
Abstract
Current pharmacological treatments for bipolar disorder are inadequate and based on serendipitously discovered drugs often with limited efficacy, burdensome side-effects, and unclear mechanisms of action. Advances in drug development for the treatment of bipolar disorder remain incremental and have come largely from repurposing drugs used for other psychiatric conditions, a strategy that has failed to find truly revolutionary therapies, as it does not target the mood instability that characterises the condition. The lack of therapeutic innovation in the bipolar disorder field is largely due to a poor understanding of the underlying disease mechanisms and the consequent absence of validated drug targets. A compelling new treatment target is the Ca2+-calmodulin dependent protein kinase kinase-2 (CaMKK2) enzyme. CaMKK2 is highly enriched in brain neurons and regulates energy metabolism and neuronal processes that underpin higher order functions such as long-term memory, mood, and other affective functions. Loss-of-function polymorphisms and a rare missense mutation in human CAMKK2 are associated with bipolar disorder, and genetic deletion of Camkk2 in mice causes bipolar-like behaviours similar to those in patients. Furthermore, these behaviours are ameliorated by lithium, which increases CaMKK2 activity. In this review, we discuss multiple convergent lines of evidence that support targeting of CaMKK2 as a new treatment strategy for bipolar disorder.
Similar content being viewed by others

Introduction
Bipolar disorder is a disabling and lifelong mental condition that affects >1% of the global population that is characterised by extreme fluctuations in mood [1]. There are four subtypes of bipolar disorder that are categorised based on the occurrence, duration, and intensity of manic and depressive episodes [2]. Bipolar 1 disorder involves at least one manic episode, with or without a depressive episode or psychosis, whereas bipolar 2 disorder involves depressive episodes with a least one current or past hypomanic episode. Cyclothymic disorder is defined by recurrent subthreshold episodes of hypomania and mild depression, and unspecified bipolar disorder involves bipolar-like symptoms that do not satisfy the diagnostic criteria for the other subtypes. Patients with bipolar disorder experience significant cognitive and functional impairments and are at high risk of premature death from drug abuse and suicide, as well as comorbidities such as the metabolic syndrome and cardiovascular diseases [3,4,5]. Consequently, life expectancy is markedly reduced (>10 years) compared with the general population [6]. The burden of disease and high mortality rate of bipolar disorder has remained unchanged for decades, demonstrating that current standard treatments for bipolar disorder are inadequate for many patients [7].
Drug therapy remains the cornerstone treatment for bipolar disorder and is mainly based on serendipitously discovered drugs that often display limited efficacy and poor tolerability [8]. Polypharmacy involving the use of combination drug therapies is standard care in the treatment of bipolar disorder, but involves higher costs and burden, as well as increased risk of drug-to-drug interactions [9]. A large proportion of bipolar disorder patients endure residual mood symptoms, and frequent switching between mood states despite treatment, which severely impacts their lives and long-term prognosis [8]. Compounding these issues, the adverse side-effects of current drugs are associated with frequent medication changes and high rates of non-adherence to treatment [10]. These unresolved problems highlight an unmet clinical need for new drugs that are effective and safe, to achieve full remission of symptoms and better mood-stabilization [11]. The failure to develop effective, mechanism-based therapies is a direct result of major gaps in our understanding of the molecular and cellular defects that cause bipolar disorder, which has prevented the identification of rational treatment targets. Here, we review an emerging treatment target for bipolar disorder, the Ca2+-calmodulin-dependent protein kinase kinase-2 (CaMKK2).
Defective Ca2+-signalling and bipolar disorder
Ca2+-signalling plays a fundamental role in the brain and regulates an array of critical functions including neuronal excitation, gene expression, neurotransmitter release, and synaptic plasticity, all of which support learning, memory and the control of mood and behaviour [12]. Defective Ca2+-signalling has long been implicated in the pathogenesis of bipolar disorder and related psychiatric conditions [13]. Mitochondria serve as important regulators of cellular Ca2+ homoeostasis and are able to modulate intracellular Ca2+-signalling due to their capability to absorb high levels of cytoplasmic Ca2+ [14]. Mitochondrial dysfunction is considered to play an underlying role in bipolar disorder, and abnormal accumulation of mitochondrial Ca2+ is a potent trigger of necrosis, apoptosis, and autophagy [15,16,17,18]. A recent systematic review and meta-analysis found evidence for increased free intracellular Ca2+ levels in patients with bipolar disorder [13]. This is supported by studies using neurons differentiated from patient-derived induced pluripotent stem cells (iPSC), which were found to display hyperexcitability and increased intracellular Ca2+ levels, as well as increased transcription of genes involved in Ca2+-signalling, compared with neurons derived from healthy unaffected controls [19, 20]. Elevated Ca2+-levels have also been reported in B-lymphoblasts and platelets extracted from bipolar disorder patients [21, 22]. The apparent increase in Ca2+ levels prompted investigations into the use of Ca2+-channel blockers, particularly drugs that block L-type voltage-gated Ca2+-channels, as potential treatments [23, 24]. However, Ca2+-channel blockers have failed to become an established treatment for bipolar disorder as there is mixed evidence on their efficacy, which perhaps argues that the observed increase in free intracellular Ca2+ in patients may simply be a marker of the underlying disturbance rather than causative [25].
An alternative view that has emerged from recent studies in mice suggest a link between reduced brain Ca2+ activity and bipolar disorder. For example, a two-photon imaging study of a ketamine and stress-induced mouse model of bipolar disorder, demonstrated that brain Ca2+ activity measured in situ was reduced in mice displaying both manic and depressive-like behaviours [26]. Similarly, conditional knockout mice lacking Cacna1c in the cerebral cortex, which encodes the L-type, voltage-dependent, Ca2+-channel, alpha 1 C subunit that is among the most commonly identified risk genes for bipolar disorder, have reduced spontaneous cortical Ca2+ activity and display hyperactive, manic-like behaviour [27, 28]. Likewise, loss-of-function mutations in another Ca2+-channel – Transient receptor potential cation channel, subfamily M, member 2 (TRPM2) – are also associated with human bipolar disorder and Trpm2 null mice display manic-like behaviours [29].
Despite a substantial body of evidence implicating abnormalities in intracellular Ca2+ dynamics in the pathophysiology of bipolar disorder, the specific Ca2+-signalling defects in the brain that underpin the characteristic manic and depressive behaviours remains unclear.
What is CaMKK2, and why is it a compelling treatment target for bipolar disorder?
Many neuronal processes regulated by Ca2+ are mediated through calmodulin, a ubiquitous Ca2+-sensing protein that binds and modifies the function of a diverse range of downstream effectors in response to increased intracellular Ca2+ [30]. CaMKK2 is a serine/threonine protein kinase and the central component of a Ca2+-calmodulin activated signalling pathway (Fig. 1) that regulates crucial brain functions including long-term memory formation, mood and emotional behaviour, and energy metabolism [31,32,33]. It is activated by voltage-dependent Ca2+-channels, as well as neurotransmitter and hormone receptors, that increase intracellular Ca2+ and cause accumulation of the Ca2+-calmodulin complex. These include CACNA1C, the NMDA receptor (NMDAR), and the Gαq-protein-coupled serotonin and ghrelin receptors [34]. Conversely, hormones that stimulate cyclic-AMP-dependent protein kinase (PKA) signalling, such as glucagon-like peptide-1 (GLP-1), inhibit CaMKK2 [35]. CAMKK2 mRNA is widely expressed in the adult human brain, with high levels in the amygdala, basal ganglia, cerebellum, cerebral cortex, hippocampus, and hypothalamus [36]. Experimental mice such as the C57BL6/6 J strain that are widely used as the genetic background for transgenic mouse models of bipolar disorder display an equivalent Camkk2 gene expression profile in the brain [37]. The level of CAMKK2 gene expression in human brain is low during early development, but expression levels markedly increase during late childhood/early adulthood which, notably, coincides with the average age-of-onset of bipolar disorder [38, 39].

CaMKK2 is activated endogenously by voltage-gated Ca2+-channels (Cav1.2), in addition to neurotransmitter and hormone receptors that increase intracellular Ca2+ and cause accumulation of the Ca2+-calmodulin (Ca2+-CaM) complex. It can also be activated exogenously by the mood-stabiliser drug, lithium (Li+). Conversely, CaMKK2 is inhibited by CDK5 and GSK3, as well as by hormones that stimulate PKA signalling such as glucagon-like peptide-1 (GLP-1). Activated CaMKK2 directly phosphorylates three known downstream effectors (CaMK1, CaMK4 and AMPK) through which it regulates a range of neuronal and metabolic processes that support brain function.
In addition to Ca2+-calmodulin regulation, CaMKK2 is also regulated by phosphorylation of the S3-node, which is a control switch in the N-terminal regulatory sequence of CaMKK2 composed of three tandem serine residues (S3) that are sequentially phosphorylated by cyclin-dependent protein kinase-5 (CDK5) and glycogen synthase kinase-3 (GSK3) [40]. The S3-node functions as a two-input logic gate that inhibits CaMKK2 activity only when both the CDK5 and GSK3 signalling pathways are activated. Notably, lithium, the mood-stabiliser and frontline treatment for bipolar disorder, increases CaMKK2 activity by blocking GSK3-mediated phosphorylation of the S3-node [33]. Once activated, CaMKK2 stimulates downstream signalling pathways and gene expression programs that regulate neurogenesis, synaptic formation and plasticity, and mitochondrial function [41,42,43,44]. For example, activation of CaMKK2 in mice increases the expression of brain-derived neurotrophic factor (BDNF), a pivotal regulator of neuronal function [45]. Several meta-analyses have reported that serum BDNF levels are significantly decreased in both the manic and depressive phases of bipolar disorder [46,47,48]. Also, BDNF expression is increased by lithium treatment in humans and mice, and may be critical to the anti-manic effects of lithium [49, 50].
In the following sections, we expand on the molecular, genetic, pharmacological, and phenomenological evidence that link CaMKK2 to bipolar disorder.
Upstream mechanisms that regulate CaMKK2 activity
The regulation of CaMKK2 activity is complex and involves an interplay between allosteric activation by Ca2+-calmodulin, autophosphorylation, and phosphorylation of regulatory sites by kinases that are coupled to signalling pathways controlled by various neurotransmitters and metabolic hormones [51]. CaMKK2 has a modular structure, composed of a catalytic kinase domain and a regulatory segment containing overlapping autoinhibitory and calmodulin-binding sequences, which are flanked by N- and C-terminal sequences of unknown function (Fig. 2) [52]. Binding of Ca2+-calmodulin to the calmodulin-binding sequence increases CaMKK2 activity by preventing the adjacent autoinhibitory sequence from hindering the catalytic site in the kinase domain [53]. In human CaMKK2, Ca2+-calmodulin binding induces autophosphorylation of a threonine residue (Thr85) located in the N-terminal regulatory sequence, which creates a molecular memory that enables CaMKK2 to remain in the activated state following an initial, transient Ca2+-signal [33]. CaMKK2 activity is also increased by autophosphorylation of another threonine residue (Thr482) located in the autoinhibitory sequence [54].

Linear schematic of the domain structure of human CaMKK2 illustrating the position of the catalytic kinase domain, the autoinhibitory (AIS) and calmodulin-binding sequences (CaMBS), and regulatory phosphorylation sites, as well as the bipolar disorder-linked T85S polymorphism and rare R311C mutation (green). Autophosphorylation of Thr85 and Thr482 (yellow) increase CaMKK2 activity. In the S3 node, phosphorylation of Ser137 (red) by CDK5 primes CaMKK2 for sequential phosphorylation on Ser133 and Ser129 (blue) by GSK3, which results in CaMKK2 inhibition. Phosphorylation of Ser100, Ser495 and Ser511 (magenta) by PKA prevents CaMKK2 activation by Ca2+-calmodulin, and causes binding of 14-3-3 adaptor proteins that keep CaMKK2 in an inactivated state.
The activation of CaMKK2 by Ca2+-calmodulin is regulated by inhibitory crosstalk with the PKA signalling pathway [35, 55, 56]. PKA phosphorylates a conserved serine residue (Ser495) in the calmodulin-binding sequence of CaMKK2 (Fig. 2), which prevents Ca2+-calmodulin binding and activation. In addition, phosphorylation of two further serine residues (Ser100 and Ser511) by PKA mediates the recruitment of 14-3-3 adaptor proteins that hold CaMKK2 in an inactivated state by preventing dephosphorylation of phospho-Ser495. The death-associated protein kinase-1 (DAPK1) has also been reported to phosphorylate Ser511 on CaMKK2 [57]. Like CaMKK2, the PKA signalling pathway is also considered to play a key role in the pathogenesis of bipolar disorder, as increased cyclic AMP levels and PKA activity have been frequently observed in post-mortem brains and peripheral cells of bipolar disorder patients [58,59,60,61]. Recently, the largest whole-exome sequencing study of bipolar disorder conducted to date demonstrated a role for rare coding variations in the A-kinase anchoring protein-11 (AKAP11) as a significant risk factor in bipolar disorder aetiology [62]. AKAP proteins function as signalling hubs and are targeted to defined subcellular locations to enable spacial integration of the PKA signalling pathway with other signal transduction networks [63]. AKAP11 binds to vesicles, peroxisomes and centrosomes and forms signalling hubs at these intracellular locations with PKA, GSK3 and Ras GTPase-activating-like protein-1 (IQGAP1), all of which directly regulate CaMKK2 activity [35, 40, 64]. PKA and GSK3 are protein kinases whereas IQGAP1 is a GTPase activating protein (GAP) for the small G-proteins, CDC42 and Rac1, both of which play essential roles in neurogenesis and display altered expression in bipolar disorder [65, 66]. These data demonstrate that CaMKK2 is a component of a signalling hub that enables crosstalk between kinase and small G-protein signalling networks that regulate brain function.
CaMKK2 activity is also modulated by a Ca2+-calmodulin-independent mechanism involving sequential phosphorylation of three tandem serine residues in the regulatory S3-node switch (Fig. 2) [40, 54]. The S3-node modulates CaMKK2 activity by regulating the interaction between the autoinhibitory sequence and the catalytic kinase domain [67]. Phosphorylation of Ser137 in the S3-node by CDK5 primes CaMKK2 for subsequent phosphorylation on Ser133 and Ser129 by GSK3, which results in inhibition of CaMKK2 activity [40]. The CDK5 priming event is critical and functions as a gatekeeping mechanism that enables GSK3 to inhibit CaMKK2. The regulation of CaMKK2 by CDK5 and GSK3 has important implications for bipolar disorder aetiology for two reasons. First, CDK5 is a regulator of circadian rhythm, and its activity oscillates over a 24-hour period [68, 69]. This is revealing, as abnormalities in circadian rhythms are considered to play a potential underlying role in bipolar disorder, as many genes associated with bipolar disorder including CLOCK, PER3 and BMAL1 are regulated in a circadian manner [70,71,72]. Disruptions in circadian rhythm have also been widely reported in patients with bipolar disorder, and sleep deprivation is a known trigger, particularly of manic and hypomanic episodes [73,74,75,76]. Second, GSK3 is regulated in a circadian manner and has been linked to bipolar disorder for over two decades since the discovery that lithium, directly and indirectly, inhibits GSK3 activity [77,78,79]. Intriguingly, a recent study found that circadian rhythms in neurons differentiated from bipolar disorder patient-derived iPSCs predict lithium response [80]. Hyperactivation of GSK3 as a result of deficient inhibitory serine phosphorylation in the N-terminal regulatory tail occurs in bipolar disorder patients and correlates with the severity of manic and depressive symptoms [81]. GSK3 activation is also associated with oxidative stress and inflammation, both of which have been reported to be elevated in bipolar disorder patients [82,83,84,85]. Significantly, neurons from mice expressing a CaMKK2 mutant that mimics constitutive GSK3 phosphorylation of the S3-node, have neurite outgrowth abnormalities and fail to establish appropriate axon-dendrite polarity [40]. In contrast, neurons expressing a CaMKK2 mutant that mimics the effect of lithium by blocking GSK3 phosphorylation of the S3-node, display normal neurite outgrowth and polarity [33, 40]. These findings imply that signalling via the S3-node in CaMKK2 is critical for regulating neuron morphology and indicates that some of the beneficial effects of lithium on neuronal health may be mediated, at least in part, via the GSK3-CaMKK2 signalling pathway.
The integration of kinase (PKA, CDK5, GSK3) and small G-protein (IQGAP1) signalling pathways through CaMKK2, all of which have demonstrated links with human bipolar disorder, points to the possibility that bipolar disorder is a signalopathy that stems from defects in this signal transduction network, and potentially explains the clinical heterogeneity and polygenic nature of the condition.
Downstream effector kinases of CaMKK2
Four known downstream effectors of CaMKK2 are the Ca2+-calmodulin-dependent protein kinases-1 and -4 (CaMK1 and CaMK4), the AMP-activated protein kinase (AMPK), and Akt/protein kinase-B (PKB) [86,87,88,89,90,91,92,93]. CaMKK2 increases the activity of each effector target by phosphorylating a highly-conserved threonine residue located within the regulatory activation loops of each of their kinase domains [51]. Through these effectors, CaMKK2 is able to regulate a variety of cellular processes that are essential for the maintenance of neuronal activity and brain function [44].
CaMK1 plays multiple roles in neuronal development and plasticity and is required for the regulation of axonal outgrowth and growth cone morphology, dendritic branching, and formation of dendritic spines and synapses [94,95,96,97,98]. Activation of CaMK1 promotes synaptogenesis via phosphorylation of p21-activated kinase interacting exchange factor (βPIX), which co-localises with the scaffolding proteins, G-protein-coupled receptor kinase-interacting protein-1 (GIT1) and Shank2, in the postsynaptic density of dendritic spines as part of a multiprotein complex that regulates actin dynamics [94, 99]. In hippocampal neurons, CaMK1 is required for NMDA-receptor mediated long-term potentiation, which is a form of synaptic plasticity that is considered a cellular basis of learning and memory formation [100]. CaMKK2 is also necessary for synaptic plasticity, as Camkk2 null mice were found to have reduced long-term potentiation at the hippocampal CA1 synapse and display long-term memory impairments [42]. Several, independent meta-analyses have reported that the majority of patients with bipolar disorder experience problems with memory loss and cognition across all phases of the disorder, even during periods of remission in a manner that increases with recurrence [101,102,103]. While the underlying mechanisms of memory loss and cognitive impairment in bipolar disorder remain uncertain, these data indicate that defects in CaMKK2-CaMK1 signalling may play a role.
The CaMK4 signalling pathway has emerged as an important regulator of homoeostatic plasticity, which is a key process by which excitatory and inhibitory signals in neurons are actively balanced to prevent the development of hyper or hypoactivity [104]. Imbalances in neuronal activity have been implicated in a wide range of neurological disorders including bipolar disorder [105]. CaMK4 regulates two crucial aspects of homoeostatic plasticity: excitatory synaptic scaling and intrinsic excitability [104, 106]. It modulates these processes by regulating the activity of the cyclic-AMP response element-binding protein (CREB), a major regulator of neuronal gene expression and a risk gene for bipolar disorder [107]. Activation of CaMK4 in cortical neurons reduces synaptic strength and spontaneous firing rates whereas CaMK4 inhibition increases both, which suggests that CaMK4 activation generates a negative feedback signal that controls neuronal firing rates [104]. Interestingly, the anti-manic effects of lithium have been proposed to occur via a mechanism that involves synaptic scaling [108]. Like CaMK4 activation, lithium treatment has been reported to reduce synaptic strength in hippocampal neurons [49, 109].
In addition to regulating homoeostatic plasticity, the CaMKK2-CaMK4 signalling pathway also regulates cerebellar function [45]. There is an increasing recognition that the cerebellum, rather than being limited to controlling motor coordination, also regulates higher-order cognitive and emotional functions [110]. Several case studies have reported the onset of mania and rapid-cycling bipolar disorder following cerebellar lesions caused by either surgery or trauma, which indicates a potential role for the cerebellum in the pathophysiology of bipolar disorder [110,111,112]. This is supported by neuroimaging studies that revealed aberrant cerebellar connectivity in bipolar disorder patients experiencing psychosis [113]. Genetic deletion of either Camkk2 or Camk4 in mice results in decreased BDNF expression in the cerebellum, reduced cerebellar volume, as well as a decreased number of Purkinje neurons within the cerebellar cortex [45, 114, 115]. Reduced numbers of cerebellar Purkinje neurons have also been reported in patients with bipolar disorder [116]. Considering the emerging link between impaired cerebellar function and mania, and that decreased serum BDNF is a biomarker of bipolar disorder, the decrease in cerebellar BDNF expression in the Camkk2 and Camk4 null mice suggests that defects in CaMKK2-CaMK4 signalling within the cerebellum may be involved in triggering manic behaviour [46,47,48,49].
AMPK is an energy-sensing protein kinase and a key regulator of cellular and whole-body energy metabolism [117]. In the brain, energy metabolism is tightly regulated as neurons are dependent on glucose as their primary energy source, but are unable to store sufficient quantities of glycogen to meet demand [118]. AMPK plays a crucial role in the regulation of glucose uptake in neurons, and mediates the translocation of the glucose transporter GLUT3 to the cell surface in response to increased energy demand driven by factors such as neurotransmission [119]. Activation of AMPK in neurons also promotes mitochondrial biogenesis and oxidative glucose metabolism by increasing the expression of the master mitochondrial regulators, peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) and mitochondrial transcription factor A (mtTFA) [120]. This is potentially important, as there is some evidence pointing to a role for mitochondrial abnormalities in bipolar disorder although this remains controversial. For example, several studies using proton magnetic resonance spectroscopy have reported reduced levels of N-acetyl aspartate, a marker of impaired mitochondrial energy production, in the brains of bipolar disorder patients compared with healthy controls [121,122,123,124,125]. However, a systematic review and meta-analysis found this is not clear cut, as indicated by a number of other studies that failed to replicate these data [126]. CaMKK2 is essential for the activation of mitochondria-localised AMPK, and genetic deletion of either Camkk2 or the AMPK catalytic α subunits (Prkaa1 and Prkaa2) results in reduced PGC-1α expression and suppression of mitochondrial respiration [43, 127,128,129,130]. Lithium has been reported to increase PGC-1α gene expression and mitochondrial biogenesis, and to increase mitochondrial respiration; however, the mechanism is poorly understood [131, 132]. Given that lithium activates CaMKK2, it is possible that some of the lithium-induced enhancements of mitochondrial function may be partly mediated by the CaMKK2-AMPK signalling pathway. In fact, metformin, an AMPK-activating drug, and frontline treatment for type 2 diabetes, also increases PGC-1α expression and mitochondrial biogenesis [133]. Intriguingly, a recent randomised controlled trial found that metformin significantly improved depressive symptoms in patients with treatment-resistant bipolar depression [134]. These observations align with a pharmaco-epidemiological study that found evidence for metformin having a protective effect against the onset of mood disorders [135]. Metformin has also been shown to have an anti-depressant effect in a chronic-restraint stress model of depression in mice, which is blocked by genetic knockdown of hippocampal AMPK α catalytic subunits (Prkaa1 and Prkaa2) [136]. Although there is no direct evidence at present to indicate whether metformin has anti-manic properties, these studies demonstrate that activation of the AMPK branch of the CaMKK2 signalling pathway can ameliorate depressive behaviours.
The Akt/PKB signalling pathway regulates numerous cellular processes in the brain including neurogenesis, neuronal differentiation, and synaptic plasticity [137,138,139]. The role of the CaMKK2-Akt/PKB signalling axis in the brain has yet to be studied; however, similar to CaMKK2, there are multiple lines of evidence that demonstrate a link between loss of Akt/PKB signalling and bipolar disorder. A recent study reported a reduction in Akt/PKB kinase activity in the dorsolateral and ventrolateral subregions of the prefrontal cortex in a large cohort of patients with bipolar disorder compared with unaffected controls [140]. Significantly, emulating this reduction in Akt/PKB signalling in the prefrontal cortex of mice resulted in cognitive impairments similar to those observed in human patients. The activation of Akt/PKB signalling has been discovered to contribute to the attenuation of manic-like behaviours in mice in response to lithium treatment [141]. This study also found that lithium responsiveness in inbred mice that differ in their response to lithium is intimately linked to the activation of Akt/PKB through the phosphorylation of Thr308. The Thr308 phosphorylation site serves as a pivotal regulatory point through which CaMKK2, as well as 3-phosphoinositide-dependent kinase-1 (PDK1), trigger the activation of Akt/PKB [87, 142]. As such, it is possible that activation of Akt/PKB signalling by lithium may involve, to some extent, activation of CaMKK2.
Polymorphisms and a rare missense mutation that impair CaMKK2 function are associated with bipolar disorder
Whole exome sequencing and genetic association studies have uncovered a link between bipolar disorder and loss-of-function polymorphisms and mutations in human CAMKK2. A rare, heterozygous, de novo mutation (rs130790572; 3.98 × 10-6 minor allele frequency) that results in a single amino acid change (R311C) in CaMKK2 was identified from a comparative whole exome sequencing study of patients with bipolar 1 disorder and their unaffected parents [143]. The R311C missense mutation maps to a highly-conserved feature called the Histidine-Arginine-Aspartate (HRD) motif located within the catalytic kinase domain of CaMKK2 (Fig. 2). The R311C mutation results in a catastrophic loss-of-function as it destabilises key electrostatic interactions within the catalytic site that are essential for CaMKK2 activity [144]. The R311C mutant also exerts a dominant-negative effect over wild-type CaMKK2 in heterozygous carriers. Strikingly, sporadic mutation of equivalent arginine residues in the HRD motifs of unrelated protein kinases are similarly catastrophic and cause rare, monogenic forms of various human diseases [145,146,147,148,149]. This is highly significant, as it reveals that rare mutations in the HRD motifs of protein kinases are highly penetrant and disease-causing, which provides a strong functional rationale for the R311C mutation in the HRD motif of CaMKK2 being considered a potential monogenic model of bipolar 1 disorder.
A missense polymorphism (rs3817190; 3.83 × 10–1 minor allele frequency) that results in a threonine to serine (T85S) substitution at the Thr85 autophosphorylation site located within the regulatory N-terminal sequence in human CaMKK2 (Fig. 2) was shown by two independent, candidate gene association studies to display statistically significant links with bipolar and anxiety disorder [150, 151]. Like wild-type CaMKK2, autophosphorylation of the T85S variant is also triggered by Ca2+-calmodulin binding, but fails to keep CaMKK2 in an activated state and instead causes a temporary loss-of-activity [33].
In terms of non-coding variations, a case-control study demonstrated an association between bipolar disorder and an intronic polymorphism in CAMKK2 (rs1063843; 1.9 × 10-1 minor allele frequency), where the minor allele was associated with reduced (>50%) CAMKK2 mRNA expression in human post-mortem brains and lymphoblastoid cells [39, 152]. The rs1063843 polymorphism was also shown by functional magnetic resonance imaging (fMRI) studies to be associated with persistent activation of the left dorsolateral prefrontal cortex (DLPFC), and with increased activation of the right DLPFC and caudate nucleus during cognitive tasks that measure attentional executive control and working memory [153]. An independent fMRI study demonstrated a similar activation of the DLPFC and caudate nucleus during a working memory task in healthy volunteers given amphetamine, a psychostimulant that induces manic-like behaviours [154]. Similar to these observations in humans, methamphetamine administration in mice caused a reduction in Camkk2 mRNA expression in the caudate nucleus [155].
Consistent with the loss-of-function effects of genetic variations in human CAMKK2, mice lacking Camkk2 display behavioural traits similar to those frequently observed in bipolar disorder [33]. For example, Camkk2 null mice are hyperactive and have deficits in pre-pulse inhibition of the startle response, both of which are seen in patients experiencing bipolar mania and psychosis, respectively [156]. Camkk2 null mice were also found to display increased cued fear conditioning and fear-potentiated startle responses, which indicates hyperactive amygdala function [33]. Deficits in emotional processing related to amygdala hyperactivity are core features of bipolar disorder and are associated with depression and increased risk of suicide [157, 158]. These data demonstrate that genetic deletion of Camkk2 in mice may model both manic and depressive-like behaviours.
Taken together, these findings support the view that loss-of-function genetic variations in human CAMKK2 are prime candidates as potential underlying causes of bipolar disorder.
Mood stabilising drugs increase CaMKK2 activity and abundance
Mood stabilisers are a class of drugs that are used to treat and manage both the manic and depressive phases of bipolar disorder [159]. Two of the most widely used drugs in this class are lithium and valproate, despite the fact that their primary targets and mechanisms of action still remain unclear. Signalling pathways and enzymes commonly affected by both lithium and valproate are more likely to be clinically relevant in bipolar disorder [160]. Therefore, identifying molecular targets that are shared between the two drugs is crucial, as it will provide some understanding of the pathogenesis of bipolar disorder, and inform the development of new, mechanism-based therapies with improved efficacy and better side-effect profiles [161].
Since the discovery of its therapeutic properties over seven decades ago, lithium remains the gold standard treatment for acute mania and prevention of recurrent bipolar disorder episodes [162, 163]. Multiple targets of lithium have been identified including inositol monophosphatase, phosphoglucomutase, and a family of four related phosphomonoesterases; however, the most convincing evidence to date suggests that GSK3 is a major effector of the therapeutic effects of lithium [79, 164, 165]. Genetic deletion of GSK3 in mice results in behaviours that mimic the mood-stabilising effects of lithium, as well as causing cognitive deficits similar to those associated with chronic lithium treatment in humans [166,167,168,169]. On the other hand, mice overexpressing GSK3 display behaviours that correlate with bipolar disorder and are desensitised to the mood-stabilising effects of lithium [170, 171]. Together, these data provide strong evidence that GSK3 is an in vivo target of lithium. GSK3 directly regulates at least forty known downstream substrates that participate in a diverse range of cellular processes [172]. The substrates that link GSK3 to bipolar disorder and lithium action are not yet clear; however, there is growing evidence that CaMKK2 could be a key effector. As described above, lithium increases CaMKK2 activity by inhibiting GSK3-mediated phosphorylation of the S3-node (Fig. 2) [33]. In addition, lithium increases CaMKK2 expression in the striatum, an area of the brain that displays structural abnormalities in bipolar disorder [173, 174]. These dual, function-enhancing effects of lithium are noteworthy, given the connection between CaMKK2 loss-of-function and bipolar disorder [33, 174]. Increased CaMKK2 activity in the brain leads to CaMK4 activation and increased expression of BDNF, which in mice is indispensable for the anti-manic actions of lithium [45, 49]. Furthermore, a recent study of genetic variants associated with lithium responsiveness in bipolar disorder patients found a significant enrichment of genes involved in glutamatergic synapse neurotransmission, which included GSK3 and CAMK4 [175]. Collectively, these data raise the possibility that the GSK3-CaMKK2-CaMK4 signalling axis is a principal site of action through which lithium increases BDNF expression and exerts its anti-manic effects. Also, since lithium increases both CaMKK2 activity and protein abundance, it is possible that CaMKK2 may be involved in mediating the acute as well as longer-term effects of lithium such as preventing episode recurrence [176].
Valproate was first used as an anticonvulsant to treat epilepsy but was subsequently discovered to also have mood-stabilising effects [177], which may be related to bipolar disorder and epilepsy sharing features, such as their episodic nature and being risk factors for one another [178]. Several direct targets of valproate have been discovered including the mitochondrial enzymes, succinate semialdehyde dehydrogenase and long-chain fatty acid-CoA ligase-4, as well as the family of nuclear-localised histone deacetylases that are involved in epigenetic regulation of gene expression [179,180,181]. However, no single target has been identified that accounts for the mood-stabilising effects of valproate. In relation to CaMKK2, a pharmacogenomic study using a methamphetamine-induced model of bipolar disorder in mice found that valproate treatment increased Camkk2 mRNA expression in the brain caudate nucleus [155]. Intriguingly, the rs1063843 polymorphism that is associated with bipolar disorder and reduced CAMKK2 mRNA expression in the human brain, is also associated with functional impairment of the caudate nucleus [153]. In addition to increasing Camkk2 mRNA expression, valproate has been reported to acutely activate AMPK, which suggests that valproate may also increase CaMKK2 activity [182].
The convergent, multi-level effects of lithium and valproate on CaMKK2 activity, mRNA expression and protein abundance reveal a functional signature that indicates CaMKK2 is likely an important mediator of their mood-stabilising effects.
CaMKK2 provides a potential link between metabolic dysfunction and bipolar disorder
Abnormalities in brain and whole-body energy metabolism are some of the most consistent features of bipolar disorder [17]. Clinically, the manic and depressive episodes of bipolar disorder closely correlate with periods of high and low energy expenditure. This is also the case phenotypically, as several studies have found objectively measured resting energy expenditure to be higher in patients with bipolar mania, and lower in patients with bipolar depression, compared with healthy controls [183,184,185,186]. Furthermore, the incidence of metabolic syndrome and type 2 diabetes is higher among patients with bipolar disorder than in the general population and is associated with a worse disease course and poor treatment outcomes [187, 188]. These discoveries have given rise to the idea that bipolar disorder is a bioenergetic and metabolic disease with psychiatric manifestations, and that disruptions in the molecular and cellular signalling networks that regulate brain energy metabolism in a biphasic manner are a primary, sufficient cause of bipolar disorder-related phenomena [189]. The energy requirements of the brain are very high due to energy intensive processes that are essential for healthy brain function, such as maintenance of ion gradients, cellular signalling, synaptic vesicle trafficking, and uptake and recycling of neurotransmitters [190]. Glucose is the major energy source, and brain activity accounts for around 25% of total body glucose consumption despite constituting just 2% of whole-body weight [191]. The disproportionate energy needs and reliance on glucose makes the brain particularly vulnerable to disruptions in the regulation of energy metabolism [192, 193].
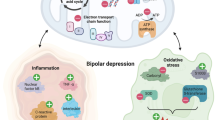
Some of the aberrations in brain energy metabolism associated with bipolar disorder are similar to the metabolic phenotype displayed by cells lacking CaMKK2. For example, multiple neuroimaging studies have reported increased brain lactate levels in patients with bipolar disorder [194,195,196,197]. Lactate is one of the oldest and best-established biomarkers in psychiatry, first documented as early as 1934, and major psychiatric disorders have been known to be associated with acidosis since 1932 [198, 199]. Lactate is also a biomarker of bipolar disorder, as lithium treatment has been found to reverse brain lactate accumulation in patients [200]. Elevated brain lactate levels indicate a pathophysiological shift in brain energy metabolism from oxidative phosphorylation to glycolysis as the major source of energy generation. CaMKK2 dysfunction may play an underlying role in this phenomenon as genetic deletion of Camkk2 in hippocampal neurons, myeloid cells and immortalized cell lines (HEK293 and HepG2) results in mitochondrial dysfunction that causes a similar switch in cellular energy generation from oxidative phosphorylation to glycolysis [43, 201, 202].
Another key metabolic feature of cells deficient in CaMKK2 is increased oxidative stress caused by accumulation of reactive oxygen species [202, 203]. Oxidative stress is a common hallmark of bipolar disorder and appears to increase with disease severity [82]. Indicators of oxidative stress such as lipid peroxidation have been shown to be elevated in serum from patients with bipolar disorder compared with healthy controls [204,205,206]. Notably, CaMKK2 suppresses lipid peroxidation by increasing the activity of NRF2, a transcription factor that promotes the expression of anti-oxidative proteins [207, 208]. Increasing NRF2 activity has been proposed as a treatment approach for bipolar disorder, and activating the CaMKK2 signalling pathway represents a potential and viable mechanism to achieve this outcome [209].
Concluding remarks and future directions
An ever-expanding body of scientific literature points to the CaMKK2 signalling pathway as a contributing factor in the pathogenesis of bipolar disorder. Given the large clinical, social and economic burden of bipolar disorder, the development of better treatment options is an urgent priority; however, drug development for bipolar disorder remains virtually stagnant [210]. We propose CaMKK2 as a rational treatment target for bipolar disorder as it links together key facets of the condition including genetic polymorphisms/mutations, defects in signal transduction, mood stabiliser action, and metabolic dysfunction. Protein kinases are highly druggable and are the second most targeted group of drug targets after G-protein-coupled receptors [211]. At least 76 drugs that target protein kinases have been approved for clinical use, and several kinase-targeted candidate therapeutics are in various stages of preclinical and clinical development for brain-related disorders [212]. A future challenge is to identify highly selective and potent, small-molecule drugs capable of activating CaMKK2 in the brain, which can then be validated preclinically in human induced pluripotent stem cell (iPSC) and animal models of bipolar disorder. High-throughput screening of small-molecule chemical libraries using fluorescence-based kinase assays followed by hit-to-lead optimisation offers a viable strategy for identifying and developing drug activators of CaMKK2. A similar approach was successfully applied to develop selective drug activators of the protein kinase AMPK, which were revealed to bind to an unexpected allosteric site on AMPK that is distinct from the canonical AMP-binding sites [213,214,215,216]. There is also a need for high-resolution structural information on CaMKK2 to facilitate fragment and structure-based drug discovery. Like the revolution in psychiatry sparked by the discovery of the effectiveness of lithium, the development of new mechanism-based therapies with superior efficacy and tolerability hold great promise to similarly transform the treatment of bipolar disorder.
References
Merikangas KR, Jin R, He JP, Kessler RC, Lee S, Sampson NA, et al. Prevalence and correlates of bipolar spectrum disorder in the world mental health survey initiative. Arch Gen Psychiatry. 2011;68:241–51.
McIntyre RS, Berk M, Brietzke E, Goldstein BI, Lopez-Jaramillo C, Kessing LV, et al. Bipolar disorders. Lancet. 2020;396:1841–56.
Elizabeth Sublette M, Carballo JJ, Moreno C, Galfalvy HC, Brent DA, Birmaher B, et al. Substance use disorders and suicide attempts in bipolar subtypes. J Psychiatr Res. 2009;43:230–8.
Fagiolini A, Frank E, Turkin S, Houck PR, Soreca I, Kupfer DJ. Metabolic syndrome in patients with bipolar disorder. J Clin Psychiatry. 2008;69:678–9.
Westman J, Hallgren J, Wahlbeck K, Erlinge D, Alfredsson L, Osby U. Cardiovascular mortality in bipolar disorder: a population-based cohort study in Sweden. BMJ Open. 2013;3:e002373.
Kessing LV, Vradi E, Andersen PK. Life expectancy in bipolar disorder. Bipolar Disord. 2015;17:543–8.
Hayes JF, Miles J, Walters K, King M, Osborn DP. A systematic review and meta-analysis of premature mortality in bipolar affective disorder. Acta Psychiatr Scand. 2015;131:417–25.
Harrison PJ, Cipriani A, Harmer CJ, Nobre AC, Saunders K, Goodwin GM, et al. Innovative approaches to bipolar disorder and its treatment. Ann NY Acad Sci. 2016;1366:76–89.
Fung VC, Overhage LN, Sylvia LG, Reilly-Harrington NA, Kamali M, Gao K, et al. Complex polypharmacy in bipolar disorder: Side effect burden, adherence, and response predictors. J Affect Disord. 2019;257:17–22.
Schloesser RJ, Martinowich K, Manji HK. Mood-stabilizing drugs: mechanisms of action. Trends Neurosci. 2012;35:36–46.
Geddes JR, Miklowitz DJ. Treatment of bipolar disorder. Lancet. 2013;381:1672–82.
Kawamoto EM, Vivar C, Camandola S. Physiology and pathology of calcium signaling in the brain. Front Pharmacol. 2012;3:61.
Harrison PJ, Hall N, Mould A, Al-Juffali N, Tunbridge EM. Cellular calcium in bipolar disorder: systematic review and meta-analysis. Mol Psychiatry. 2021;26:4106–16.
Giorgi C, Marchi S, Pinton P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol. 2018;19:713–30.
Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, et al. Mitochondrial Ca(2+) and apoptosis. Cell Calcium. 2012;52:36–43.
Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018;69:62–72.
Morris G, Walder K, McGee SL, Dean OM, Tye SJ, Maes M, et al. A model of the mitochondrial basis of bipolar disorder. Neurosci Biobehav Rev. 2017;74:1–20.
Rimessi A, Bonora M, Marchi S, Patergnani S, Marobbio CM, Lasorsa FM, et al. Perturbed mitochondrial Ca2+ signals as causes or consequences of mitophagy induction. Autophagy. 2013;9:1677–86.
Chen HM, DeLong CJ, Bame M, Rajapakse I, Herron TJ, McInnis MG, et al. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients. Transl Psychiatry. 2014;4:e375.
Mertens J, Wang QW, Kim Y, Yu DX, Pham S, Yang B, et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature. 2015;527:95–9.
Berk M, Bodemer W, van Oudenhove T, Butkow N. Dopamine increases platelet intracellular calcium in bipolar affective disorder and controls. Int Clin Psychopharmacol. 1994;9:291–3.
Perova T, Wasserman MJ, Li PP, Warsh JJ. Hyperactive intracellular calcium dynamics in B lymphoblasts from patients with bipolar I disorder. Int J Neuropsychopharmacol. 2008;11:185–96.
Levy NA, Janicak PG. Calcium channel antagonists for the treatment of bipolar disorder. Bipolar Disord. 2000;2:108–19.
Walton SA, Berk M, Brook S. Superiority of lithium over verapamil in mania: a randomized, controlled, single-blind trial. J Clin Psychiatry. 1996;57:543–6.
Cipriani A, Saunders K, Attenburrow MJ, Stefaniak J, Panchal P, Stockton S, et al. A systematic review of calcium channel antagonists in bipolar disorder and some considerations for their future development. Mol Psychiatry. 2016;21:1324–32.
Chen M, Tian H, Huang G, Fang T, Lin X, Shan J, et al. Calcium imaging reveals depressive- and manic-phase-specific brain neural activity patterns in a murine model of bipolar disorder: a pilot study. Transl Psychiatry. 2021;11:619.
Moon AL, Haan N, Wilkinson LS, Thomas KL, Hall J. CACNA1C: association with psychiatric disorders, behavior, and neurogenesis. Schizophr Bull. 2018;44:958–65.
Smedler E, Louhivuori L, Romanov RA, Masini D, Dehnisch Ellstrom I, Wang C, et al. Disrupted Cacna1c gene expression perturbs spontaneous Ca(2+) activity causing abnormal brain development and increased anxiety. Proc Natl Acad Sci USA. 2022;119:e2108768119.
Jang Y, Lee SH, Lee B, Jung S, Khalid A, Uchida K, et al. TRPM2, a susceptibility gene for bipolar disorder, regulates glycogen synthase kinase-3 activity in the brain. J Neurosci. 2015;35:11811–23.
Chin D, Means AR. Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 2000;10:322–8.
Anderson KA, Ribar TJ, Lin F, Noeldner PK, Green MF, Muehlbauer MJ, et al. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 2008;7:377–88.
Mizuno K, Antunes-Martins A, Ris L, Peters M, Godaux E, Giese KP. Calcium/calmodulin kinase kinase beta has a male-specific role in memory formation. Neuroscience. 2007;145:393–402.
Scott JW, Park E, Rodriguiz RM, Oakhill JS, Issa SM, O’Brien MT, et al. Autophosphorylation of CaMKK2 generates autonomous activity that is disrupted by a T85S mutation linked to anxiety and bipolar disorder. Sci Rep. 2015;5:14436.
Racioppi L, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J Biol Chem. 2012;287:31658–65.
Langendorf CG, O’Brien MT, Ngoei KRW, McAloon LM, Dhagat U, Hoque A, et al. CaMKK2 is inactivated by cAMP-PKA signaling and 14-3-3 adaptor proteins. J Biol Chem. 2020;295:16239–50.
Sjostedt E, Zhong W, Fagerberg L, Karlsson M, Mitsios N, Adori C, et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science. 2020;367:eaay5947.
Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–76.
Berk M, Dodd S, Callaly P, Berk L, Fitzgerald P, de Castella AR, et al. History of illness prior to a diagnosis of bipolar disorder or schizoaffective disorder. J Affect Disord. 2007;103:181–6.
Luo XJ, Li M, Huang L, Steinberg S, Mattheisen M, Liang G, et al. Convergent lines of evidence support CAMKK2 as a schizophrenia susceptibility gene. Mol Psychiatry. 2014;19:774–83.
Green MF, Scott JW, Steel R, Oakhill JS, Kemp BE, Means AR. Ca2+-Calmodulin-dependent protein kinase kinase beta is regulated by multisite phosphorylation. J Biol Chem. 2011;286:28066–79.
Fortin DA, Srivastava T, Dwarakanath D, Pierre P, Nygaard S, Derkach VA, et al. Brain-derived neurotrophic factor activation of CaM-kinase kinase via transient receptor potential canonical channels induces the translation and synaptic incorporation of GluA1-containing calcium-permeable AMPA receptors. J Neurosci. 2012;32:8127–37.
Peters M, Mizuno K, Ris L, Angelo M, Godaux E, Giese KP. Loss of Ca2+/calmodulin kinase kinase beta affects the formation of some, but not all, types of hippocampus-dependent long-term memory. J Neurosci. 2003;23:9752–60.
Sabbir MG, Taylor CG, Zahradka P. CAMKK2 regulates mitochondrial function by controlling succinate dehydrogenase expression, post-translational modification, megacomplex assembly, and activity in a cell-type-specific manner. Cell Commun Signal. 2021;19:98.
Wayman GA, Lee YS, Tokumitsu H, Silva AJ, Soderling TR. Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron. 2008;59:914–31.
Kokubo M, Nishio M, Ribar TJ, Anderson KA, West AE, Means AR. BDNF-mediated cerebellar granule cell development is impaired in mice null for CaMKK2 or CaMKIV. J Neurosci. 2009;29:8901–13.
Fernandes BS, Gama CS, Cereser KM, Yatham LN, Fries GR, Colpo G, et al. Brain-derived neurotrophic factor as a state-marker of mood episodes in bipolar disorders: a systematic review and meta-regression analysis. J Psychiatr Res. 2011;45:995–1004.
Fernandes BS, Molendijk ML, Kohler CA, Soares JC, Leite CM, Machado-Vieira R, et al. Peripheral brain-derived neurotrophic factor (BDNF) as a biomarker in bipolar disorder: a meta-analysis of 52 studies. BMC Med. 2015;13:289.
Munkholm K, Vinberg M, Kessing LV. Peripheral blood brain-derived neurotrophic factor in bipolar disorder: a comprehensive systematic review and meta-analysis. Mol Psychiatry. 2016;21:216–28.
Gideons ES, Lin PY, Mahgoub M, Kavalali ET, Monteggia LM. Chronic lithium treatment elicits its antimanic effects via BDNF-TrkB dependent synaptic downscaling. Elife. 2017;6:e25480.
Suwalska A, Sobieska M, Rybakowski JK. Serum brain-derived neurotrophic factor in euthymic bipolar patients on prophylactic lithium therapy. Neuropsychobiology. 2010;62:229–34.
Marcelo KL, Means AR, York B. The Ca2+/Calmodulin/CaMKK2 axis: nature’s metabolic CaMshaft. Trends Endocrinol Metab. 2016;27:706–18.
Tokumitsu H, Wayman GA, Muramatsu M, Soderling TR. Calcium/calmodulin-dependent protein kinase kinase: identification of regulatory domains. Biochemistry. 1997;36:12823–7.
Kylarova S, Psenakova K, Herman P, Obsilova V, Obsil T. CaMKK2 kinase domain interacts with the autoinhibitory region through the N-terminal lobe including the RP insert. Biochim Biophys Acta Gen Subj. 2018;1862:2304–13.
Tokumitsu H, Hatano N, Fujimoto T, Yurimoto S, Kobayashi R. Generation of autonomous activity of Ca(2+)/calmodulin-dependent protein kinase kinase beta by autophosphorylation. Biochemistry. 2011;50:8193–201.
Psenakova K, Petrvalska O, Kylarova S, Lentini Santo D, Kalabova D, Herman P, et al. 14-3-3 protein directly interacts with the kinase domain of calcium/calmodulin-dependent protein kinase kinase (CaMKK2). Biochim Biophys Acta Gen Subj. 2018;1862:1612–25.
Takabatake S, Ohtsuka S, Sugawara T, Hatano N, Kanayama N, Magari M, et al. Regulation of Ca(2+)/calmodulin-dependent protein kinase kinase beta by cAMP signaling. Biochim Biophys Acta Gen Subj. 2019;1863:672–80.
Schumacher AM, Schavocky JP, Velentza AV, Mirzoeva S, Watterson DM. A calmodulin-regulated protein kinase linked to neuron survival is a substrate for the calmodulin-regulated death-associated protein kinase. Biochemistry. 2004;43:8116–24.
Fields A, Li PP, Kish SJ, Warsh JJ. Increased cyclic AMP-dependent protein kinase activity in postmortem brain from patients with bipolar affective disorder. J Neurochem. 1999;73:1704–10.
Karege F, Schwald M, Papadimitriou P, Lachausse C, Cisse M. The cAMP-dependent protein kinase A and brain-derived neurotrophic factor expression in lymphoblast cells of bipolar affective disorder. J Affect Disord. 2004;79:187–92.
Rahman S, Li PP, Young LT, Kofman O, Kish SJ, Warsh JJ. Reduced [3H]cyclic AMP binding in postmortem brain from subjects with bipolar affective disorder. J Neurochem. 1997;68:297–304.
Tardito D, Mori S, Racagni G, Smeraldi E, Zanardi R, Perez J. Protein kinase A activity in platelets from patients with bipolar disorder. J Affect Disord. 2003;76:249–53.
Palmer DS, Howrigan DP, Chapman SB, Adolfsson R, Bass N, Blackwood D, et al. Exome sequencing in bipolar disorder identifies AKAP11 as a risk gene shared with schizophrenia. Nat Genet. 2022;54:541–7.
Omar MH, Scott JD. AKAP signaling islands: venues for precision pharmacology. Trends Pharmacol Sci. 2020;41:933–46.
Hedman AC, Li Z, Gorisse L, Parvathaneni S, Morgan CJ, Sacks DB. IQGAP1 binds AMPK and is required for maximum AMPK activation. J Biol Chem. 2021;296:100075.
Folsom TD, Thuras PD, Fatemi SH. Protein expression of targets of the FMRP regulon is altered in brains of subjects with schizophrenia and mood disorders. Schizophr Res. 2015;165:201–11.
Padmos RC, Hillegers MH, Knijff EM, Vonk R, Bouvy A, Staal FJ, et al. A discriminating messenger RNA signature for bipolar disorder formed by an aberrant expression of inflammatory genes in monocytes. Arch Gen Psychiatry. 2008;65:395–407.
Tokumitsu H, Iwabu M, Ishikawa Y, Kobayashi R. Differential regulatory mechanism of Ca2+/calmodulin-dependent protein kinase kinase isoforms. Biochemistry. 2001;40:13925–32.
Brenna A, Olejniczak I, Chavan R, Ripperger JA, Langmesser S, Cameroni E, et al. Cyclin-dependent kinase 5 (CDK5) regulates the circadian clock. Elife. 2019;8:e50925.
Kwak Y, Jeong J, Lee S, Park YU, Lee SA, Han DH, et al. Cyclin-dependent kinase 5 (Cdk5) regulates the function of CLOCK protein by direct phosphorylation. J Biol Chem. 2013;288:36878–89.
Benedetti F, Serretti A, Colombo C, Barbini B, Lorenzi C, Campori E, et al. Influence of CLOCK gene polymorphism on circadian mood fluctuation and illness recurrence in bipolar depression. Am J Med Genet B Neuropsychiatr Genet. 2003;123B:23–6.
Mansour HA, Wood J, Logue T, Chowdari KV, Dayal M, Kupfer DJ, et al. Association study of eight circadian genes with bipolar I disorder, schizoaffective disorder and schizophrenia. Genes Brain Behav. 2006;5:150–7.
Nievergelt CM, Kripke DF, Barrett TB, Burg E, Remick RA, Sadovnick AD, et al. Suggestive evidence for association of the circadian genes PERIOD3 and ARNTL with bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:234–41.
Geoffroy PA, Lajnef M, Bellivier F, Jamain S, Gard S, Kahn JP, et al. Genetic association study of circadian genes with seasonal pattern in bipolar disorders. Sci Rep. 2015;5:10232.
Geoffroy PA, Scott J, Boudebesse C, Lajnef M, Henry C, Leboyer M, et al. Sleep in patients with remitted bipolar disorders: a meta-analysis of actigraphy studies. Acta Psychiatr Scand. 2015;131:89–99.
Lyall LM, Wyse CA, Graham N, Ferguson A, Lyall DM, Cullen B, et al. Association of disrupted circadian rhythmicity with mood disorders, subjective wellbeing, and cognitive function: a cross-sectional study of 91 105 participants from the UK Biobank. Lancet Psychiatry. 2018;5:507–14.
Ng TH, Chung KF, Ho FY, Yeung WF, Yung KP, Lam TH. Sleep-wake disturbance in interepisode bipolar disorder and high-risk individuals: a systematic review and meta-analysis. Sleep Med Rev. 2015;20:46–58.
Besing RC, Paul JR, Hablitz LM, Rogers CO, Johnson RL, Young ME, et al. Circadian rhythmicity of active GSK3 isoforms modulates molecular clock gene rhythms in the suprachiasmatic nucleus. J Biol Rhythms. 2015;30:155–60.
Freland L, Beaulieu JM. Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front Mol Neurosci. 2012;5:14.
Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–9.
Mishra HK, Ying NM, Luis A, Wei H, Nguyen M, Nakhla T, et al. Circadian rhythms in bipolar disorder patient-derived neurons predict lithium response: preliminary studies. Mol Psychiatry. 2021;26:3383–94.
Polter A, Beurel E, Yang S, Garner R, Song L, Miller CA, et al. Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology. 2010;35:1761–74.
Akarsu S, Bolu A, Aydemir E, Zincir SB, Kurt YG, Zincir S, et al. The Relationship between the Number of Manic Episodes and Oxidative Stress Indicators in Bipolar Disorder. Psychiatry Investig. 2018;15:514–9.
Brown NC, Andreazza AC, Young LT. An updated meta-analysis of oxidative stress markers in bipolar disorder. Psychiatry Res. 2014;218:61–8.
Cheng Y, Pardo M, Armini RS, Martinez A, Mouhsine H, Zagury JF, et al. Stress-induced neuroinflammation is mediated by GSK3-dependent TLR4 signaling that promotes susceptibility to depression-like behavior. Brain Behav Immun. 2016;53:207–22.
Dargel AA, Godin O, Kapczinski F, Kupfer DJ, Leboyer M. C-reactive protein alterations in bipolar disorder: a meta-analysis. J Clin Psychiatry. 2015;76:142–50.
Anderson KA, Means RL, Huang QH, Kemp BE, Goldstein EG, Selbert MA, et al. Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase beta. J Biol Chem. 1998;273:31880–9.
Gocher AM, Azabdaftari G, Euscher LM, Dai S, Karacosta LG, Franke TF, et al. Akt activation by Ca(2+)/calmodulin-dependent protein kinase kinase 2 (CaMKK2) in ovarian cancer cells. J Biol Chem. 2017;292:14188–204.
Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19.
Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–6.
Kitani T, Okuno S, Fujisawa H. Molecular cloning of Ca2+/calmodulin-dependent protein kinase kinase beta. J Biochem. 1997;122:243–50.
Tokumitsu H, Brickey DA, Glod J, Hidaka H, Sikela J, Soderling TR. Activation mechanisms for Ca2+/calmodulin-dependent protein kinase IV. Identification of a brain CaM-kinase IV kinase. J Biol Chem. 1994;269:28640–7.
Tokumitsu H, Enslen H, Soderling TR. Characterization of a Ca2+/calmodulin-dependent protein kinase cascade. Molecular cloning and expression of calcium/calmodulin-dependent protein kinase kinase. J Biol Chem. 1995;270:19320–4.
Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33.
Saneyoshi T, Wayman G, Fortin D, Davare M, Hoshi N, Nozaki N, et al. Activity-dependent synaptogenesis: regulation by a CaM-kinase kinase/CaM-kinase I/betaPIX signaling complex. Neuron. 2008;57:94–107.
Takemoto-Kimura S, Ageta-Ishihara N, Nonaka M, Adachi-Morishima A, Mano T, Okamura M, et al. Regulation of dendritogenesis via a lipid-raft-associated Ca2+/calmodulin-dependent protein kinase CLICK-III/CaMKIgamma. Neuron. 2007;54:755–70.
Wayman GA, Davare M, Ando H, Fortin D, Varlamova O, Cheng HY, et al. An activity-regulated microRNA controls dendritic plasticity by down-regulating p250GAP. Proc Natl Acad Sci USA. 2008;105:9093–8.
Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, et al. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50:897–909.
Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, et al. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J Neurosci. 2004;24:3786–94.
Park E, Na M, Choi J, Kim S, Lee JR, Yoon J, et al. The Shank family of postsynaptic density proteins interacts with and promotes synaptic accumulation of the beta PIX guanine nucleotide exchange factor for Rac1 and Cdc42. J Biol Chem. 2003;278:19220–9.
Schmitt JM, Guire ES, Saneyoshi T, Soderling TR. Calmodulin-dependent kinase kinase/calmodulin kinase I activity gates extracellular-regulated kinase-dependent long-term potentiation. J Neurosci. 2005;25:1281–90.
Bortolato B, Miskowiak KW, Kohler CA, Vieta E, Carvalho AF. Cognitive dysfunction in bipolar disorder and schizophrenia: a systematic review of meta-analyses. Neuropsychiatr Dis Treat. 2015;11:3111–25.
Bourne C, Aydemir O, Balanza-Martinez V, Bora E, Brissos S, Cavanagh JT, et al. Neuropsychological testing of cognitive impairment in euthymic bipolar disorder: an individual patient data meta-analysis. Acta Psychiatr Scand. 2013;128:149–62.
Robinson LJ, Thompson JM, Gallagher P, Goswami U, Young AH, Ferrier IN, et al. A meta-analysis of cognitive deficits in euthymic patients with bipolar disorder. J Affect Disord. 2006;93:105–15.
Joseph A, Turrigiano GG. All for one but not one for all: excitatory synaptic scaling and intrinsic excitability are coregulated by CaMKIV, whereas inhibitory synaptic scaling is under independent control. J Neurosci. 2017;37:6778–85.
Wondolowski J, Dickman D. Emerging links between homeostatic synaptic plasticity and neurological disease. Front Cell Neurosci. 2013;7:223.
Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–26.
Xiao X, Zhang C, Grigoroiu-Serbanescu M, Wang L, Li L, Zhou D, et al. The cAMP responsive element-binding (CREB)-1 gene increases risk of major psychiatric disorders. Mol Psychiatry. 2018;23:1957–67.
Kavalali ET, Monteggia LM. Targeting homeostatic synaptic plasticity for treatment of mood disorders. Neuron. 2020;106:715–26.
Wei J, Liu W, Yan Z. Regulation of AMPA receptor trafficking and function by glycogen synthase kinase 3. J Biol Chem. 2010;285:26369–76.
Lupo M, Olivito G, Siciliano L, Masciullo M, Molinari M, Cercignani M, et al. Evidence of Cerebellar Involvement in the Onset of a Manic State. Front Neurol. 2018;9:774.
Bottemanne H, Tang J, Claret A. Rapid-cycling bipolar disorder and cerebellar cognitive affective syndrome associated with cerebellum and frontal neurosurgical lesions. Prim Care Companion CNS Disord. 2021;23:20cr02901.
Drange OK, Vaaler AE, Morken G, Andreassen OA, Malt UF, Finseth PI. Clinical characteristics of patients with bipolar disorder and premorbid traumatic brain injury: a cross-sectional study. Int J Bipolar Disord. 2018;6:19.
Shinn AK, Roh YS, Ravichandran CT, Baker JT, Ongur D, Cohen BM. Aberrant cerebellar connectivity in bipolar disorder with psychosis. Biol Psychiatry Cogn Neurosci Neuroimaging. 2017;2:438–48.
Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, et al. Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci. 2000;20:6459–72.
Ribar TJ, Rodriguiz RM, Khiroug L, Wetsel WC, Augustine GJ, Means AR. Cerebellar defects in Ca2+/calmodulin kinase IV-deficient mice. J Neurosci. 2000;20:RC107.
Maloku E, Covelo IR, Hanbauer I, Guidotti A, Kadriu B, Hu Q, et al. Lower number of cerebellar Purkinje neurons in psychosis is associated with reduced reelin expression. Proc Natl Acad Sci USA. 2010;107:4407–11.
Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase–development of the energy sensor concept. J Physiol. 2006;574:7–15.
Vilchez D, Ros S, Cifuentes D, Pujadas L, Valles J, Garcia-Fojeda B, et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci. 2007;10:1407–13.
Weisova P, Concannon CG, Devocelle M, Prehn JH, Ward MW. Regulation of glucose transporter 3 surface expression by the AMP-activated protein kinase mediates tolerance to glutamate excitation in neurons. J Neurosci. 2009;29:2997–3008.
Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci USA. 2007;104:7217–22.
Bertolino A, Frye M, Callicott JH, Mattay VS, Rakow R, Shelton-Repella J, et al. Neuronal pathology in the hippocampal area of patients with bipolar disorder: a study with proton magnetic resonance spectroscopic imaging. Biol Psychiatry. 2003;53:906–13.
Cecil KM, DelBello MP, Morey R, Strakowski SM. Frontal lobe differences in bipolar disorder as determined by proton MR spectroscopy. Bipolar Disord. 2002;4:357–65.
Chang K, Adleman N, Dienes K, Barnea-Goraly N, Reiss A, Ketter T. Decreased N-acetylaspartate in children with familial bipolar disorder. Biol Psychiatry. 2003;53:1059–65.
Deicken RF, Pegues MP, Anzalone S, Feiwell R, Soher B. Lower concentration of hippocampal N-acetylaspartate in familial bipolar I disorder. Am J Psychiatry. 2003;160:873–82.
Winsberg ME, Sachs N, Tate DL, Adalsteinsson E, Spielman D, Ketter TA. Decreased dorsolateral prefrontal N-acetyl aspartate in bipolar disorder. Biol Psychiatry. 2000;47:475–81.
Kraguljac NV, Reid M, White D, Jones R, den Hollander J, Lowman D, et al. Neurometabolites in schizophrenia and bipolar disorder - a systematic review and meta-analysis. Psychiatry Res. 2012;203:111–25.
Anderson KA, Lin F, Ribar TJ, Stevens RD, Muehlbauer MJ, Newgard CB, et al. Deletion of CaMKK2 from the liver lowers blood glucose and improves whole-body glucose tolerance in the mouse. Mol Endocrinol. 2012;26:281–91.
Marinangeli C, Didier S, Ahmed T, Caillerez R, Domise M, Laloux C, et al. AMP-activated protein kinase is essential for the maintenance of energy levels during synaptic activation. iScience. 2018;9:1–13.
Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, et al. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. 2017;21:1–9.
Schmitt DL, Curtis SD, Lyons AC, Zhang JF, Chen M, He CY, et al. Spatial regulation of AMPK signaling revealed by a sensitive kinase activity reporter. Nat Commun. 2022;13:3856.
Martin SA, Souder DC, Miller KN, Clark JP, Sagar AK, Eliceiri KW, et al. GSK3beta regulates brain energy metabolism. Cell Rep. 2018;23:1922–31 e4.
Osete JR, Akkouh IA, de Assis DR, Szabo A, Frei E, Hughes T, et al. Lithium increases mitochondrial respiration in iPSC-derived neural precursor cells from lithium responders. Mol Psychiatry. 2021;26:6789–805.
Kang H, Khang R, Ham S, Jeong GR, Kim H, Jo M, et al. Activation of the ATF2/CREB-PGC-1alpha pathway by metformin leads to dopaminergic neuroprotection. Oncotarget. 2017;8:48603–18.
Calkin CV, Chengappa KNR, Cairns K, Cookey J, Gannon J, Alda M, et al. Treating insulin resistance with metformin as a strategy to improve clinical outcomes in treatment-resistant bipolar depression (the TRIO-BD Study): a randomized, quadruple-masked, placebo-controlled clinical trial. J Clin Psychiatry. 2022;83:21m14022.
Lake J, Bortolasci CC, Stuart AL, Pasco JA, Kidnapillai S, Spolding B, et al. Metformin is protective against the development of mood disorders. Pharmacopsychiatry. 2023;56:25–31.
Fang W, Zhang J, Hong L, Huang W, Dai X, Ye Q, et al. Metformin ameliorates stress-induced depression-like behaviors via enhancing the expression of BDNF by activating AMPK/CREB-mediated histone acetylation. J Affect Disord. 2020;260:302–13.
Levenga J, Wong H, Milstead RA, Keller BN, LaPlante LE, Hoeffer CA. AKT isoforms have distinct hippocampal expression and roles in synaptic plasticity. Elife. 2017;6:e30640.
Sun L, Cui K, Xing F, Liu X. Akt dependent adult hippocampal neurogenesis regulates the behavioral improvement of treadmill running to mice model of post-traumatic stress disorder. Behav Brain Res. 2020;379:112375.
Vojtek AB, Taylor J, DeRuiter SL, Yu JY, Figueroa C, Kwok RP, et al. Akt regulates basic helix-loop-helix transcription factor-coactivator complex formation and activity during neuronal differentiation. Mol Cell Biol. 2003;23:4417–27.
Vanderplow AM, Eagle AL, Kermath BA, Bjornson KJ, Robison AJ, Cahill ME. Akt-mTOR hypoactivity in bipolar disorder gives rise to cognitive impairments associated with altered neuronal structure and function. Neuron. 2021;109:1479–96 e6.
Pan JQ, Lewis MC, Ketterman JK, Clore EL, Riley M, Richards KR, et al. AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology. 2011;36:1397–411.
Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–9.
Kataoka M, Matoba N, Sawada T, Kazuno AA, Ishiwata M, Fujii K, et al. Exome sequencing for bipolar disorder points to roles of de novo loss-of-function and protein-altering mutations. Mol Psychiatry. 2016;21:885–93.
Ling NXY, Langendorf CG, Hoque A, Galic S, Loh K, Kemp BE, et al. Functional analysis of an R311C variant of Ca(2+) -calmodulin dependent protein kinase kinase-2 (CaMKK2) found as a de novo mutation in a patient with bipolar disorder. Bipolar Disord. 2020;22:841–8.
Abdalla SA, Cymerman U, Johnson RM, Deber CM, Letarte M. Disease-associated mutations in conserved residues of ALK-1 kinase domain. Eur J Hum Genet. 2003;11:279–87.
George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304:1325–8.
Hagemann TL, Chen Y, Rosen FS, Kwan SP. Genomic organization of the Btk gene and exon scanning for mutations in patients with X-linked agammaglobulinemia. Hum Mol Genet. 1994;3:1743–9.
Longo N, Wang Y, Pasquali M. Progressive decline in insulin levels in Rabson-Mendenhall syndrome. J Clin Endocrinol Metab. 1999;84:2623–9.
Longo N, Wang Y, Smith SA, Langley SD, DiMeglio LA, Giannella-Neto D. Genotype-phenotype correlation in inherited severe insulin resistance. Hum Mol Genet. 2002;11:1465–75.
Barden N, Harvey M, Gagne B, Shink E, Tremblay M, Raymond C, et al. Analysis of single nucleotide polymorphisms in genes in the chromosome 12Q24.31 region points to P2RX7 as a susceptibility gene to bipolar affective disorder. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:374–82.
Erhardt A, Lucae S, Unschuld PG, Ising M, Kern N, Salyakina D, et al. Association of polymorphisms in P2RX7 and CaMKKb with anxiety disorders. J Affect Disord. 2007;101:159–68.
Atakhorrami M, Rahimi-Aliabadi S, Jamshidi J, Moslemi E, Movafagh A, Ohadi M, et al. A genetic variant in CAMKK2 gene is possibly associated with increased risk of bipolar disorder. J Neural Transm (Vienna). 2016;123:323–8.
Yu P, Chen X, Zhao W, Zhang Z, Zhang Q, Han B, et al. Effect of rs1063843 in the CAMKK2 gene on the dorsolateral prefrontal cortex. Hum Brain Mapp. 2016;37:2398–406.
O’Daly OG, Joyce D, Stephan KE, Murray RM, Shergill SS. Functional magnetic resonance imaging investigation of the amphetamine sensitization model of schizophrenia in healthy male volunteers. Arch Gen Psychiatry. 2011;68:545–54.
Ogden CA, Rich ME, Schork NJ, Paulus MP, Geyer MA, Lohr JB, et al. Candidate genes, pathways and mechanisms for bipolar (manic-depressive) and related disorders: an expanded convergent functional genomics approach. Mol Psychiatry. 2004;9:1007–29.
Dunayevich E, Keck PE Jr. Prevalence and description of psychotic features in bipolar mania. Curr Psychiatry Rep. 2000;2:286–90.
Hariri AR. The highs and lows of amygdala reactivity in bipolar disorders. Am J Psychiatry. 2012;169:780–3.
Wang L, Zhao Y, Edmiston EK, Womer FY, Zhang R, Zhao P, et al. Structural and functional abnormities of amygdala and prefrontal cortex in major depressive disorder with suicide attempts. Front Psychiatry. 2019;10:923.
Miura T, Noma H, Furukawa TA, Mitsuyasu H, Tanaka S, Stockton S, et al. Comparative efficacy and tolerability of pharmacological treatments in the maintenance treatment of bipolar disorder: a systematic review and network meta-analysis. Lancet Psychiatry. 2014;1:351–9.
Lee RS, Pirooznia M, Guintivano J, Ly M, Ewald ER, Tamashiro KL, et al. Search for common targets of lithium and valproic acid identifies novel epigenetic effects of lithium on the rat leptin receptor gene. Transl Psychiatry. 2015;5:e600.
Phiel CJ, Klein PS. Molecular targets of lithium action. Annu Rev Pharmacol Toxicol. 2001;41:789–813.
Cade JF. Lithium salts in the treatment of psychotic excitement. Med J Aust. 1949;2:349–52.
Volkmann C, Bschor T, Kohler S. Lithium treatment over the lifespan in bipolar disorders. Front Psychiatry. 2020;11:377.
Acharya JK, Labarca P, Delgado R, Jalink K, Zuker CS. Synaptic defects and compensatory regulation of inositol metabolism in inositol polyphosphate 1-phosphatase mutants. Neuron. 1998;20:1219–29.
Hallcher LM, Sherman WR. The effects of lithium ion and other agents on the activity of myo-inositol-1-phosphatase from bovine brain. J Biol Chem. 1980;255:10896–901.
Beaulieu JM, Zhang X, Rodriguiz RM, Sotnikova TD, Cools MJ, Wetsel WC, et al. Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci USA. 2008;105:1333–8.
Kaidanovich-Beilin O, Lipina TV, Takao K, van Eede M, Hattori S, Laliberte C, et al. Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol Brain. 2009;2:35.
Kimura T, Yamashita S, Nakao S, Park JM, Murayama M, Mizoroki T, et al. GSK-3beta is required for memory reconsolidation in adult brain. PLoS One. 2008;3:e3540.
O’Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo S, et al. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24:6791–8.
O’Brien WT, Huang J, Buccafusca R, Garskof J, Valvezan AJ, Berry GT, et al. Glycogen synthase kinase-3 is essential for beta-arrestin-2 complex formation and lithium-sensitive behaviors in mice. J Clin Invest. 2011;121:3756–62.
Prickaerts J, Moechars D, Cryns K, Lenaerts I, van Craenendonck H, Goris I, et al. Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J Neurosci. 2006;26:9022–9.
Sutherland C. What Are the bona fide GSK3 Substrates? Int J Alzheimers Dis. 2011;2011:505607.
Marchand WR, Yurgelun-Todd D. Striatal structure and function in mood disorders: a comprehensive review. Bipolar Disord. 2010;12:764–85.
Rushlow WJ, Seah C, Sutton LP, Bjelica A, Rajakumar N. Antipsychotics affect multiple calcium calmodulin dependent proteins. Neuroscience. 2009;161:877–86.
Nunes A, Stone W, Ardau R, Berghofer A, Bocchetta A, Chillotti C, et al. Exemplar scoring identifies genetically separable phenotypes of lithium responsive bipolar disorder. Transl Psychiatry. 2021;11:36.
Young AH, Newham JI. Lithium in maintenance therapy for bipolar disorder. J Psychopharmacol. 2006;20:17–22.
Bowden CL, Brugger AM, Swann AC, Calabrese JR, Janicak PG, Petty F, et al. Efficacy of divalproex vs lithium and placebo in the treatment of mania. The Depakote Mania Study Group. JAMA. 1994;271:918–24.
Bialer M. Why are antiepileptic drugs used for nonepileptic conditions? Epilepsia. 2012;53:26–33.
Bazinet RP, Weis MT, Rapoport SI, Rosenberger TA. Valproic acid selectively inhibits conversion of arachidonic acid to arachidonoyl-CoA by brain microsomal long-chain fatty acyl-CoA synthetases: relevance to bipolar disorder. Psychopharmacology (Berl). 2006;184:122–9.
El-Habr EA, Dubois LG, Burel-Vandenbos F, Bogeas A, Lipecka J, Turchi L, et al. A driver role for GABA metabolism in controlling stem and proliferative cell state through GHB production in glioma. Acta Neuropathol. 2017;133:645–60.
Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–41.
Avery LB, Bumpus NN. Valproic acid is a novel activator of AMP-activated protein kinase and decreases liver mass, hepatic fat accumulation, and serum glucose in obese mice. Mol Pharmacol. 2014;85:1–10.
Caliyurt O, Altiay G. Resting energy expenditure in manic episode. Bipolar Disord. 2009;11:102–6.
Faurholt-Jepsen M, Brage S, Vinberg M, Christensen EM, Knorr U, Jensen HM, et al. Differences in psychomotor activity in patients suffering from unipolar and bipolar affective disorder in the remitted or mild/moderate depressive state. J Affect Disord. 2012;141:457–63.
Faurholt-Jepsen M, Vinberg M, Frost M, Debel S, Margrethe Christensen E, Bardram JE, et al. Behavioral activities collected through smartphones and the association with illness activity in bipolar disorder. Int J Methods Psychiatr Res. 2016;25:309–23.
Vancampfort D, Sienaert P, Wyckaert S, Probst M, De Herdt A, De Hert M, et al. Cardiorespiratory fitness in outpatients with bipolar disorder versus matched controls: An exploratory study. J Affect Disord. 2016;199:1–5.
Brietzke E, Kapczinski F, Grassi-Oliveira R, Grande I, Vieta E, McIntyre RS. Insulin dysfunction and allostatic load in bipolar disorder. Expert Rev Neurother. 2011;11:1017–28.
Vancampfort D, Vansteelandt K, Correll CU, Mitchell AJ, De Herdt A, Sienaert P, et al. Metabolic syndrome and metabolic abnormalities in bipolar disorder: a meta-analysis of prevalence rates and moderators. Am J Psychiatry. 2013;170:265–74.
Mansur RB, Lee Y, McIntyre RS, Brietzke E. What is bipolar disorder? A disease model of dysregulated energy expenditure. Neurosci Biobehav Rev. 2020;113:529–45.
Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–38.
Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 2013;36:587–97.
Bullmore E, Sporns O. The economy of brain network organization. Nat Rev Neurosci. 2012;13:336–49.
Tomasi D, Wang GJ, Volkow ND. Energetic cost of brain functional connectivity. Proc Natl Acad Sci USA. 2013;110:13642–7.
Chu WJ, Delbello MP, Jarvis KB, Norris MM, Kim MJ, Weber W, et al. Magnetic resonance spectroscopy imaging of lactate in patients with bipolar disorder. Psychiatry Res. 2013;213:230–4.
Dager SR, Friedman SD, Parow A, Demopulos C, Stoll AL, Lyoo IK, et al. Brain metabolic alterations in medication-free patients with bipolar disorder. Arch Gen Psychiatry. 2004;61:450–8.
Kim DJ, Lyoo IK, Yoon SJ, Choi T, Lee B, Kim JE, et al. Clinical response of quetiapine in rapid cycling manic bipolar patients and lactate level changes in proton magnetic resonance spectroscopy. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1182–8.
Xu J, Dydak U, Harezlak J, Nixon J, Dzemidzic M, Gunn AD, et al. Neurochemical abnormalities in unmedicated bipolar depression and mania: a 2D 1H MRS investigation. Psychiatry Res. 2013;213:235–41.
Hurst RH. p(H) of blood of psychotics measured by the glass electrode. Biochem J. 1932;26:1536–41.
Looney JM, Childs HM. The lactic acid and glutathione content of the blood of Schizophrenic patients. J Clin Invest. 1934;13:963–8.
Machado-Vieira R, Zanetti MV, Otaduy MC, De Sousa RT, Soeiro-de-Souza MG, Costa AC, et al. Increased brain lactate during depressive episodes and reversal effects by lithium monotherapy in drug-naive bipolar disorder: a 3-T 1H-MRS study. J Clin Psychopharmacol. 2017;37:40–5.
He C, Gao P, Cui Y, Li Q, Li Y, Lu Z, et al. Low-glucose-sensitive TRPC6 dysfunction drives hypoglycemia-induced cognitive impairment in diabetes. Clin Transl Med. 2020;10:e205.
Huang W, Liu Y, Luz A, Berrong M, Meyer JN, Zou Y, et al. Calcium/calmodulin dependent protein kinase kinase 2 regulates the expansion of tumor-induced myeloid-derived suppressor cells. Front Immunol. 2021;12:754083.
Ying L, Li N, He Z, Zeng X, Nan Y, Chen J, et al. Fibroblast growth factor 21 Ameliorates diabetes-induced endothelial dysfunction in mouse aorta via activation of the CaMKK2/AMPKalpha signaling pathway. Cell Death Dis. 2019;10:665.
Cudney LE, Sassi RB, Behr GA, Streiner DL, Minuzzi L, Moreira JC, et al. Alterations in circadian rhythms are associated with increased lipid peroxidation in females with bipolar disorder. Int J Neuropsychopharmacol. 2014;17:715–22.
Kuloglu M, Ustundag B, Atmaca M, Canatan H, Tezcan AE, Cinkilinc N. Lipid peroxidation and antioxidant enzyme levels in patients with schizophrenia and bipolar disorder. Cell Biochem Funct. 2002;20:171–5.
Versace A, Andreazza AC, Young LT, Fournier JC, Almeida JR, Stiffler RS, et al. Elevated serum measures of lipid peroxidation and abnormal prefrontal white matter in euthymic bipolar adults: toward peripheral biomarkers of bipolar disorder. Mol Psychiatry. 2014;19:200–8.
Cuadrado A, Manda G, Hassan A, Alcaraz MJ, Barbas C, Daiber A, et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: a systems medicine approach. Pharmacol Rev. 2018;70:348–83.
Wang S, Yi X, Wu Z, Guo S, Dai W, Wang H, et al. CAMKK2 defines ferroptosis sensitivity of melanoma cells by regulating AMPK‒NRF2 pathway. J Invest Dermatol. 2022;142:189–200 e8.
Morris G, Walker AJ, Walder K, Berk M, Marx W, Carvalho AF, et al. Increasing Nrf2 activity as a treatment approach in neuropsychiatry. Mol Neurobiol. 2021;58:2158–82.
Truong TTT, Panizzutti B, Kim JH, Walder K. Repurposing drugs via network analysis: opportunities for psychiatric disorders. Pharmaceutics. 2022;14:1464.
Bhullar KS, Lagaron NO, McGowan EM, Parmar I, Jha A, Hubbard BP, et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer. 2018;17:48.
Cohen P, Cross D, Janne PA. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat Rev Drug Discov. 2021;20:551–69.
Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–16.
Ngoei KRW, Langendorf CG, Ling NXY, Hoque A, Varghese S, Camerino MA, et al. Structural determinants for small-molecule activation of skeletal muscle AMPK alpha2beta2gamma1 by the glucose importagog SC4. Cell Chem Biol. 2018;25:728–37 e9.
Scott JW, van Denderen BJ, Jorgensen SB, Honeyman JE, Steinberg GR, Oakhill JS, et al. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol. 2008;15:1220–30.
Xiao B, Sanders MJ, Carmena D, Bright NJ, Haire LF, Underwood E, et al. Structural basis of AMPK regulation by small molecule activators. Nat Commun. 2013;4:3017.
Acknowledgements
This work was supported by an Ideas Grant from the National Health and Medical Research Council (NHMRC) of Australia (2001817) to JWS and ALG. JK is the recipient of an International PhD Scholarship from the Australian Catholic University. AGM is supported by the Cyril Tonkin Scholarship from Monash University. MB and MAF are supported by NHMRC Senior Principal Research Fellowships and Leadership 3 Investigator Grants (MB; 1156072 and 2017131) and (MAF; 1116936 and 1194141). JMM acknowledges an NHMRC Leadership 1 Investigator Grant (1172929) and NHMRC Infrastructure Support (9000719). AJH is supported by an NHMRC Principal Research Fellowship. JMM and AJH acknowledge the Victorian State Government operational infrastructure support program. The figures were created with Biorender.com.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Contributions
JWS, ALG and JK conceived the manuscript topic and structural design. JK and CRH prepared the figures. JWS, ALG and JK drafted and finalised the manuscript with critical input and revisions from KN, LMM, CRH, OKF, AGM, DGW, ARM, KW, MB, AJH, JMM and MAF.
Corresponding author
Ethics declarations
Competing interests
MAF is a founder and shareholder of Celesta Therapeutics. All other authors declare no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaiser, J., Nay, K., Horne, C.R. et al. CaMKK2 as an emerging treatment target for bipolar disorder. Mol Psychiatry 28, 4500–4511 (2023). https://doi.org/10.1038/s41380-023-02260-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-023-02260-3
