillie, S. et al. Targeting ferroptosis protects against experimental (multi)organ dysfunction and death. Nat. Commun. 13, 1046 (2022).
Xu, S., Min, J. & Wang, F. Ferroptosis: an emerging player in immune cells. Sci. Bull. 66, 2257–2260 (2021).
Matsushita, M. et al. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 212, 555–568 (2015).
Muri, J., Thut, H., Bornkamm, G. W. & Kopf, M. B1 and marginal zone B cells but not follicular B2 cells require Gpx4 to prevent lipid peroxidation and ferroptosis. Cell Rep. 29, 2731–2744.e2734 (2019).
Kapralov, A. A. et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 16, 278–290 (2020).
Li, P. et al. Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat. Immunol. 22, 1107–1117 (2021).
Dar, H. H. et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J. Clin. Invest. 128, 4639–4653 (2018).
Amaral, E. P. et al. A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J. Exp. Med. 216, 556–570 (2019).
Li, W. et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Invest. 129, 2293–2304 (2019).
Bai, T., Li, M., Liu, Y., Qiao, Z. & Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic. Biol. Med. 160, 92–102 (2020).
Richardson, D. R. & Ponka, P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim. Biophys. Acta 1331, 1–40 (1997).
Hentze, M. W., Muckenthaler, M. U., Galy, B. & Camaschella, C. Two to tango: regulation of Mammalian iron metabolism. Cell 142, 24–38 (2010).
Ohgami, R. S. et al. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet 37, 1264–1269 (2005).
Fleming, M. D. et al. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc. Natl Acad. Sci. USA 95, 1148–1153 (1998).
Liuzzi, J. P., Aydemir, F., Nam, H., Knutson, M. D. & Cousins, R. J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl Acad. Sci. USA 103, 13612–13617 (2006).
Kristiansen, M. et al. Identification of the haemoglobin scavenger receptor. Nature 409, 198–201 (2001).
Hvidberg, V. et al. Identification of the receptor scavenging hemopexin-heme complexes. Blood 106, 2572–2579 (2005).
Torti, F. M. & Torti, S. V. Regulation of ferritin genes and protein. Blood 99, 3505–3516 (2002).
Donovan, A. et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 1, 191–200 (2005).
Nemeth, E. et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093 (2004).
Jiang, L. et al. RNF217 regulates iron homeostasis through its E3 ubiquitin ligase activity by modulating ferroportin degradation. Blood 138, 689–705 (2021).
Kontoghiorghes, G. J., Eracleous, E., Economides, C. & Kolnagou, A. Advances in iron overload therapies. prospects for effective use of deferiprone (L1), deferoxamine, the new experimental chelators ICL670, GT56-252, L1NA11 and their combinations. Curr. Med. Chem. 12, 2663–2681 (2005).
Kontoghiorghes, G. J., Pattichi, K., Hadjigavriel, M. & Kolnagou, A. Transfusional iron overload and chelation therapy with deferoxamine and deferiprone (L1). Transfus. Sci. 23, 211–223 (2000).
Richardson, D. R. Iron chelators as therapeutic agents for the treatment of cancer. Crit. Rev. Oncol. Hematol. 42, 267–281 (2002).
Keberle, H. The biochemistry of desferrioxamine and its relation to iron metabolism. Ann. N. Y. Acad. Sci. 119, 758–768 (1964).
Li, J. et al. Ferroptosis: past, present and future. Cell Death Dis. 11, 88 (2020).
Qi, J., Kim, J. W., Zhou, Z., Lim, C. W. & Kim, B. Ferroptosis affects the progression of nonalcoholic steatohepatitis via the modulation of lipid peroxidation-mediated cell death in mice. Am. J. Pathol. 190, 68–81 (2020).
Porter, J. B., Taher, A. T., Cappellini, M. D. & Vichinsky, E. P. Ethical issues and risk/benefit assessment of iron chelation therapy: advances with deferiprone/deferoxamine combinations and concerns about the safety, efficacy and costs of deferasirox. Hemoglobin 32, 601–607 (2008).
Kontoghiorghes, G. J. Design, properties, and effective use of the oral chelator L1 and other alpha-ketohydroxypyridines in the treatment of transfusional iron overload in thalassemia. Ann. N. Y Acad. Sci. 612, 339–350 (1990).
Hider, R. C. & Hoffbrand, A. V. The role of deferiprone in iron chelation. N. Engl. J. Med 379, 2140–2150 (2018).
Fredenburg, A. M., Sethi, R. K., Allen, D. D. & Yokel, R. A. The pharmacokinetics and blood-brain barrier permeation of the chelators 1,2 dimethly-, 1,2 diethyl-, and 1-[ethan-1’ol]-2-methyl-3-hydroxypyridin-4-one in the rat. Toxicology 108, 191–199 (1996).
Kontoghiorghes, G. J. Iron mobilization from transferrin and non-transferrin-bound-iron by deferiprone. Implications in the treatment of thalassemia, anemia of chronic disease, cancer and other conditions. Hemoglobin 30, 183–200 (2006).
Kakhlon, O. et al. Cell functions impaired by frataxin deficiency are restored by drug-mediated iron relocation. Blood 112, 5219–5227 (2008).
Sauerbeck, A., Schonberg, D. L., Laws, J. L. & McTigue, D. M. Systemic iron chelation results in limited functional and histological recovery after traumatic spinal cord injury in rats. Exp. Neurol. 248, 53–61 (2013).
Manabe, E. et al. Reduced lifespan of erythrocytes in Dahl/Salt sensitive rats is the cause of the renal proximal tubule damage. Sci. Rep. 10, 22023 (2020).
Imai, T. et al. Deferasirox, a trivalent iron chelator, ameliorates neuronal damage in hemorrhagic stroke models. Naunyn Schmiedebergs Arch. Pharm. 394, 73–84 (2021).
Ichikawa, Y. et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Invest 124, 617–630 (2014).
Chen, W. et al. CN128: A new orally active hydroxypyridinone iron chelator. J. Med Chem. 63, 4215–4226 (2020).
Eberhard, Y. et al. Chelation of intracellular iron with the antifungal agent ciclopirox olamine induces cell death in leukemia and myeloma cells. Blood 114, 3064–3073 (2009).
Shen, T. & Huang, S. Repositioning the old fungicide ciclopirox for new medical uses. Curr. Pharm. Des. 22, 4443–4450 (2016).
Wu, J. et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406 (2019).
Feng, H. et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 30, 3411–3423.e3417 (2020).
Horonchik, L. & Wessling-Resnick, M. The small-molecule iron transport inhibitor ferristatin/NSC306711 promotes degradation of the transferrin receptor. Chem. Biol. 15, 647–653 (2008).
Wetli, H. A., Buckett, P. D. & Wessling-Resnick, M. Small-molecule screening identifies the selanazal drug ebselen as a potent inhibitor of DMT1-mediated iron uptake. Chem. Biol. 13, 965–972 (2006).
Zhang, Z. et al. Discovery of benzylisothioureas as potent divalent metal transporter 1 (DMT1) inhibitors. Bioorg. Med Chem. Lett. 22, 5108–5113 (2012).
Katsarou, A. & Pantopoulos, K. Hepcidin therapeutics. Pharmaceuticals (Basel) 11, 127 (2018).
Chung, B. et al. Oncostatin M is a potent inducer of hepcidin, the iron regulatory hormone. FASEB J. 24, 2093–2103 (2010).
Boyce, M. et al. Safety, pharmacokinetics and pharmacodynamics of the anti-hepcidin Spiegelmer lexaptepid pegol in healthy subjects. Br. J. Pharm. 173, 1580–1588 (2016).
Vadhan-Raj, S. et al. A first-in-human phase 1 study of a hepcidin monoclonal antibody, LY2787106, in cancer-associated anemia. J. Hematol. Oncol. 10, 73 (2017).
Mancias, J. D., Wang, X., Gygi, S. P., Harper, J. W. & Kimmelman, A. C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109 (2014).
Sui, S. et al. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis. 10, 331 (2019).
Huang, T. et al. Growth inhibition of a novel iron chelator, DpdtC, against hepatoma carcinoma cell lines partly attributed to ferritinophagy-mediated lysosomal ROS generation. Oxid. Med Cell Longev. 2018, 4928703 (2018).
Li, J. et al. Small-molecule MMRi62 induces ferroptosis and inhibits metastasis in pancreatic cancer via degradation of ferritin heavy chain and mutant p53. Mol. Cancer Ther. 21, 535–545 (2022).
Fang, Y. et al. Inhibiting ferroptosis through disrupting the NCOA4-FTH1 interaction: a new mechanism of action. ACS Cent. Sci. 7, 980–989 (2021).
Muckenthaler, M. U., Rivella, S., Hentze, M. W. & Galy, B. A red carpet for iron metabolism. Cell 168, 344–361 (2017).
Otterbein, L. E., Soares, M. P., Yamashita, K. & Bach, F. H. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 24, 449–455 (2003).
Suttner, D. M. & Dennery, P. A. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J. 13, 1800–1809 (1999).
Hassannia, B., Vandenabeele, P. & Vanden Berghe, T. Targeting ferroptosis to iron out cancer. Cancer Cell 35, 830–849 (2019).
Campbell, N. K., Fitzgerald, H. K. & Dunne, A. Regulation of inflammation by the antioxidant haem oxygenase 1. Nat. Rev. Immunol. 21, 411–425 (2021).
Vreman, H. J., Ekstrand, B. C. & Stevenson, D. K. Selection of metalloporphyrin heme oxygenase inhibitors based on potency and photoreactivity. Pediatr. Res 33, 195–200 (1993).
Morisawa, T., Wong, R. J., Bhutani, V. K., Vreman, H. J. & Stevenson, D. K. Inhibition of heme oxygenase activity in newborn mice by azalanstat. Can. J. Physiol. Pharm. 86, 651–659 (2008).
Bannai, S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem. 261, 2256–2263 (1986).
Hayano, M., Yang, W. S., Corn, C. K., Pagano, N. C. & Stockwell, B. R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 23, 270–278 (2016).
Aoyama, K. et al. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 9, 119–126 (2006).
Chairoungdua, A. et al. Identification of an amino acid transporter associated with the cystinuria-related type II membrane glycoprotein. J. Biol. Chem. 274, 28845–28848 (1999).
Stipanuk, M. H., Dominy, J. E. Jr., Lee, J. I. & Coloso, R. M. Mammalian cysteine metabolism: new insights into regulation of cysteine metabolism. J. Nutr. 136, 1652S–1659S (2006).
Combs, J. A. & DeNicola, G. M. The non-essential amino acid cysteine becomes essential for tumor proliferation and survival. Cancers (Basel) 11, 678 (2019).
Wang, Y. et al. SLC25A39 is necessary for mitochondrial glutathione import in mammalian cells. Nature 599, 136–140 (2021).
Dixon, S. J. & Stockwell, B. R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10, 9–17 (2014).
Larraufie, M. H. et al. Incorporation of metabolically stable ketones into a small molecule probe to increase potency and water solubility. Bioorg. Med. Chem. Lett. 25, 4787–4792 (2015).
Zhang, Y. et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem. Biol. 26, 623–633.e629 (2019).
Codenotti, S. et al. Cell growth potential drives ferroptosis susceptibility in rhabdomyosarcoma and myoblast cell lines. J. Cancer Res. Clin. Oncol. 144, 1717–1730 (2018).
Liu, Q. & Wang, K. The induction of ferroptosis by impairing STAT3/Nrf2/GPx4 signaling enhances the sensitivity of osteosarcoma cells to cisplatin. Cell Biol. Int. 43, 1245–1256 (2019).
Chen, L. et al. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-gamma-lyase function. Oncol. Rep. 33, 1465–1474 (2015).
Yu, Y. et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell Oncol. 2, e1054549 (2015).
Yan, R. et al. The structure of erastin-bound xCT-4F2hc complex reveals molecular mechanisms underlying erastin-induced ferroptosis. Cell Res. 32, 687–690 (2022).
Gout, P. W., Buckley, A. R., Simms, C. R. & Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia 15, 1633–1640 (2001).
Robe, P. A. et al. In vitro and in vivo activity of the nuclear factor-kappaB inhibitor sulfasalazine in human glioblastomas. Clin. Cancer Res. 10, 5595–5603 (2004).
Doxsee, D. W. et al. Sulfasalazine-induced cystine starvation: potential use for prostate cancer therapy. Prostate 67, 162–171 (2007).
Guan, J. et al. The xc- cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: use of sulfasalazine. Cancer Chemother. Pharm. 64, 463–472 (2009).
Lo, M., Ling, V., Low, C., Wang, Y. Z. & Gout, P. W. Potential use of the anti-inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Curr. Oncol. 17, 9–16 (2010).
Timmerman, L. A. et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 24, 450–465 (2013).
Robe, P. A. et al. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 9, 372 (2009).
Sleire, L. et al. Drug repurposing: sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene 34, 5951–5959 (2015).
Rodman, S. N. et al. Enhancement of radiation response in breast cancer stem cells by inhibition of thioredoxin- and glutathione-dependent metabolism. Radiat. Res. 186, 385–395 (2016).
Nagane, M. et al. Sulfasalazine, an inhibitor of the cystine-glutamate antiporter, reduces DNA damage repair and enhances radiosensitivity in murine B16F10 melanoma. PLoS One 13, e0195151 (2018).
Zhang, P. et al. xCT expression modulates cisplatin resistance in Tca8113 tongue carcinoma cells. Oncol. Lett. 12, 307–314 (2016).
Wada, F. et al. High expression of CD44v9 and xCT in chemoresistant hepatocellular carcinoma: Potential targets by sulfasalazine. Cancer Sci. 109, 2801–2810 (2018).
Ogihara, K. et al. Sulfasalazine could modulate the CD44v9-xCT system and enhance cisplatin-induced cytotoxic effects in metastatic bladder cancer. Cancer Sci. 110, 1431–1441 (2019).
Takayama, T. et al. Potential of sulfasalazine as a therapeutic sensitizer for CD44 splice variant 9-positive urogenital cancer. Med Oncol. 33, 45 (2016).
Otsuki, Y. et al. Vasodilator oxyfedrine inhibits aldehyde metabolism and thereby sensitizes cancer cells to xCT-targeted therapy. Cancer Sci. 111, 127–136 (2020).
Jiang, Q. et al. Platelet membrane-camouflaged magnetic nanoparticles for ferroptosis-enhanced cancer immunotherapy. Small 16, e2001704 (2020).
Dixon, S. J. et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523 (2014).
Louandre, C. et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 356, 971–977 (2015).
Bai, T. et al. Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem Biophys. Res Commun. 491, 919–925 (2017).
Li, Y. et al. Sorafenib induces mitochondrial dysfunction and exhibits synergistic effect with cysteine depletion by promoting HCC cells ferroptosis. Biochem Biophys. Res Commun. 534, 877–884 (2021).
Tang, H. et al. Targeted manganese doped silica nano GSH-cleaner for treatment of liver cancer by destroying the intracellular redox homeostasis. Theranostics 10, 9865–9887 (2020).
Tang, H. et al. Dual GSH-exhausting sorafenib loaded manganese-silica nanodrugs for inducing the ferroptosis of hepatocellular carcinoma cells. Int J. Pharm. 572, 118782 (2019).
Zheng, J. et al. Sorafenib fails to trigger ferroptosis across a wide range of cancer cell lines. Cell Death Dis. 12, 698 (2021).
Shaw, A. T. et al. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc. Natl Acad. Sci. USA 108, 8773–8778 (2011).
Yamada, N., Karasawa, T. & Takahashi, M. Role of ferroptosis in acetaminophen-induced hepatotoxicity. Arch. Toxicol. 94, 1769–1770 (2020).
Vad, N. M., Yount, G., Moore, D., Weidanz, J. & Moridani, M. Y. Biochemical mechanism of acetaminophen (APAP) induced toxicity in melanoma cell lines. J. Pharm. Sci. 98, 1409–1425 (2009).
Gai, C. et al. Acetaminophen sensitizing erastin-induced ferroptosis via modulation of Nrf2/heme oxygenase-1 signaling pathway in non-small-cell lung cancer. J. Cell Physiol. 235, 3329–3339 (2020).
Yamashita, M. Auranofin: Past to present, and repurposing. Int Immunopharmacol. 101, 108272 (2021).
Chepikova, O. E. et al. Lysine oxidase exposes a dependency on the thioredoxin antioxidant pathway in triple-negative breast cancer cells. Breast Cancer Res. Treat. 183, 549–564 (2020).
Fang, X. et al. Malic enzyme 1 as a novel anti-ferroptotic regulator in hepatic ischemia/reperfusion injury. Adv. Sci. (Weinh.) 10, e2205436 (2023).
Fruehauf, J. P. et al. Selective and synergistic activity of L-S,R-buthionine sulfoximine on malignant melanoma is accompanied by decreased expression of glutathione-S-transferase. Pigment. Cell Res. 10, 236–249 (1997).
Bailey, H. H. et al. Phase I clinical trial of intravenous L-buthionine sulfoximine and melphalan: an attempt at modulation of glutathione. J. Clin. Oncol. 12, 194–205 (1994).
Nishizawa, S. et al. Low tumor glutathione level as a sensitivity marker for glutamate-cysteine ligase inhibitors. Oncol. Lett. 15, 8735–8743 (2018).
Kehrer, J. P. The effect of BCNU (carmustine) on tissue glutathione reductase activity. Toxicol. Lett. 17, 63–68 (1983).
Xu, F. L. et al. SLC27A5 promotes sorafenib-induced ferroptosis in hepatocellular carcinoma by downregulating glutathione reductase. Cell Death Dis. 14, 22 (2023).
Gordillo, G. M. et al. Multidrug resistance-associated Protein-1 (MRP-1)-dependent glutathione disulfide (GSSG) efflux as a critical survival factor for oxidant-enriched tumorigenic endothelial cells. J. Biol. Chem. 291, 10089–10103 (2016).
Michaelis, M. et al. Glycyrrhizin exerts antioxidative effects in H5N1 influenza A virus-infected cells and inhibits virus replication and pro-inflammatory gene expression. PLoS One 6, e19705 (2011).
Monti, D. A. et al. N-Acetyl cysteine may support dopamine neurons in Parkinson’s disease: preliminary clinical and cell line data. PLoS One 11, e0157602 (2016).
Hendrickson, R. G. What is the most appropriate dose of N-acetylcysteine after massive acetaminophen overdose? Clin. Toxicol. (Philos.) 57, 686–691 (2019).
Ates, B., Abraham, L. & Ercal, N. Antioxidant and free radical scavenging properties of N-acetylcysteine amide (NACA) and comparison with N-acetylcysteine (NAC). Free Radic. Res. 42, 372–377 (2008).
He, R. et al. Pharmacokinetic profile of N-acetylcysteine amide and its main metabolite in mice using new analytical method. Eur. J. Pharm. Sci. 143, 105158 (2020).
Sunitha, K. et al. N-Acetylcysteine amide: a derivative to fulfill the promises of N-Acetylcysteine. Free Radic. Res. 47, 357–367 (2013).
Wen, Q., Liu, J., Kang, R., Zhou, B. & Tang, D. The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 510, 278–283 (2019).
Hassannia, B. et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Invest. 128, 3341–3355 (2018).
Zhang, X. et al. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J. Cell Physiol. 235, 3425–3437 (2020).
Shin, D., Kim, E. H., Lee, J. & Roh, J. L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 129, 454–462 (2018).
Bai, Y. et al. Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 508, 997–1003 (2019).
Deng, F., Sharma, I., Dai, Y., Yang, M. & Kanwar, Y. S. Myo-inositol oxygenase expression profile modulates pathogenic ferroptosis in the renal proximal tubule. J. Clin. Invest. 129, 5033–5049 (2019).
Baba, Y. et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 314, H659–H668 (2018).
Fischer, W., Currais, A., Liang, Z., Pinto, A. & Maher, P. Old age-associated phenotypic screening for Alzheimer’s disease drug candidates identifies sterubin as a potent neuroprotective compound from Yerba santa. Redox Biol. 21, 101089 (2019).
Weiwer, M. et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg. Med. Chem. Lett. 22, 1822–1826 (2012).
Eaton, J. K., Ruberto, R. A., Kramm, A., Viswanathan, V. S. & Schreiber, S. L. Diacylfuroxans are masked nitrile oxides that inhibit GPX4 covalently. J. Am. Chem. Soc. 141, 20407–20415 (2019).
Eaton, J. K. et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat. Chem. Biol. 16, 497–506 (2020).
Shimada, K. et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503 (2016).
Verma, N. et al. Synthetic lethal combination targeting BET uncovered intrinsic susceptibility of TNBC to ferroptosis. Sci. Adv. 6, eaba8968 (2020).
Gaschler, M. M. et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 14, 507–515 (2018).
Wu, K. et al. Creatine kinase B suppresses ferroptosis by phosphorylating GPX4 through a moonlighting function. Nat. Cell Biol. 25, 714–725 (2023).
Hangauer, M. J. et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017).
Hatfield, D. L. & Gladyshev, V. N. How selenium has altered our understanding of the genetic code. Mol. Cell Biol. 22, 3565–3576 (2002).
Wang, D. et al. Antiferroptotic activity of non-oxidative dopamine. Biochem. Biophys. Res. Commun. 480, 602–607 (2016).
Li, C. et al. Novel allosteric activators for ferroptosis regulator glutathione peroxidase 4. J. Med. Chem. 62, 266–275 (2019).
Wang, S., Liu, W., Wang, J. & Bai, X. Curculigoside inhibits ferroptosis in ulcerative colitis through the induction of GPX4. Life Sci. 259, 118356 (2020).
Zhou, Y. X., Zhang, H. & Peng, C. Puerarin: a review of pharmacological effects. Phytother. Res. 28, 961–975 (2014).
Liu, B. et al. Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochem. Biophys. Res Commun. 497, 233–240 (2018).
Hayes, J. D. & Dinkova-Kostova, A. T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39, 199–218 (2014).
Boettler, U. et al. Coffee constituents as modulators of Nrf2 nuclear translocation and ARE (EpRE)-dependent gene expression. J. Nutr. Biochem. 22, 426–440 (2011).
Kong, L. et al. Sitagliptin activates the p62-Keap1-Nrf2 signalling pathway to alleviate oxidative stress and excessive autophagy in severe acute pancreatitis-related acute lung injury. Cell Death Dis. 12, 928 (2021).
Liu, N., Lin, X. & Huang, C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br. J. Cancer 122, 279–292 (2020).
Wang, L. et al. A pharmacological probe identifies cystathionine beta-synthase as a new negative regulator for ferroptosis. Cell Death Dis. 9, 1005 (2018).
Martinez, A. M., Mirkovic, J., Stanisz, Z. A., Patwari, F. S. & Yang, W. S. NSC-34 motor neuron-like cells are sensitized to ferroptosis upon differentiation. FEBS Open Bio. 9, 582–593 (2019).
Kang, E. S. et al. xCT deficiency aggravates acetaminophen-induced hepatotoxicity under inhibition of the transsulfuration pathway. Free Radic. Res. 51, 80–90 (2017).
Wu, M., Xu, L. G., Li, X., Zhai, Z. & Shu, H. B. AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J. Biol. Chem. 277, 25617–25623 (2002).
Yoshioka, H. et al. Identification of a small molecule that enhances ferroptosis via inhibition of ferroptosis suppressor protein 1 (FSP1). ACS Chem. Biol. 17, 483–491 (2022).
Hendricks, J. M. et al. Identification of structurally diverse FSP1 inhibitors that sensitize cancer cells to ferroptosis. Cell Chem. Biol. 30, 1–14 (2023).
Fang, Y., Tan, Q., Zhou, H., Gu, Q. & Xu, J. Discovery of novel diphenylbutene derivative ferroptosis inhibitors as neuroprotective agents. Eur. J. Med. Chem. 231, 114151 (2022).
Hensley, C. T., Wasti, A. T. & DeBerardinis, R. J. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Invest. 123, 3678–3684 (2013).
Luo, M. et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 25, 1457–1472 (2018).
Shin, D., Lee, J., You, J. H., Kim, D. & Roh, J. L. Dihydrolipoamide dehydrogenase regulates cystine deprivation-induced ferroptosis in head and neck cancer. Redox Biol. 30, 101418 (2020).
Lv, C. et al. Low-dose paclitaxel inhibits tumor cell growth by regulating glutaminolysis in colorectal carcinoma cells. Front. Pharm. 8, 244 (2017).
Ye, J., Jiang, X., Dong, Z., Hu, S. & Xiao, M. Low-concentration PTX and RSL3 inhibits tumor cell growth synergistically by inducing ferroptosis in mutant p53 hypopharyngeal squamous carcinoma. Cancer Manag. Res. 11, 9783–9792 (2019).
Jiang, L. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015).
Ou, Y., Wang, S. J., Li, D., Chu, B. & Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl Acad. Sci. USA 113, E6806–E6812 (2016).
Tarangelo, A. et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 22, 569–575 (2018).
Xie, Y. et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 20, 1692–1704 (2017).
Vasan, K., Werner, M. & Chandel, N. S. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 32, 341–352 (2020).
Xiong, R. et al. Novel and potent inhibitors targeting DHODH are broad-spectrum antivirals against RNA viruses including newly-emerged coronavirus SARS-CoV-2. Protein Cell 11, 723–739 (2020).
Gong, M. et al. Novel quinolone derivatives targeting human dihydroorotate dehydrogenase suppress Ebola virus infection in vitro. Antivir. Res. 194, 105161 (2021).
Chen, X., Li, J., Kang, R., Klionsky, D. J. & Tang, D. Ferroptosis: machinery and regulation. Autophagy 17, 2054–2081 (2021).
Kuhn, H. et al. Structural biology of mammalian lipoxygenases: enzymatic consequences of targeted alterations of the protein structure. Biochem. Biophys. Res. Commun. 338, 93–101 (2005).
Angeli, J. P. F., Shah, R., Pratt, D. A. & Conrad, M. Ferroptosis inhibition: mechanisms and opportunities. Trends Pharm. Sci. 38, 489–498 (2017).
Haeggstrom, J. Z. & Funk, C. D. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem. Rev. 111, 5866–5898 (2011).
Kuhn, H., Banthiya, S. & van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim Biophys. Acta 1851, 308–330 (2015).
Pergola, C. et al. Cinnamyl-3,4-dihydroxy-alpha-cyanocinnamate is a potent inhibitor of 5-lipoxygenase. J. Pharm. Exp. Ther. 338, 205–213 (2011).
Gregus, A. M. et al. Systematic analysis of rat 12/15-lipoxygenase enzymes reveals critical role for spinal eLOX3 hepoxilin synthase activity in inflammatory hyperalgesia. FASEB J. 27, 1939–1949 (2013).
Probst, L., Dachert, J., Schenk, B. & Fulda, S. Lipoxygenase inhibitors protect acute lymphoblastic leukemia cells from ferroptotic cell death. Biochem. Pharm. 140, 41–52 (2017).
Liu, Y. et al. The 5-lipoxygenase inhibitor zileuton confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis. Biol. Pharm. Bull. 38, 1234–1239 (2015).
Luci, D. K. et al. Synthesis and structure-activity relationship studies of 4-((2-hydroxy-3-methoxybenzyl)amino)benzenesulfonamide derivatives as potent and selective inhibitors of 12-lipoxygenase. J. Med. Chem. 57, 495–506 (2014).
Adili, R. et al. First selective 12-LOX inhibitor, ML355, impairs thrombus formation and vessel occlusion in vivo with minimal effects on hemostasis. Arterioscler Thromb. Vasc. Biol. 37, 1828–1839 (2017).
Ma, K. et al. 12-Lipoxygenase inhibitor improves functions of cytokine-treated human islets and type 2 diabetic islets. J. Clin. Endocrinol. Metab. 102, 2789–2797 (2017).
Zhang, X. J. et al. A small molecule targeting ALOX12-ACC1 ameliorates nonalcoholic steatohepatitis in mice and macaques. Sci. Transl. Med. 13, eabg8116 (2021).
Zhao, J., Wu, Y., Liang, S. & Piao, X. Activation of SSAT1/ALOX15 axis aggravates cerebral ischemia/reperfusion injury via triggering neuronal ferroptosis. Neuroscience 485, 78–90 (2022).
Ma, X. H. et al. ALOX15-launched PUFA-phospholipids peroxidation increases the susceptibility of ferroptosis in ischemia-induced myocardial damage. Signal Transduct. Target Ther. 7, 288 (2022).
Walters, J. L. H. et al. Pharmacological inhibition of arachidonate 15-lipoxygenase protects human spermatozoa against oxidative stress. Biol. Reprod. 98, 784–794 (2018).
Cai, W. et al. Alox15/15-HpETE aggravates myocardial ischemia-reperfusion injury by promoting cardiomyocyte ferroptosis. Circulation 147, 1444–1460 (2023).
Zou, Y. et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 16, 302–309 (2020).
Devisscher, L. et al. Discovery of novel, drug-Like ferroptosis inhibitors with in vivo efficacy. J. Med. Chem. 61, 10126–10140 (2018).
Zilka, O. et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent. Sci. 3, 232–243 (2017).
Hanthorn, J. J., Valgimigli, L. & Pratt, D. A. Incorporation of ring nitrogens into diphenylamine antioxidants: striking a balance between reactivity and stability. J. Am. Chem. Soc. 134, 8306–8309 (2012).
Shah, R., Margison, K. & Pratt, D. A. The potency ofdiarylamine radical-trapping antioxidants as inhibitors of ferroptosis underscores the role of autoxidation in the mechanism of cell death. ACS Chem. Biol. 12, 2538–2545 (2017).
Mishima, E. et al. Drugs repurposed as antiferroptosis agents suppress organ damage, including AKI, by functioning as lipid peroxyl radical scavengers. J. Am. Soc. Nephrol. 31, 280–296 (2020).
Homma, T., Kobayashi, S., Sato, H. & Fujii, J. Edaravone, a free radical scavenger, protects against ferroptotic cell death in vitro. Exp. Cell Res. 384, 111592 (2019).
Zilka, O., Poon, J. F. & Pratt, D. A. Radical-trapping antioxidant activity of copper and nickel bis(thiosemicarbazone) complexes underlies their potency as inhibitors of ferroptotic cell death. J. Am. Chem. Soc. 143, 19043–19057 (2021).
Wu, Z. et al. Hydropersulfides inhibit lipid peroxidation and protect cells from ferroptosis. J. Am. Chem. Soc. 144, 15825–15837 (2022).
Barayeu, U. et al. Hydropersulfides inhibit lipid peroxidation and ferroptosis by scavenging radicals. Nat. Chem. Biol. 19, 28–37 (2023).
Askari, B. et al. Rosiglitazone inhibits acyl-CoA synthetase activity and fatty acid partitioning to diacylglycerol and triacylglycerol via a peroxisome proliferator-activated receptor-gamma-independent mechanism in human arterial smooth muscle cells and macrophages. Diabetes 56, 1143–1152 (2007).
Shah, R., Shchepinov, M. S. & Pratt, D. A. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 4, 387–396 (2018).
Shchepinov, M. S. et al. Isotopic reinforcement of essential polyunsaturated fatty acids diminishes nigrostriatal degeneration in a mouse model of Parkinson’s disease. Toxicol. Lett. 207, 97–103 (2011).
Cotticelli, M. G., Crabbe, A. M., Wilson, R. B. & Shchepinov, M. S. Insights into the role of oxidative stress in the pathology of Friedreich ataxia using peroxidation resistant polyunsaturated fatty acids. Redox Biol. 1, 398–404 (2013).
Magtanong, L. et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem. Biol. 26, 420–432.e429 (2019).
Xu, S. et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 54, 1561–1577.e1567 (2021).
Hess, D., Chisholm, J. W. & Igal, R. A. Inhibition of stearoylCoA desaturase activity blocks cell cycle progression and induces programmed cell death in lung cancer cells. PLoS One 5, e11394 (2010).
Batchuluun, B., Pinkosky, S. L. & Steinberg, G. R. Lipogenesis inhibitors: therapeutic opportunities and challenges. Nat. Rev. Drug Discov. 21, 283–305 (2022).
Minami, J. K. et al. CDKN2A deletion remodels lipid metabolism to prime glioblastoma for ferroptosis. Cancer Cell 41, 1048–1060.e9 (2023).
Di Stasi, S. L., MacLeod, T. D., Winters, J. D. & Binder-Macleod, S. A. Effects of statins on skeletal muscle: a perspective for physical therapists. Phys. Ther. 90, 1530–1542 (2010).
Skottheim, I. B., Gedde-Dahl, A., Hejazifar, S., Hoel, K. & Asberg, A. Statin induced myotoxicity: the lactone forms are more potent than the acid forms in human skeletal muscle cells in vitro. Eur. J. Pharm. Sci. 33, 317–325 (2008).
Licarete, E., Sesarman, A. & Banciu, M. Exploitation of pleiotropic actions of statins by using tumour-targeted delivery systems. J. Microencapsul. 32, 619–631 (2015).
Yuan, B. et al. Dihydroartemisinin inhibits the proliferation, colony formation and induces ferroptosis of lung cancer cells by inhibiting PRIM2/SLC7A11 axis. Onco Targets Ther. 13, 10829–10840 (2020).
Lin, R. et al. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 381, 165–175 (2016).
Yi, R. et al. Dihydroartemisinin initiates ferroptosis in glioblastoma through GPX4 inhibition. Biosci. Rep. 40, BSR20193314 (2020).
Du, J. et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic. Biol. Med. 131, 356–369 (2019).
Mishra, D., Jain, N., Rajoriya, V. & Jain, A. K. Glycyrrhizin conjugated chitosan nanoparticles for hepatocyte-targeted delivery of lamivudine. J. Pharm. Pharm. 66, 1082–1093 (2014).
Ojha, S., Javed, H., Azimullah, S., Abul Khair, S. B. & Haque, M. E. Glycyrrhizic acid attenuates neuroinflammation and oxidative stress in rotenone model of Parkinson’s disease. Neurotox. Res. 29, 275–287 (2016).
Wang, X. R., Hao, H. G. & Chu, L. Glycyrrhizin inhibits LPS-induced inflammatory mediator production in endometrial epithelial cells. Micro. Pathog. 109, 110–113 (2017).
Wang, Y., Chen, Q., Shi, C., Jiao, F. & Gong, Z. Mechanism of glycyrrhizin on ferroptosis during acute liver failure by inhibiting oxidative stress. Mol. Med. Rep. 20, 4081–4090 (2019).
Huang, Y., Tsang, S. Y., Yao, X. & Chen, Z. Y. Biological properties of baicalein in cardiovascular system. Curr. Drug Targets Cardiovasc Haematol. Disord. 5, 177–184 (2005).
Afanas’ev, I. B., Dorozhko, A. I., Brodskii, A. V., Kostyuk, V. A. & Potapovitch, A. I. Chelating and free radical scavenging mechanisms of inhibitory action of rutin and quercetin in lipid peroxidation. Biochem. Pharm. 38, 1763–1769 (1989).
Patel, H. H. et al. 12-lipoxygenase in opioid-induced delayed cardioprotection: gene array, mass spectrometric, and pharmacological analyses. Circ. Res 92, 676–682 (2003).
van Leyen, K. et al. Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke 37, 3014–3018 (2006).
Xie, Y. et al. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem. Biophys. Res. Commun. 473, 775–780 (2016).
Shen, Z. et al. Fenton-reaction-acceleratable magnetic nanoparticles for ferroptosis therapy of orthotopic brain tumors. ACS Nano 12, 11355–11365 (2018).
Lu, X., Gao, S., Lin, H. & Shi, J. Single-atom catalysts for nanocatalytic tumor therapy. Small 17, e2004467 (2021).
Zhang, P. et al. Coordinatively unsaturated Fe(3+) based activatable probes for enhanced MRI and therapy of tumors. Angew. Chem. Int Ed. Engl. 58, 11088–11096 (2019).
Zhu, J. et al. Upconverting nanocarriers enable triggered microtubule inhibition and concurrent ferroptosis induction for selective treatment of triple-negative breast cancer. Nano Lett. 20, 6235–6245 (2020).
Xu, X., Chen, Y., Zhang, Y., Yao, Y. & Ji, P. Highly stable and biocompatible hyaluronic acid-rehabilitated nanoscale MOF-Fe(2+) induced ferroptosis in breast cancer cells. J. Mater. Chem. B 8, 9129–9138 (2020).
Hu, Z., Wang, S., Dai, Z., Zhang, H. & Zheng, X. A novel theranostic nano-platform (PB@FePt-HA-g-PEG) for tumor chemodynamic-photothermal co-therapy and triple-modal imaging (MR/CT/PI) diagnosis. J. Mater. Chem. B 8, 5351–5360 (2020).
Zhang, D. et al. Tumor microenvironment responsive FePt/MoS2 nanocomposites with chemotherapy and photothermal therapy for enhancing cancer immunotherapy. Nanoscale 11, 19912–19922 (2019).
Huo, M., Wang, L., Chen, Y. & Shi, J. Tumor-selective catalytic nanomedicine by nanocatalyst delivery. Nat. Commun. 8, 357 (2017).
Bao, W. et al. Nanolongan with multiple on-demand conversions for ferroptosis-apoptosis combined anticancer therapy. ACS Nano 13, 260–273 (2019).
Xue, C. C. et al. Tumor microenvironment-activatable Fe-doxorubicin preloaded amorphous CaCO3 nanoformulation triggers ferroptosis in target tumor cells. Sci. Adv. 6, eaax1346 (2020).
Ma, P. et al. Enhanced cisplatin chemotherapy by iron oxide nanocarrier-mediated generation of highly toxic reactive oxygen species. Nano Lett. 17, 928–937 (2017).
Wang, S. et al. Arginine-rich manganese silicate nanobubbles as a ferroptosis-inducing agent for tumor-targeted theranostics. ACS Nano 12, 12380–12392 (2018).
Liu, T. et al. Ferrous-supply-regeneration nanoengineering for cancer-cell-specific ferroptosis in combination with imaging-guided photodynamic therapy. ACS Nano 12, 12181–12192 (2018).
Guan, Q. et al. Mesoporous polydopamine carrying sorafenib and SPIO nanoparticles for MRI-guided ferroptosis cancer therapy. J. Control Release 320, 392–403 (2020).
Li, K. et al. Multienzyme-like reactivity cooperatively impairs glutathione peroxidase 4 and ferroptosis suppressor protein 1 pathways in triple-negative breast cancer for sensitized ferroptosis therapy. ACS Nano 16, 2381–2398 (2022).
Kim, S. E. et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 11, 977–985 (2016).
Zheng, D. W. et al. Switching apoptosis to ferroptosis: metal-organic network for high-efficiency anticancer therapy. Nano Lett. 17, 284–291 (2017).
Gao, M. et al. Triggered ferroptotic polymer micelles for reversing multidrug resistance to chemotherapy. Biomaterials 223, 119486 (2019).
Li, Y. et al. Nanoparticle ferritin-bound erastin and rapamycin: a nanodrug combining autophagy and ferroptosis for anticancer therapy. Biomater. Sci. 7, 3779–3787 (2019).
Zhao, Y., Zhao, W., Lim, Y. C. & Liu, T. Salinomycin-loaded gold nanoparticles for treating cancer stem cells by ferroptosis-induced cell death. Mol. Pharm. 16, 2532–2539 (2019).
Xiao, K. et al. Pro-oxidant response and accelerated ferroptosis caused by synergetic Au(I) release in hypercarbon-centered gold(I) cluster prodrugs. Nat. Commun. 13, 4669 (2022).
Sang, M. et al. Mitochondrial membrane anchored photosensitive nano-device for lipid hydroperoxides burst and inducing ferroptosis to surmount therapy-resistant cancer. Theranostics 9, 6209–6223 (2019).
He, S., Jiang, Y., Li, J. & Pu, K. Semiconducting polycomplex nanoparticles for photothermal ferrotherapy of cancer. Angew. Chem. Int. Ed. Engl. 59, 10633–10638 (2020).
Zhu, T. et al. Ferroptosis promotes photodynamic therapy: supramolecular photosensitizer-inducer nanodrug for enhanced cancer treatment. Theranostics 9, 3293–3307 (2019).
Xu, T. et al. Enhanced ferroptosis by oxygen-boosted phototherapy based on a 2-in-1 nanoplatform of ferrous hemoglobin for tumor synergistic therapy. ACS Nano 14, 3414–3425 (2020).
Meng, X. et al. Triggered all-active metal organic framework: ferroptosis machinery contributes to the apoptotic photodynamic antitumor therapy. Nano Lett. 19, 7866–7876 (2019).
Nie, T. et al. Bioactive iridium nanoclusters with glutathione depletion ability for enhanced sonodynamic-triggered ferroptosis-like cancer cell death. Adv. Mater. 34, e2206286 (2022).
Liu, C. et al. Biodegradable biomimic copper/manganese silicate nanospheres for chemodynamic/photodynamic synergistic therapy with simultaneous glutathione depletion and hypoxia relief. ACS Nano 13, 4267–4277 (2019).
Zhang, F. et al. Engineering magnetosomes for ferroptosis/immunomodulation synergism in cancer. ACS Nano 13, 5662–5673 (2019).
Wan, X. et al. Tumor-targeted cascade nanoreactor based on metal-organic frameworks for synergistic ferroptosis-starvation anticancer therapy. ACS Nano 14, 11017–11028 (2020).
Yang, Z. et al. Tumor-killing nanoreactors fueled by tumor debris can enhance radiofrequency ablation therapy and boost antitumor immune responses. Nat. Commun. 12, 4299 (2021).
Yao, X. et al. Multifunctional nanoplatform for photoacoustic imaging-guided combined therapy enhanced by CO induced ferroptosis. Biomaterials 197, 268–283 (2019).
Li, L. et al. Polystyrene nanoparticles reduced ROS and inhibited ferroptosis by triggering lysosome stress and TFEB nucleus translocation in a size-dependent manner. Nano Lett. 19, 7781–7792 (2019).
Dharmalingam, P. et al. Pervasive genomic damage in experimental intracerebral hemorrhage: therapeutic potential of a mechanistic-based carbon nanoparticle. ACS Nano 14, 2827–2846 (2020).
Hopkins, A. L. & Groom, C. R. The druggable genome. Nat. Rev. Drug Discov. 1, 727–730 (2002).
Luo, T. et al. Intracellular delivery of glutathione peroxidase degrader induces ferroptosis in vivo. Angew. Chem. Int Ed. Engl. 61, e202206277 (2022).
Liu, S. et al. PDTAC: targeted photodegradation of GPX4 triggers ferroptosis and potent antitumor immunity. J. Med. Chem. 65, 12176–12187 (2022).
Banik, S. M. et al. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297 (2020).
Takahashi, D. et al. AUTACs: cargo-specific degraders using selective autophagy. Mol. Cell 76, 797–810.e710 (2019).
Hanan, E. J. et al. Monomeric targeted protein degraders. J. Med. Chem. 63, 11330–11361 (2020).
Liang, J. et al. GDC-9545 (Giredestrant): a potent and orally bioavailable selective estrogen receptor antagonist and degrader with an exceptional preclinical profile for ER+ breast cancer. J. Med. Chem. 64, 11841–11856 (2021).
Domostegui, A., Nieto-Barrado, L., Perez-Lopez, C. & Mayor-Ruiz, C. Chasing molecular glue degraders: screening approaches. Chem. Soc. Rev. 51, 5498–5517 (2022).
Fischer, E. S. et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 (2014).
Yang, X., Wang, Y., Byrne, R., Schneider, G. & Yang, S. Concepts of artificial intelligence for computer-assisted drug discovery. Chem. Rev. 119, 10520–10594 (2019).
Gupta, R. et al. Artificial intelligence to deep learning: machine intelligence approach for drug discovery. Mol. Divers 25, 1315–1360 (2021).
Li, G., Lin, P., Wang, K., Gu, C. C. & Kusari, S. Artificial intelligence-guided discovery of anticancer lead compounds from plants and associated microorganisms. Trends Cancer 8, 65–80 (2022).
Bhinder, B., Gilvary, C., Madhukar, N. S. & Elemento, O. Artificial intelligence in cancer research and precision medicine. Cancer Discov. 11, 900–915 (2021).
Wu, M. & Zhang, Y. Combining bioinformatics, network pharmacology and artificial intelligence to predict the mechanism of celastrol in the treatment of type 2 diabetes. Front. Endocrinol. (Lausanne) 13, 1030278 (2022).
Miao, L., Zhang, Y. & Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 20, 41 (2021).
Tian, Y., Deng, Z. & Yang, P. mRNA vaccines: a novel weapon to control infectious diseases. Front. Microbiol. 13, 1008684 (2022).
Lee, N. et al. xCT-driven expression of GPX4 determines sensitivity of breast cancer cells to ferroptosis inducers. Antioxidants (Basel) 10, 317 (2021).
El-Benhawy, S. A. et al. Studying ferroptosis and iron metabolism pre- and post-radiotherapy treatment in breast cancer patients. J. Egypt Natl Cancer Inst. 35, 4 (2023).
Yang, J., Wei, X., Hu, F., Dong, W. & Sun, L. Development and validation of a novel 3-gene prognostic model for pancreatic adenocarcinoma based on ferroptosis-related genes. Cancer Cell Int. 22, 21 (2022).
Ayton, S. et al. Cerebral quantitative susceptibility mapping predicts amyloid-beta-related cognitive decline. Brain 140, 2112–2119 (2017).
Dexter, D. T. et al. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 114, 1953–1975 (1991).
Simmons, D. A. et al. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia 55, 1074–1084 (2007).
Kitsugi, K. et al. Simvastatin inhibits hepatic stellate cells activation by regulating the ferroptosis signaling pathway. Biochim. Biophys. Acta Mol. Basis Dis. 1869, 166750 (2023).
Sun, S. et al. AMPK activation alleviated DSS-induced colitis by inhibiting ferroptosis. J. Dig. Dis. 24, 213–223 (2023).
Ousingsawat, J., Schreiber, R., Gulbins, E., Kamler, M. & Kunzelmann, K. P. aeruginosa induced lipid peroxidation causes ferroptotic cell death in airways. Cell Physiol. Biochem. 55, 590–604 (2021).
Yang, J. et al. Metformin induces ferroptosis by inhibiting UFMylation of SLC7A11 in breast cancer. J. Exp. Clin. Cancer Res 40, 206 (2021).
Du, J. et al. DHA exhibits synergistic therapeutic efficacy with cisplatin to induce ferroptosis in pancreatic ductal adenocarcinoma via modulation of iron metabolism. Cell Death Dis. 12, 705 (2021).
Alborzinia, H. et al. MYCN mediates cysteine addiction and sensitizes neuroblastoma to ferroptosis. Nat. Cancer 3, 471–485 (2022).
Yang, S. et al. Salidroside attenuates neuronal ferroptosis by activating the Nrf2/HO1 signaling pathway in Abeta(1-42)-induced Alzheimer’s disease mice and glutamate-injured HT22 cells. Chin. Med. 17, 82 (2022).
Huang, Z. et al. Moxibustion protects dopaminergic neurons in Parkinson’s disease through antiferroptosis. Evid. Based Complement Altern. Med. 2021, 6668249 (2021).
Puylaert, P. et al. Effect of erythrophagocytosis-induced ferroptosis during angiogenesis in atherosclerotic plaques. Angiogenesis https://doi.org/10.1007/s10456-023-09877-6 (2023).
Xiao, Z. et al. Reduction of lactoferrin aggravates neuronal ferroptosis after intracerebral hemorrhagic stroke in hyperglycemic mice. Redox Biol. 50, 102256 (2022).
Wang, Y. et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J. Adv. Res. 28, 231–243 (2021).
Tadokoro, T. et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 5, e132747 (2020).
Liu, H. et al. A novel function of ATF3 in suppression of ferroptosis in mouse heart suffered ischemia/reperfusion. Free Radic. Biol. Med. 189, 122–135 (2022).
Roh, J. L., Kim, E. H., Jang, H. & Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 11, 254–262 (2017).
Garcia-Bermudez, J. et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 567, 118–122 (2019).
Fan, Z. et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 6, e371 (2017).
Tonnus, W. et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat. Commun. 12, 4402 (2021).
Soula, M. et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 16, 1351–1360 (2020).
Radadiya, P. S. et al. Ciclopirox olamine induces ferritinophagy and reduces cyst burden in polycystic kidney disease. JCI Insight 6, e141299 (2021).
Fernandez-Mendivil, C. et al. Protective role of microglial HO-1 blockade in aging: Implication of iron metabolism. Redox Biol. 38, 101789 (2021).
Chen, Y., Zhang, P., Chen, W. & Chen, G. Ferroptosis mediated DSS-induced ulcerative colitis associated with Nrf2/HO-1 signaling pathway. Immunol. Lett. 225, 9–15 (2020).
Li, W., Yu, J., Jin, B., Zhang, H. & Zhang, J. Protective effects of aminooxyacetic acid on colitis induced in mice with dextran sulfate sodium. Biomed. Res. Int. 2021, 1477345 (2021).
Du, A. et al. Effects of aminooxyacetic acid on hippocampal mitochondria in rats with chronic alcoholism: the analysis of learning and memory-related genes. J. Integr. Neurosci. 18, 451–462 (2019).
Dang, R. et al. Edaravone ameliorates depressive and anxiety-like behaviors via Sirt1/Nrf2/HO-1/Gpx4 pathway. J. Neuroinflammation 19, 41 (2022).
Zeng, Y. et al. Edaravone attenuated particulate matter-induced lung inflammation by inhibiting ROS-NF-kappaB signaling pathway. Oxid. Med. Cell Longev. 2022, 6908884 (2022).
Yang, Y. et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 11, 433 (2020).
Hu, M. et al. Suppression of uterine and placental ferroptosis by N-acetylcysteine in a rat model of polycystic ovary syndrome. Mol. Hum. Reprod. 27, gaab067 (2021).
Li, Q. et al. NAC alleviative ferroptosis in diabetic nephropathy via maintaining mitochondrial redox homeostasis through activating SIRT3-SOD2/Gpx4 pathway. Free Radic. Biol. Med. 187, 158–170 (2022).
Huang, J. et al. The role of ferroptosis and endoplasmic reticulum stress in intermittent hypoxia-induced myocardial injury. Sleep Breath 27, 1005–1011 (2022).
Fan, Z., Cai, L., Wang, S., Wang, J. & Chen, B. Baicalin prevents myocardial ischemia/reperfusion injury through inhibiting ACSL4 mediated ferroptosis. Front. Pharm. 12, 628988 (2021).
Lee, M. S., Kim, D., Jo, K. & Hwang, J. K. Nordihydroguaiaretic acid protects against high-fat diet-induced fatty liver by activating AMP-activated protein kinase in obese mice. Biochem. Biophys. Res. Commun. 401, 92–97 (2010).
Ninomiya, I. et al. Pioglitazone inhibits the proliferation and metastasis of human pancreatic cancer cells. Oncol. Lett. 8, 2709–2714 (2014).
Takano, S. et al. Pioglitazone, a ligand for peroxisome proliferator-activated receptor-gamma acts as an inhibitor of colon cancer liver metastasis. Anticancer Res. 28, 3593–3599 (2008).
Castillo, A. F. et al. New inhibitor targeting Acyl-CoA synthetase 4 reduces breast and prostate tumor growth, therapeutic resistance and steroidogenesis. Cell Mol. Life Sci. 78, 2893–2910 (2021).
Fujita, M., Hasegawa, A., Yamamori, M. & Okamura, N. In vitro and in vivo cytotoxicity of troglitazone in pancreatic cancer. J. Exp. Clin. Cancer Res. 36, 91 (2017).
Lee, J. J. et al. 5-Lipoxygenase inhibition protects retinal pigment epithelium from sodium iodate-induced ferroptosis and prevents retinal degeneration. Oxid. Med. Cell Longev. 2022, 1792894 (2022).
Acknowledgements
The authors received support from the National Natural Science Foundation of China (31970689 to J.M.; 32330047, 31930057 to F.W.; and 32100941 to S.S.) and the National Key R&D Program (2018YFA0507801 to J.M. and 2018YFA0507802 to F.W.).
Author information
Authors and Affiliations
Contributions
All the authors contributed substantially to all aspects of the article. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, S., Shen, J., Jiang, J. et al. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Sig Transduct Target Ther 8, 372 (2023). https://doi.org/10.1038/s41392-023-01606-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01606-1
This article is cited by
-
Ferroptosis in cancer: From molecular mechanisms to therapeutic strategies
Signal Transduction and Targeted Therapy (2024)
-
A New Perspective in the Treatment of Ischemic Stroke: Ferroptosis
Neurochemical Research (2024)
