
Abstract
Meningiomas are the most common primary brain tumor and their incidence and prevalence is increasing. This review summarizes current evidence regarding the embryogenesis of the human meninges in the context of meningioma pathogenesis and anatomical distribution. Though not mutually exclusive, chromosomal instability and pathogenic variants affecting the long arm of chromosome 22 (22q) result in meningiomas in neural-crest cell-derived meninges, while variants affecting Hedgehog signaling, PI3K signaling, TRAF7, KLF4, and POLR2A result in meningiomas in the mesodermal-derived meninges of the midline and paramedian anterior, central, and ventral posterior skull base. Current evidence regarding the common pathways for genetic pathogenesis and the anatomical distribution of meningiomas is presented alongside existing understanding of the embryological origins for the meninges prior to proposing next steps for this work.
Similar content being viewed by others

Introduction
Meningiomas are the most common primary brain tumor, representing 37% of all intra-cranial tumors with an annual incidence of 4.5 per 100,000 people with a lifetime risk of around 1 in 280, and their incidence and prevalence is increasing [1,2,3]. Incidence over a 14-year period (1999–2013) of diagnoses and surgical resection of meningiomas have increased by 52% and 58% respectively [4]. Skull base meningiomas represent up to half of all meningiomas requiring surgery [5]. Due to their proximity to cranial nerves, brainstem, upper cervical spinal cord, and critical cerebral vasculature, they are challenging to resect completely (Fig. 1); consequently recurrence rates can be as high as 29% [6, 7]. Around a third of recurrences are of a higher tumor grade (World Health Organization (WHO) grade II and III [8]. Patients with atypical (WHO grade II) and malignant (WHO grade III) meningioma suffer from a high morbidity and mortality, with reported 10-year survival of 63% and 15% respectively, in spite of a relatively young mean age at diagnosis [1, 4]. Aside from radiotherapy which has a limited evidence base, there are scarce alternative therapies with proven efficacy [9].

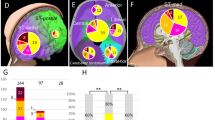
A Anatomical depiction of meninges with brain and spinal cord removed displaying skull base, sagittal, and convexity regions including tentorium cerebelli on the right side. B distribution of meningioma by pathogenic variant gene pathway. C meningeal embryonic development by the tissue of origin.
The cell of origin of a meningioma is frequently reported to be the arachnoid cap cell, primarily due to cytological similarity [2]. It is however more probable meningiomas develop both from dural border cells and arachnoid barrier cells based on the shared expression of prostaglandin D2 synthase (PGDS) in these cellular layers and meningiomas [10, 11]. This may also explain the broad spectrum of histologically distinct variants in the classification of meningiomas, which remain classified solely according to histological appearances (Table 1) [2]. Robust epidemiological data of the incidence of meningioma by histological subtype has not been reported. Based on genomic data from a recently published cohort, meningothelial (41%) and transitional (17%) subtypes represent the most common variants [12]. A study of registry data reported 80.6% of meningioma were WHO grade I, 17.6% WHO grade II, and 1.7% WHO grade III [1].
Cranial meningiomas most commonly develop in the convexity and parasagittal regions and in the skull base in relation to the sphenoid (Table 2) [13]. Registry data from the USA reported that 79.8% of meningiomas were located in the cranial meninges compared to 4.2% located in spinal meninges (the remainder were unknown) [1]. In an alternative study of 25,694 surgically treated meningiomas in England, 92.3% were located in the cranial meninges compared to 7.7% located in the spinal meninges [4].
Neurofibromatosis type 2 (NF2) is the most common and first identified driver gene associated with meningioma. NF2 encodes the protein Merlin, which was initially found to interact with CD44 during contact inhibition of cell proliferation, but additionally inhibits PI3K/mTORC1/Akt signaling pathways and activates the mammalian Hippo pathway [14]. This has traditionally been associated with convexity meningiomas in patients with NF2, a tumor suppressor syndrome where patients have a 50–75% lifetime risk of developing a meningioma [15,16,17]. However, pathogenic variants in NF2 have been identified in 40–60% of sporadic meningiomas [2]. Genomic analysis of NF2 and non-NF2 sporadic meningiomas have identified further pathogenic variants in AKT1 [18], AKT3 [19], BRCA1-associated protein 1 (BAP1) [20, 21], Kruppel-like factor 4 (KLF4) [18], PIK3CA [22], PIK3R1 [19], POLR2A [19], PRKA-R1A [19], SMARCB1 [23,24,25], subfamily B, member 1 (SMARCE1) [26], smoothened (SMO) [27], SUFU [28], and TNF receptor associated factor 7 (TRAF7) [18]. Epidemiological molecular data are lacking; based on the largest genomic study to date, NF2 remains the most commonly affected gene in meningiomas (Table 3) [12].
This review presents existing evidence of the relationship between the histological subtype, WHO grade, and genomic alterations of meningioma and the location of their development. The embryology of the meninges is presented, summarizing the hypothesis that the cephalic mesoderm contributes to the meninges of the midline and paramedian ventral posterior and central skull base. A synthesis is summarized including evidence of meningioma pathogenesis through interruption of genes in key developmental pathways. Implications for the utility of therapies used in other tumor types, development of in vitro and in vivo modeling of meningioma genetic pathogenesis, and patient selection for trials, are discussed.
Meningioma and its location—genomics and histology
With the advent of next-generation sequencing, significant advances have been made in the last decade in identifying pathogenic variants in meningioma tumorigenesis. These have been categorized into major gene pathways which demonstrate striking mutual exclusivity across multiple studies [3, 19]. These are summarized in Table 4.
22q deletion (NF2, SMARCB1)
Pathogenic variants in NF2 are associated with somatic loss of the second chromosome 22 allele [27, 29, 30], and are strongly though not exclusively associated with fibrous, psammomatous, transitional, atypical, and anaplastic meningioma. [27, 31,32,33] NF2-mutated meningioma constitute the majority of meningiomas located in the falx cerebri, tentorium cerebelli, and cerebral and cerebellar convexities [12, 18, 32]. In large-scale genomic studies of meningioma, higher grade (WHO grade II and III) meningioma were in some studies exclusively related to pathogenic variants in NF2, associated with mutations in the TERT promoter, and deletion of 1p and CDKN2A [30, 34]. SWI/SNF related, matrix associated, actin dependent regulator of chromatin, SMARCB1, adjacent to NF2 on chromosome 22q, has been identified to contribute to meningioma tumorigenesis with somatic missense mutations identified in exon 9 [24], and a germline variant in exon 2 [35]. In patients with NF2, those with truncating NF2 mutations towards the 5′end of the gene were associated with a higher prevalence and lifetime risk of meningioma [15]. A four-hit mechanism has been proposed resulting in tumor suppressor gene inactivation and the development of familial multiple meningiomas [25]. It is of interest that few variants associated with SMARCB1 related schwannomatosis have been associated with meningioma risk and overall the chances of developing meningioma in SMARCB1 related schwannomatosis without these specific missense variants is low [23, 36].
PI3K-AKT-mTOR pathway (AKT1, AKT3, PIK3CA, PIK3R1)
Pathogenic variants in AKT serine/threonine kinase 1 (AKT1), an oncogenic component of the (PI3K)-AKT-mTOR pathway, have been identified in non-NF2 meningiomas of the medial skull base [18, 27, 37]. Subsequent studies have additionally identified an association of AKT1 pathogenic variants with benign WHO grade I meningothelial subtype lacking genomic instability [18, 30, 34]. Mutually exclusive somatic pathogenic variants have additionally been identified in AKT3 [19], phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K), the catalytic subunit alpha (PI3KCA) and regulatory subunit alpha (PI3KR1) have also been identified in meningiomas with WHO grade I meningothelial or transitional histology arising from the medial skull base [19, 22].
Hedgehog signaling pathway (SMO, SUFU, PRKA-R1A)
SMO, a G-protein coupled receptor and key transmembrane protein member of the Hedgehog signaling pathway, was identified in several studies of non-NF2 meningiomas [18, 27]. SUFU (suppressor of fused homolog) protein acts downstream of SMO and loss of SUFU function has been implicated in familial multiple meningioma [28]. Pathogenic variants in PRKA-R1A have additionally been identified in a small proportion of meningiomas [19]. PRKA-R1A is a critical component of type I protein kinase A (PKA) and pathogenic variants result in increased PKA activity and subsequently increased SMO cell surface accumulation thus contributes to Hedgehog signaling [19, 38]. Meningiomas harboring pathogenic variants in the Hedgehog signaling pathway are more likely to develop as a WHO grade I meningothelial subtype in the midline anterior fossa floor of the skull base [12, 18, 27]. Across multiple studies it has been additionally demonstrated that meningiomas with pathogenic variants in the Hedgehog signaling pathway are not associated with genomic instability [18, 30, 34].
Other pathogenic variants (KLF4, TRAF7, SMARCE1, BAP1)
KLF4 is a transcription factor known to induce pluripotency in adult fibroblast cultures [39]. The role of KLF4 is context-specific, with evidence of its function both as an oncogene and tumor suppressor in cancer [40]. A highly recurrent p.Lys409Gln mutation was identified in the first of three zinc fingers pivotal for DNA binding [41]. Meningiomas with pathogenic variants involving KLF4 were more commonly identified in the skull base away from the midline [12]. Secretory meningiomas have been defined based on combined pathogenic variants of KLF4 and TRAF7 mutually exclusive of the PI3K pathway or NF2 [41]. Hallmarks of secretory meningioma, hyaline periodic acid-Schiff-positive globules, and peritumoral edema, are suspected mechanistically to be associated to KLF4 signaling as a result of regulatory of cytokeratins 4 and 19 and activation of the bradykinin B2 receptor [41].
TRAF7 has been reported as the most common pathogenic variant identified in non-NF2 meningioma [18, 19, 22]. Pathogenic variants in have been reported to occur in combination with AKT1, PIK3CA, PIK3R1, and KLF4 [12, 18, 19, 41]. Multiple somatic mutations were identified in an intronic hot spot of TRAF7 related to the first WD40 domain which plays an important regulatory role in the NF-κB pathway [18, 42]. Meningiomas with TRAF7 pathogenic variants alone or in combination commonly develop in the skull base, with isolated TRAF7-mutated meningioma associated with a microcystic histological subtype [12].
Recurrent pathogenic variants in polymerase (RNA) II (DNA directed) polypeptide A (POLR2A) are characterized by mutations localized to the dock domain involved in formation of the pre-initiation complex [19]. Meningiomas with identified pathogenic variants in POLR2A are most commonly mutually exclusive, genomically stable, and associated with benign meningiomas in the midline skull base, in particular the region of the tuberculum sellae [19].
BAP1 is involved in the response to DNA damage as a tumor suppressor gene functioning as a ubiquitin carboxy-terminal hydrolase [21]. Germline mutations in BAP1 result in a cancer syndrome involving the development of BAP1-mutated melanocytic skin tumors and a high incidence of mesothelioma [43]. All tumors share a common histological rhabdoid morphology. Of the six tumors with BAP1 mutations and BAP1 loss on immunohistochemistry, four were located in the convexity regions and two in the skull base [21]. There is currently insufficient evidence to demonstrate a spatial phenotype of these tumors.
SWI/SNF related, matrix associated, actin dependent regulator of chromatin, SMARCE1 pathogenic variants have been specifically associated with heritable clear cell meningiomas [26, 44, 45]. Initially suspected to present exclusively as multiple spinal meningiomas [26], cases of cranial meningiomas with pathogenic variants in SMARCE1 have subsequently been identified [44]. Clear cells are characterized by vacuolated cytoplasm and bland nuclei in a whorled, syncytial architecture, a likely consequence of SMARCE1 protein loss [46]. While the histology is diagnostic of WHO grade II clear cell subtype, of the few cases reported they have included meningiomas of the spine, convexity, and skull base regions without a propensity to a specific location [45]. A simplified summary of the above pathogenic variant categories and their relationship with histological subtype and the location of meningioma tumorigenesis is shown in Fig. 1B.
Meningioma and its location—embryological origin of the meninges
The most comprehensive early study of the development of the meninges was undertaken by O’Rahilly and Muller in 1986 [47]. This study of cranial meninges involved the serial sectioning of 61 human embryos. At Carnegie stage (hereafter stage) 11 (24 postovulatory days), the pia mater is first identified at the caudal medulla while elsewhere a thick mesenchyme surrounds the developing brain. This thick mesenchyme is derived from a combination of neural crest cell mesoectoderm and neurilemmal cells, the prechordal plate, the unsegmented paraxial mesoderm, and the segmented paraxial (somitic) mesoderm. By stage 15 (33 postovulatory days) this mesenchyme surrounds most of the brain and is called the primary meninx. Subsequently, the primary meninx differentiates into the pachymeninges (later dura mater) and leptomeninges (later arachnoid and pia mater) [47, 48]. Similar work was undertaken by Sensenig in characterizing the embryological origin of the spinal meninges, where paraxial somitic mesodermal and neural crest cells were concluded to contribute to the dura and arachnoid mater (mesodermal) and pia mater (neural crest), respectively [49].
The development of quail-chick chimeras resulted in the ability to track the migration of neural crest cells, demonstrating the contribution of the neural crest to the meninges of the forebrain while the meninges of the brainstem derive from cephalic mesoderm [50, 51]. HNK1 expression was used to further demonstrate a contribution of the neural crest to the spinal meninges, in contrast with earlier studies demonstrating an exclusively mesodermal contribution [52, 53].
The use of permanent molecular markers for neural crest cells and developmental stage-specific conditional knockout mice has resulted in significant progress with characterization of the embryonic origin of the cranial bones and meninges [10, 54,55,56,57]. The use of X-gal staining and Dil labeling has been used in transgenic mice with in vivo permanent labeling of neural crest and mesoderm (Wnt1-Cre/R26R and Mesp1-Cre/R26R strains, respectively) [54, 56]. A further PGDS transgenic Cre strain was developed based on PGDS representing a specific marker of arachnoidal cells. The PDGS positive meningeal cell was identified as a common precursor to both the dural border cells and arachnoid border cells [11, 58]. Collectively, these models have demonstrated that the meninges at the skull base derive from mesoderm, while the meninges covering the cerebral and cerebellar hemispheres derive from the neural crest [10, 54, 55, 59].
Most recently, single-cell transcriptomic analyses of meningeal fibroblasts in the forebrain have identified fibroblast populations that are transcriptionally distinct between brain regions, particularly in pia mater [60]. The authors state that anterior meninges arise from the neural crest, whereas posterior meninges originate from the mesoderm, and conclude that due to this mixed contribution there is regionalization of gene expression. Of particular relevance is the M3 subcluster, an arachnoid cell cluster, where in the embryonic day 14.5 (E14.5) mouse embryo in in situ validation there was patchy expression of Ptgds, the gene encoding PGDS, in the dorsal telencephalon contrasting with high expression throughout the skull base surrounding the midbrain and hindbrain regions [60].
Notable similarities have been identified between the development of the meninges and the skull bones. Animals with mutations in Foxc1, an identified gene crucial in meningeal development, develop significant meningeal and calvarial defects [61, 62]. Furthermore, intramembranous ossification of mesodermal bone requires interaction with neural crest-derived meninges [56, 63]. In the transgenic mouse models, the frontal, ethmoid, presphenoid, squamous temporal, and interparietal bones were identified as neural crest derived. Conversely, parietal, non-squamous temporal, and basioccipital bones are derive from mesoderm [55, 56, 59]. The middle of the basisphenoid, corresponding to the sella turcica in the adult skull base, marks the demarcation between bone derived from neural crest and mesoderm with the notable exception of the post-optic root of the presphenoid bone which is derived from mesoderm [55, 64].
The identified junction at which cranial bones are derived from mesoderm and neural crest are species-specific, and it is probable that this is also observed in the meninges [65]. Overall, the likely development of the adult human meninges is a complex interplay between neural crest and mesodermally derived cells resulting in differentiation into cytologically similar meninges in the adult (Fig. 1).
Meningioma genetic pathogenesis and embryological development—a synthesis
The above evidence demonstrates a clear, reproducible correlation between the location of a meningioma and the types of underlying pathogenic variants identified as driving tumorigenesis. There is evidence that the spatial contribution of the mesoderm and neural crest to the meninges correlates with the locations of commonly identified pathogenic variants in meningioma. In molecular profiling of 86 sequenced, spatially distinct meningiomas, expression of neural crest genes have been implicated in meningeal tumorigenesis [66], suggesting that meningioma tumorigenesis capitalizes on gene regulatory networks with subsequent misactivation of a developmental cell population [67, 68]. However, there still remains a great deal that is unknown; in the largest study of 1970 meningiomas with targeted and/or whole exome sequencing, 667 (26.1%) did not have an identified mutation [12]. While mechanistic explanations have now been provided for the development of multiple histological subtypes, the underlying genomic characteristics of microcystic grade I and chordoid grade II meningioma also remain unknown [12, 69]. This section reviews the underlying mechanisms of tumorigenesis in the meninges and its relationship to developmental pathways.
The neural crest, NF2-Hippo, and the SWI/SNF complex
The development of neural-crest-derived tissues is dependent on a process balancing proliferation, migration, and pluripotency that share many characteristics with tumorigenesis [70]. The development of NF2 knockout mice resulted in greater understanding of the role of the gene during development. NF2 null mice die during embryonic development due to a failure to initiate gastrulation [71], while heterozygous models resulted in widespread tumor development [72], and conditional NF2 gene inactivation in leptomeningeal cells resulted in the development of meningiomas [73]. The use of a β‐gal reporter under the control of an NF2 promoter in transgenic mice identified intense β‐gal staining in forebrain and telencephalon extending caudally in the later covered by pia mater, consistent with meningeal layers derived from the neural crest and the most common locations for the development of NF2-mutated meningioma [74].
Merlin is known to have multiple functions, but with respect to meningioma pathogenesis it notable for its role as a tumor suppressor regulating proliferation and apoptosis through Hippo signaling [75], and in cellular motility, spreading and attachment through mediation of the actin cytoskeleton [76]. Yap, a component of the Hippo pathway and inhibited upstream by Merlin, has been implicated in neural crest cell fate and migration [77, 78]. SMARCB1 loss in the early neural crest results in the development of human rhabdoid tumors, while induced loss at a later stage results in Schwannomatosis [79]. Overall, the SWI/SNF complex is strongly linked to mammalian differentiation and is a critical regulator of pluripotency in embryonic stem cells, with SMARCB1 essential for neural induction but non-essential for mesodermal differentiation [67, 80].
The relationship between Hedgehog signaling and meningioma pathogenesis is perhaps the most convincing. In a zebrafish model, Hedgehog signaling is required for cranial morphogenesis and chondrogenesis in the midline of the zebrafish skull [81]. Dysregulation results in craniofacial defects including holoprosencephaly and hypotelorism [82]. SMO-mutated meningiomas occur predominantly in the midline anterior skull base. Conditional activation of SMO in developing mouse embryos resulted in the development of meningothelial meningiomas in the ventral skull base, with similar location and histological appearances to SMO-mutated meningioma [83].
The role of the cranial neural crest and mesoderm in craniofacial development is not mutually exclusive, with interdependence identified in the patterning of facial tissues and chondrogenesis [56, 63, 84]. Manipulation of migratory and proliferative behaviors reveals crucial interactions between the two cell populations in normal embryogenesis. Although a simplification, on review of the embryological origin of the meninges, there is reason to hypothesize that the relative contribution of cell types to different layers and regions of the meninges may contribute to an explanation for these spatial phenotypes.
Future directions
Existing in vitro models use highly malignant immortalized meningioma cells that do not represent the diverse genomic characteristics of meningiomas, particularly of the skull base [33]. Challenges exist due to the senescence of in vitro cell lines of benign meningiomas [2]. The different developmental progenitors of meninges of the skull base should be considered in any future meningioma models including pathogenic variants commonly found in this region. Pathogenic variants associated with specific histological subtypes will likely be incorporated into future classification guidance, and it is recommended that location is included as part of this. Where whole genome sequencing is not possible for every patient, targeted sequencing of pathogenic variants corresponding to the location of the resected meningioma will be crucial to facilitate participation in trials with targeted therapy and for prognostic information for patients. The development of a molecularly driven trial of patients given targeted therapy based on their AKT1, SMO, and NF2 pathogenic variant status is an ideal example of the future for clinical trials in patients with meningioma (NCT02523014) [85].
There are still conflicting accounts regarding the cell of origin of the meningioma, with arachnoid cap cells, arachnoid barrier cells and dural border cells candidates. There has been limited consideration to tumor heterogeneity although limited unpublished evidence suggests this could be substantial [66]. The tumor microenvironment should be examined in the context of extended understanding of the heterogeneity of meningiomas, and the gene regulatory networks underlying meningeal development given its correlation with the genomic signatures of meningiomas. DNA methylation profiling has been used successfully to predict risk of recurrence and prognosis across multiple studies, and demonstrates the significance of epigenetic modifications in tumor pathogenesis, particularly given the relative lack of chromosomal instability in meningiomas of the skull base [30, 86]. Capitalizing on spatial and temporal transcriptomics will facilitate greater understanding in both animal models and tumor samples of remaining candidate pathogenic variants responsible for meningioma pathogenesis and identify developmental pathways that could be modulated resulting in more effective targeted therapies for these tumors.
With the advent of immunotherapy, understanding of the tumor microenvironment has become pivotal in identifying potential immune-mediated mechanisms for treatments. Understanding of the tumor microenvironment in meningioma is comparatively understudied, with no existing single-cell transcriptomic immune cell profiling currently. T cell repertoire characterization of 28 meningiomas of all grades identified populations of CD4+ and CD8+ T cells, regulatory T cells, and T cells expressing PD-1 (Programmed cell death protein 1) indicative of exhaustion [87]. A study of bulk transcriptomic data from 107 meningiomas identified immune processes to be the sole biological mechanism correlated with anatomical location after correcting for the WHO grade in the tumor [88]. Whereas oncolytic gamma-delta T cells dominate skull base meningiomas, mast cells and neutrophils were more prominent in convexity meningiomas [88]. Conversely, a study of tumor-associated macrophage infiltration in meningioma found no significant differences in the macrophage number or ratio of M1 to M2 phenotype between the skull base and convexity meningioma samples [89].
Given the increasing importance of location in the understanding of tumor biology and immune microenvironment, biologically and clinically meaningful and accurate classification is essential. Despite the numerous surgical classifications of meningioma subtypes [90,91,92,93], and classifications in studies including genomic characteristics [12], there is currently no international consensus regarding the reporting of a location of a meningioma, particularly classification of meningiomas of the skull base. Reaching a consensus will facilitate cross-study comparisons and drive standardization in the investigation and reporting of meningioma pathogenesis.
Conclusions
In summary, there is emerging evidence of a correlation between location, phenotype and genotype in meningioma and such correlation has its basis on the embryology of meninges. A combination of temporal and spatial epigenetic and genetic analyses is required to better characterize the developing meninges, the arachnoid from which meningiomas are thought to derive, and meningiomas themselves to advance our understanding of these tumors for further biomarker and therapy discovery and implementation.
Change history
14 September 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41388-021-01700-0
References
Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol. 2018;20:iv1–86.
Riemenschneider MJ, Perry A, Reifenberger G. Histological classification and molecular genetics of meningiomas. Lancet Neurol. 2006;5:1045–54.
Smith MJ. Germline and somatic mutations in meningiomas. Cancer Genet. 2015;208:107–14.
Brodbelt AR, Barclay ME, Greenberg D, Williams M, Jenkinson MD, Karabatsou K. The outcome of patients with surgically treated meningioma in England: 1999–2013. A cancer registry data analysis The outcome of patients with surgically treated meningioma in England: 1999–2013. A cancer registry data analysis. 2019. https://doi.org/10.1080/02688697.2019.1661965.
Magill ST, Young JS, Chae R, Aghi MK, Theodosopoulos PV, McDermott MW. Relationship between tumor location, size, and WHO grade in meningioma. Neurosurg Focus. 2018;44. https://doi.org/10.3171/2018.1.FOCUS17752.
Nanda A, Vannemreddy P. Recurrence and outcome in skull base meningiomas: do they differ from other intracranial meningiomas? Skull Base. 2008;18:243–52.
Chen C-M, Huang AP-H, Kuo L-T, Tu Y-K. Contemporary surgical outcome for skull base meningiomas. Neurosurg Rev. 2011;34:281–96.
Magill ST, Dalle Ore CL, Diaz MA, Jalili DD, Raleigh DR, Aghi MK, et al. Surgical outcomes after reoperation for recurrent skull base meningiomas. J Neurosurg. 2018;130:876–83.
Brastianos PK, Galanis E, Butowski N, Chan JW, Dunn IF, Goldbrunner R, et al. Advances in multidisciplinary therapy for meningiomas. Neuro Oncol. 2019;21:i18–31.
Kalamarides M, Stemmer-Rachamimov AO, Niwa-Kawakita M, Chareyre F, Taranchon E, Han Z-Y, et al. Identification of a progenitor cell of origin capable of generating diverse meningioma histological subtypes. Oncogene. 2011;30:2333–44.
Yamashima T, Sakuda K, Tohma Y, Yamashita J, Oda H, Irikura D, et al. Prostaglandin D synthase (β-Trace) in human arachnoid and meningioma cells: roles as a cell marker or in cerebrospinal fluid absorption, tumorigenesis, and calcification process. J Neurosci. 1997;17:2376–82.
Youngblood MW, Duran D, Montejo JD, Li C, Omay SB, Özduman K, et al. Correlations between genomic subgroup and clinical features in a cohort of more than 3000 meningiomas. J Neurosurg. 2019;1–10. Online ahead of print.
Ketter R, Kim Y, Feiden W. Correspondence of tumor localization with tumor recurrence and cytogenetic progression in meningiomas. Neurosurgery. 2008;62:61–70.
Petrilli AM, Fernández-Valle C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene. 2016;35:537–48.
Smith MJ, Higgs JE, Bowers NL, Halliday D, Paterson J, Gillespie J, et al. Cranial meningiomas in 411 neurofibromatosis type 2 (NF2) patients with proven gene mutations: Clear positional effect of mutations, but absence of female severity effect on age at onset. J Med Genet. 2011;48:261–5.
Evans DG, Huson SM, Donnai D, Neary W, Blair V, Teare D, et al. A genetic study of type 2 neurofibromatosis in the United Kingdom. I. Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet. 1992;29:841.
Aavikko M, Li SP, Saarinen S, Alhopuro P, Kaasinen E, Morgunova E, et al. Supplementary material for loss of SUFU function in familial multiple meningioma. Am J Hum Genet. 2012;91:520–6.
Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Ozduman K, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339:1077–80.
Clark VE, Harmancl AS, Bai H, Youngblood MW, Lee TI, Baranoski JF, et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat Genet. 2016;48:1253–9.
Abdel-Rahman MH, Pilarski R, Cebulla CM, Massengill JB, Christopher BN, Boru G, et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011;48:856–9.
Shankar GM, Abedalthagafi M, Vaubel RA, Merrill PH, Nayyar N, Gill CM, et al. Germline and somatic BAP1 mutations in high-grade rhabdoid meningiomas. Neuro Oncol. 2017;19:535–45.
Abedalthagafi M, Bi WL, Aizer AA, Merrill PH, Brewster R, Agarwalla PK, et al. Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro Oncol. 2016;18:649–55.
Hadfield KD, Smith MJ, Trump D, Newman WG, Evans DG. SMARCB1 mutations are not a common cause of multiple meningiomas. J Med Genet. 2010;47:567–8.
Schmitz U, Mueller W, Weber M, Sévenet N, Delattre O, Deimling Avon. INI1 mutations in meningiomas at a potential hotspot in exon 9. Br J Cancer. 2001;84:199–201.
Christiaans I, Kenter SB, Brink HC, Van Os TAM, Baas F, Van Den Munckhof P, et al. Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J Med Genet. 2011;48:93–97.
Smith MJ, O’Sullivan J, Bhaskar SS, Hadfield KD, Poke G, Caird J, et al. Loss-of-function mutations in SMARCE1 cause an inherited disorder of multiple spinal meningiomas. Nat Genet. 2013;45:295–8.
Brastianos PK, Horowitz PM, Santagata S, Jones RT, Mckenna A, Getz G, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet. 2013;45:285–9.
Aavikko M, Li SP, Saarinen S, Alhopuro P, Kaasinen E, Morgunova E, et al. Loss of SUFU function in familial multiple meningioma. Am J Hum Genet. 2012;91:520–6.
Mawrin C, Perry A. Pathological classification and molecular genetics of meningiomas. J Neurooncol. 2010;99:379–91.
Sahm F, Schrimpf D, Stichel D, Jones DTW, Hielscher T, Schefzyk S, et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. 2017;18:682–94.
Wellenreuther R, Kraus JA, Lenartz D, Menon AG, Schramm J, Louis DN, et al. Analysis of the neurofibromatosis 2 gene reveals molecular variants of meningioma. 1995. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1869258/pdf/amjpathol00052-0049.pdf. Accessed 28 Jun 2020.
Kros J, De Greve K, Van Tilborg A, Hop W, Pieterman H, Avezaat C, et al. NF2 status of meningiomas is associated with tumour localization and histology. J Pathol. 2001;194:367–72.
Preusser M, Brastianos PK, Mawrin C. Advances in meningioma genetics: novel therapeutic opportunities. Nat Rev Neurol. 2018;14:106–15.
Patel AJ, Wan YW, Al-Ouran R, Revelli JP, Cardenas MF, Oneissi M, et al. Molecular profiling predicts meningioma recurrence and reveals loss of DREAM complex repression in aggressive tumors. Proc Natl Acad Sci USA. 2019;116:21715–26.
van den Munckhof P, Christiaans I, Kenter SB, Baas F, Hulsebos TJM. Germline SMARCB1 mutation predisposes to multiple meningiomas and schwannomas with preferential location of cranial meningiomas at the falx cerebri. Neurogenetics. 2012;13:1–7.
Smith MJ, Wallace AJ, Bowers NL, Rustad CF, Woods CG, Leschziner GD, et al. Frequency of SMARCB1 mutations in familial and sporadic schwannomatosis. Neurogenetics. 2012;13:141–5.
Sahm F, Bissel J, Koelsche C, Schweizer L, Capper D, Reuss D, et al. AKT1E17K mutations cluster with meningothelial and transitional meningiomas and can be detected by SFRP1 immunohistochemistry. Acta Neuropathol. 2013;126:757–62.
Jia J, Tong C, Wang B, Luo L, Jiang J. Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature. 2004;432:1045–50.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76.
Rowland BD, Peeper DS. KLF4, p21 and context-dependent opposing forces in cancer. Nat Rev Cancer. 2006;6:11–23.
Reuss DE, Piro RM, Jones DTW, Simon M, Ketter R, Kool M, et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol. 2013;125:351–8.
Bouwmeester T, Bauch A, Ruffner H, Angrand P-O, Bergamini G, Croughton K, et al. A physical and functional map of the human TNF-α/NF-κB signal transduction pathway. Nat Cell Biol. 2004;6:97–105.
Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer. Nat Rev Cancer. 2013;13:153–9.
Smith MJ, Wallace AJ, Bennett C, Hasselblatt M, Elert-Dobkowska E, Evans LT, et al. Germline SMARCE1 mutations predispose to both spinal and cranial clear cell meningiomas. J Pathol. 2014;234:436–40.
Smith MJ, Ahn S, Lee JIL, Bulman M, Plessis Ddu, Suh YL. SMARCE1 mutation screening in classification of clear cell meningiomas. Histopathology. 2017;70:814–20.
Imlay SP, Snider TE, Raab SS. Clear-cell meningioma: diagnosis by fine-needle aspiration biopsy. Diagn Cytopathol. 1998;18:131–6.
O’Rahilly R, Müller F. The meninges in human development. J Neuropathol Exp Neurol 1986;45:588–608.
Lopes MBS. Meninges: Embryology. In: Meningiomas. London: Springer London; 2009. pp 25–29.
Sensenig E. The early development of the meninges of the spinal cord in human embryos. Contrib Embryol. 1951;228:145–157.
Catalá M. Embryonic and fetal development of structures associated with the cerebro-spinal fluid in man and other species. Part I: The ventricular system, meninges and choroid plexuses. undefined 1998. https://www.semanticscholar.org/paper/Embryonic-and-fetal-development-of-structures-with-Catalá/1ead37fe2af4bcd55608f8d6225ad6c80916b9bc. Accessed 21 Jul 2020.
Couly GF, Coltey PM, Le Douarin NM. The developmental fate of the cephalic mesoderm in quail-chick chimeras. 1992. https://dev.biologists.org/content/develop/114/1/1.full.pdf. Accessed 29 Jun 2020.
Batarfi M, Valasek P, Krejci E, Huang R, Patel. The development and origins of vertebrate meninges. Biol Commun. 2017;55–6273–81.
Halata Z, Grim M, Christ B. Origin of spinal cord meninges, sheaths of peripheral nerves, and cutaneous receptors including Merkel cells. Anat Embryol. 1990;182:529–37.
Yoshida T, Vivatbutsiri P, Morriss-Kay G, Saga Y, Iseki S. Cell lineage in mammalian craniofacial mesenchyme. Mech Dev. 2008;125:797–808.
McBratney-Owen B, Iseki S, Bamforth SD, Olsen BR, Morriss-Kay GM. Development and tissue origins of the mammalian cranial base. Dev Biol. 2008;322:121–32.
Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM. Tissue origins and interactions in the mammalian skull vault. Dev Biol. 2002;241:106–16.
Jiang X, Rowitch DH, Soriano P, Mcmahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. 2000;1616:1607–16.
Yamashima T. Human meninges: anatomy and its role in meningioma pathogenesis. In: Meningiomas. London: Springer London; 2009, p. 15–24.
Dasgupta K, Jeong J. Developmental biology of the meninges. Genesis. 2019;57:1–12.
DeSisto J, O’Rourke R, Jones HE, Pawlikowski B, Malek AD, Bonney S, et al. Single-cell transcriptomic analyses of the developing meninges reveal meningeal fibroblast diversity and function. Dev Cell. 2020;54:43–59.e4.
Kume T, Deng KY, Winfrey V, Gould DB, Walter MA, Hogan BL. The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell. 1998;93:985–96.
Siegenthaler JA, Ashique AM, Zarbalis K, Patterson KP, Hecht JH, Kane MA, et al. Retinoic acid from the meninges regulates cortical neuron generation. Cell. 2009;139:597–609.
Siegenthaler JA, Pleasure SJ. We have got you ‘covered’: how the meninges control brain development. Curr Opin Genet Dev. 2011;21:249–55.
Nie X. Cranial base in craniofacial development: developmental features, influence on facial growth, anomaly, and molecular basis. Acta Odontol Scand. 2005;63:127–35.
Pan A, Chang L, Nguyen A, James AW, Bronner-Fraser M, Cobourne M, et al. A review of hedgehog signaling in cranial bone development. 2013. https://doi.org/10.3389/fphys.2013.00061.
Magill S, Vasudevan H, Seo K, John Liu S, Hilz S, Villanueva-Meyer J, et al. TMOD-27. a neural crest cell subpopulation underlies intratumor heterogeneity in meningioma. Neuro Oncol. 2019;21:vi268.
Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene. 2009;28:1653–68.
Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461–73.
Goldbrunner R, Minniti G, Preusser M, Jenkinson MD, Sallabanda K, Houdart E, et al. EANO guidelines for the diagnosis and treatment of meningiomas. Lancet Oncol. 2016;17:e383–91.
Maguire LH, Thomas AR, Goldstein AM. Tumors of the neural crest: common themes in development and cancer. Dev Dyn. 2015;244:311–22.
McClatchey AI, Saotome I, Ramesh V, Gusella JF, Jacks T. The Nf2 tumor suppressor gene product is essential for extraembryonic development immediately prior to gastrulation. Genes Dev. 1997;11:1253–65.
McClatchey AI, Saotome I, Mercer K, Crowley D, Gusella JF, Bronson RT, et al. Mice heterozygous for a mutation at the Nf2 tumor suppressor locus develop a range of highly metastatic tumors. Genes Dev. 1998;12:1121–33.
Kalamarides M, Niwa-Kawakita M, Leblois H, Abramowski V, Perricaudet M, Janin A, et al. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. 2002. https://doi.org/10.1101/gad.226302.
Akhmametyeva EM, Mihaylova MM, Luo H, Kharzai S, Welling DB, Chang L-S. Regulation of the neurofibromatosis 2 gene promoter expression during embryonic development. Dev Dyn. 2006;235:2771–85.
Gutmann DH, Giovannini M. Mouse models of neurofibromatosis 1 and 2. Neoplasia. 2002;4:279–90.
Laulajainen M, Muranen T, Carpén O, Grönholm M. Protein kinase A-mediated phosphorylation of the NF2 tumor suppressor protein merlin at serine 10 affects the actin cytoskeleton. Oncogene. 2008;27:3233–43.
Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. 2010. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2925178/pdf/nihms-225649.pdf. Accessed 24 Jul 2020.
Hindley CJ, Condurat AL, Menon V, Thomas R, Azmitia LM, Davis JA, et al. The Hippo pathway member YAP enhances human neural crest cell fate and migration. Sci Rep. 2016;6:23208.
Vitte J, Gao F, Coppola G, Judkins AR, Giovannini M. Timing of Smarcb1 and Nf2 inactivation determines schwannoma versus rhabdoid tumor development. Nat Commun. 2017;8:1–13.
Langer LF, Ward JM, Archer TK. Tumor suppressor SMARCB1 suppresses super-enhancers to govern hESC lineage determination. Elife. 2019;8. https://doi.org/10.7554/eLife.45672.
Wada N, Javidan Y, Nelson S, Carney TJ, Kelsh RN, Schilling TF. Hedgehog signaling is required for cranial neural crest morphogenesis and chondrogenesis at the midline in the zebrafish skull. Development. 2005;132:3977–88.
Belloni E, Muenke M, Roessler E, Traverse G, Siegel-Bartelt J, Frumkin A, et al. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet. 1996;14:353–6.
Boetto J, Apra C, Bielle F, Peyre M, Kalamarides M. Selective vulnerability of the primitive meningeal layer to prenatal Smo activation for skull base meningothelial meningioma formation. Oncogene. 2018;37:4955–63.
Trainor P, Krumlauf R. Plasticity in mouse neural crest cells reveals a new patterning role for cranial mesoderm. Nat Cell Biol. 2000;2:96–102.
Brastianos PK. Vismodegib and FAK Inhibitor GSK2256098 in treating patients with progressive meningiomas. Webpage. 2017;2–5.
Nassiri F, Mamatjan Y, Suppiah S, Badhiwala JH, Mansouri S, Karimi S, et al. DNA methylation profiling to predict recurrence risk in meningioma: Development and validation of a nomogram to optimize clinical management. Neuro Oncol. 2019;21:901–10.
Fang L, Lowther DE, Meizlish ML, Anderson RCE, Bruce JN, Devine L, et al. The immune cell infiltrate populating meningiomas is composed of mature, antigen-experienced T and B cells. https://doi.org/10.1093/neuonc/not110.
Zador Z, Landry AP, Balas M, Cusimano MD. Landscape of immune cell gene expression is unique in predominantly WHO grade 1 skull base meningiomas when compared to convexity. Sci Rep. 2020;10:9065.
Proctor DT, Huang J, Lama S, Albakr A, Van Marle G, Sutherland GR. Tumor-associated macrophage infiltration in meningioma. Neuro-Oncology Adv. 2019;1. https://doi.org/10.1093/noajnl/vdz018.
Almefty R, Dunn IF, Pravdenkova S, Abolfotoh M, Al-Mefty O. True petroclival meningiomas: results of surgical management. Clinical article. J Neurosurg. 2014;120:40–51.
Al-Mefty O. Clinoidal meningiomas. J Neurosurg. 1990;73:840–9.
Bonnal J, Thibaut A, Brotchi J, Born J. Invading meningiomas of the sphenoid ridge. J Neurosurg. 1980;53:587–99.
DeMonte F, McDermott MW, Al-Mefty O Al-Mefty’s meningiomas. Thieme, 2011.
Funding
DMF is supported by the National Institute for Health Research (NIHR) and a Cancer Research UK (CRUK) Predoctoral Research Bursary (C72069/A30348). DGE and MJS are supported by the all Manchester National Institute for Health Research Biomedical Research Centre (IS-BRC-1215-20007).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: The funding statement was missing.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fountain, D.M., Smith, M.J., O’Leary, C. et al. The spatial phenotype of genotypically distinct meningiomas demonstrate potential implications of the embryology of the meninges. Oncogene 40, 875–884 (2021). https://doi.org/10.1038/s41388-020-01568-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-020-01568-6
This article is cited by
-
Metastatic meningioma: a case series and systematic review
Acta Neurochirurgica (2023)
-
Clinical significance of NF2 alteration in grade I meningiomas revisited; prognostic impact integrated with extent of resection, tumour location, and Ki-67 index
Acta Neuropathologica Communications (2022)
