
Abstract
High thymidylate synthase (TS) level in cancer tissue is considered to result in resistance to pemetrexed therapy for advanced stages of nonsquamous non-small cell lung cancers. To further investigate the mechanism of pemetrexed resistance and potential prognostic outcomes in lung cancer, we established pemetrexed-resistant lung adenocarcinoma cell sublines from CL1 harboring a mutated TP53 gene (R248W) and A549 harboring wild-type TP53. We found the TS expression is upregulated in both pemetrexed-resistant sublines and the reduced TS level achieved through shRNA inhibition resulted in higher pemetrexed sensitivity. We also demonstrated that the acquisitions of pemetrexed resistance enhances epithelial–mesenchymal transition (EMT) in vivo with a mice animal model and in vitro with CL1 and A549 sublines, which was associated with upregulation of ZEB1 which, in turn, downregulates E-cadherin and upregulates fibronectin. When ERK1/2 phosphorylation was reduced by an inhibitor (U0126) or siRNA inhibition, both pemetrexed-resistant sublines reduced their migration and invasion abilities. Therefore, the ERK-mediated pathways induce apoptosis with pemetrexed treatment, and may in turn mediate EMT when cancer cells are resistant to pemetrexed. We further demonstrated that the growth of pemetrexed-resistant tumors could be inhibited by vinblastine in vivo and vincristine in vitro. Our data indicate that pemetrexed resistance could be relieved by non-cross-resistant chemotherapeutic drugs such as vinca alkaloids and might be independent to TP53 status. Furthermore, the phosphorylation of ERK was reduced by vincristine. This finding provides a new insight for overcoming pemetrexed resistance and metastasis by application of vinca alkaloids.
Similar content being viewed by others


Introduction
Pemetrexed (LY231514) inhibits at least three key enzymes including thymidylate synthase (TS), dihydrofolate reductase (DHFR) and glycinamide ribonucleotide formyltransferase (GARFT).1, 2 Inhibition of TS results in decreased turnover of dUMP to dTMP, which is necessary for DNA synthesis.3 These targets are related to the cytotoxicity of pemetrexed, because both thymidine and hypoxanthine are required to circumvent cellular death caused by pemetrexed.4 Intracellularly, pemetrexed is polyglutamated to the active pentaglutamide by a reaction catalyzed by folylpolyglutamate synthase (FPGS). The enzyme counteracting antifolate polyglutamylation is γ-glutamyl hydrolase (GGH).
Recently, it has been reported that TS gene expression is significantly increased in pemetrexed-resistant small cell lung cancer cell lines.5 Knockdown of TS expression using siRNA has enhanced pemetrexed cytotoxicity in PC6/MTA-4.0 cells. Zhang et al.6 have shown TS and DHFR were significantly increased in the four pemetrexed-resistant A549 sublines. Chen et al. have suggested that TS expression, rather than DHFR, may be an important predictive factor of the treatment efficacy of pemetrexed in non-small cell lung cancer (NSCLC) patients.7 Another report concluded that better response usually appears in patients with a lower expression of TS by meta-analysis8 with a significant association between TS expression and outcomes of pemetrexed-based chemotherapy for NSCLC. Therefore, it can be concluded that upregulation of TS gene expression may have an important role in pemetrexed resistance.
Multiple studies have revealed that chemoresistance cells often acquire an epithelial–mesenchymal transition (EMT)-like phenotype.9 During the acquisition of EMT characteristics, epithelial cancer cells lose the expression of proteins that promote cell–cell contact, such as E-cadherin and β-catenin, and gain the expression of mesenchymal markers, such as fibronectin, vimentin and N-cadherin, leading to remodeling of the cytoskeleton and enhancement of cancer cell migration and invasion. Recently, an EMT phenotype was observed in gemcitabine-resistant pancreatic cancer cells,10, 11 gefitinib-resistant NSCLC,12 oxaliplatin-resistant colorectal cancer cells,13 paclitaxel-resistant ovarian cancer cells14 and tamoxifen-resistant breast cancer cells.15 The association of pemetrexed resistance with EMT alteration has not been reported yet and the question of how EMT is mechanistically involved in pemetrexed resistance has not been answered.
TGF-β triggers diverse cellular processes including growth arrest, tissue fibrosis and EMT.16, 17 As a result, TGF-β activates R-Smads (Smad2 and Smad3) via phosphorylation at their C-terminal serine residues. R-Smads form a heterocomplex with Smad4 and translocate into the nucleus to regulate gene expression.18, 19, 20, 21, 22 These pathways are referred to as Smad-dependent pathways. Snail and Slug, key regulators of TGF-β-induced EMT, are sufficient for the induction of single-cell invasion.23 In addition to the Smad signaling pathways, TGF-β also elicits diverse types of non-Smad signaling pathways. Among them, activation of Ras, mitogen-activated protein kinases (MAPKs) such as ERK and p38MAPK (p38), Rho GTPases, and PI3K/Akt signaling has been linked to TGF-β-induced EMT.24, 25, 26 Recent studies have identified the crucial role of TGF-β signaling pathways in inducing EMT through the Smad-dependent and Smad-independent pathways.17, 27, 28, 29
Transcription factors involved in EMT such as Snail, Slug, TWIST and the ZEB families mainly repress expression of E-cadherin during EMT.30, 31, 32, 33 The transcription factor ZEB1 can be activated by the TGF-β, TNF-α and IGF1 signaling pathways. A correlation of ZEB1 expression and loss of E-cadherin has been demonstrated in tumor cell lines of lung adenocarcinomas.34 Therefore, ZEB1 is also a crucial mediator of EMT, exerting its effects on induction of EMT by inhibiting expression of E-cadherin.
Finding a way to control the growth of pemetrexed-induced resistance in lung cancer cells is clinically important. However, it has been reported that pemetrexed-resistant lung cancer sublines show cross-resistance to cisplatin, but not to docetaxel, vinorelbine and 5-fluorouracil,6 which points to the possibility of reversing pemetrexed resistance by using another clinically chemotheraputic drug. In this study, we have identified the signaling pathway that controls pemetrexed-induced EMT. Furthermore, we also provide evidence that vinca alkaloids, a group of clinically used anti-cancer drugs, reversed the pemetrexed resistance and EMT. These findings may be applied immediately to overcome pemetrexed resistance.
Results
Establishment of pemetrexed resistant CL1 and A549 lung cancer sublines
According to MTT sensitivity assay, the established A549/A400, CL1/A100 and CL1/A200 sublines (Table 1) revealed their drug sensitivities in terms of IC50 (inhibition concentration). The A549/A400, CL1/A100 and CL1/A200 sublines have 37.8-fold, 22.9-fold and 86.5-fold resistance to pemetrexed, respectively, when compared with parental cells. To investigate whether pemetrexed resistance may result in cross-resistance to other antimetabolic chemotherapy, the sensitivities to MTX and 5-FU were also determined and only minimal resistances were detected. For example, the CL1/A200 subline is 2.5-fold resistant to MTX and 0.6-fold resistant to 5-FU. The drug sensitivity profiles of the A549 and CL1 sublines in our study exhibit similar results to those previously reported for PC-9 and A549 pemetrexed-resistant sublines.6 Interestingly, all three pemetrexed-resistant sublines have only a low degree of cross-resistance to docetaxel (1.1–1.9-fold). Moreover, the pemetrexed-resistant sublines all showed a significant sensitivity to vincristine when compared with parental A549 (0.3-fold) and CL1 (0.6–0.8-fold) cells.
Characterization of the pemetrexed-regulated genes
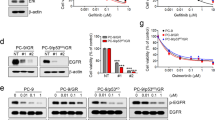
To investigate the mechanism of the pemetrexed resistance, we compared the expression of pemetrexed-target proteins TS and DHFR, as well as the expression of the enzymes FPGS and GGH by western blot analysis. The protein levels of TS and DHFR significantly increased in A549/A400 (Figure 1a), CL1/A100 and CL1/A200 sublines (Figure 1b). The levels of FPGS were slightly increased in all three sublines. Upregulated GGH levels in A549/A400 cells and downregulated GGH levels in CL1 sublines were observed. It has been reported that the level of TS protein determines pemetrexed sensitivity in cell lines6, 35 and lung cancer tissues.7 Therefore, to demonstrate the dominant role of TS overexpression in the acquired pemetrexed resistance, the sublines were infected with the shRNA expressing lentivirus that targets TS mRNA. Three shTS constructs significantly inhibited TS expression when compared with the shGFP control and the proteins of TS were detected by western blotting. The pemetrexed sensitivities were enhanced in shTS-infected CL1/A100 (Figure 1c), and CL1/A200 (Figure 1d) sublines were measured by MTT cell survival assay. When TS protein expression was reduced, the viabilities of CL1/A100 and CL1/A200 cells were coordinately reduced with pemetrexed treatment. We also further investigated whether the 5-FU and MTX sensitivities could be affected by TS inhibition. From the results of MTT assay, the drug sensitivities of MTX (Figure 1e) and 5-FU (Figure 1f) in the TS-inhibited CL1/A200 subline were not altered significantly. Our data demonstrate that high levels of TS expression are associated with pemetrexed resistance. Inhibition of TS expression by shRNA enhances pemetrexed sensitivity, but not sensitivity to MTX and 5-FU.

Characterization of the pemetrexed-resistant A549 and CL1 lung cancer cells. The expression of TS, DHFR, FPGS and GGH were detected by western blot assay in A549 (a) and CL1 (b) sublines. Inhibition of TS by shRNA in CL1/A100 (c) and CL1/A200 (d) sublines were followed by MTT assay. The sensitivities of TS-inhibited CL1 sublines to MTX treatment were examined by MTX (e) and 5-FU (f). *P<0.05 was considered statistically significant. Individual clones are identified by their unique TRC number, which were purchased from National RNAi Core Facility at Academic Sinica, Taiwan. To inhibit TS expression, the CL1 sublines (5 × 104 per well) were sub-cultured onto 60-mm plates. After 16 h of incubation at 37 °C, cells were infected with recombinant lentivirus vectors with protamine sulfate added (0.8 μg/μl) to help infection. The next day, the medium was removed and the cells were selected by 2 μg/ml puromycin (Sigma, P8833) at 37 °C for 48 h, followed by MTT assay or protein detection. The clone of shGFP (TRCN0000072178) for vector control targeted to GFP, whereas the clones of shTS-1 (TRCN0000291720), shTS-2 (TRCN0000291655) and shTS-3 (TRCN0000045665) targeted to TS.
Acquisition of pemetrexed resistance promotes metastasis in vivo
To answer whether pemetrexed resistance may result in lung cancer metastasis, we injected the A549/A400 and parental A549 cells through the tail vein of SCID mice. Although all A549-injected and A549/A400-injected animals developed lung tumors, a greater number and larger size of tumors were found in the animal group injected with A549/A400 cells and the weights of the tumors were significantly higher (Figure 2a) in the pemetrexed-resistant A549/A400-injected animals. The lung tissues were further examined with hematoxylin and eosin (H&E) staining for tumor verification and higher TS expressions were detected in A549/A400 tumors by immunohistochemistry (IHC; Figure 2b).

Investigation of the pemetrexed-induced metastasis by a mice metastasis model. The cells of A549/A400 and A549 were tail-vein injected into mice followed by monitoring for 55 days until sacrifice. The lung tissues were examined for tumor metastasis and the weights of the two groups were recorded and listed (a). H&E and IHC of TS staining were applied to verify the tumors and TS overexpression followed by pERK, ZEB1, E cadherin and fibronectin detection (b). All five CL1-injected mice were free of tumors and two of the CL1/A200-injected mice established metastatic tumors, which were stained with H&E and IHC with anti-TS antibody (c).
The CL1 and CL1/A200 cells were also injected into SCID mice to test their metastatic potential. As low metastasis of the CL1 cells has been demonstrated,36 it is not surprising that none of the CL1-injected animals developed metastasis and only two of the CL1/A200-injected animals developed lung metastasis at 75 days when compared with the lungs of CL1-injected animals (Figure 2c). The H&E staining and high TS expression confirmed the CL1/A200 cells have metastatic potential.
Pemetrexed-resistant sublines show changed EMT molecular markers in vitro and in vivo
In order to further support the metastatic potential of pemetrexed-resistant sublines detected in the mice model, in vitro assays were performed using transwell cell culture chambers to compare the migration and invasion abilities of parental cells and sublines. The pemetrexed-resistant A549/A400 cells showed a 3.5-fold increase in migration with 4 h incubation (Figure 3a) and a 3.2-fold increase in invasion with 16 h incubation (Figure 3b) through the transwell membrane when compared with parental cells. Similar results also revealed that CL1/A100 and CL1/A200 have significant 2.7-fold increases in migration with 24 h incubation (Figure 3c). Both CL1 sublines also have 2.8-fold and 3.7-fold increases in invasion abilities, respectively, with 48 h incubation (Figure 3d). Therefore, we examined whether the induced EMT found in pemetrexed-resistant sublines came through Smad-dependent or Smad-independent pathways by western blot analysis in all three sublines and compared the results with those of their parental cells with known EMT markers (Figure 3e). According to the data, the expression of E-cadherin protein levels were downregulated but the phosphorylated Smad2 and Smad3 levels were not significantly increased in pemetrexed-resistant sublines, nor were the protein levels of Slug and Snail. Only phosphoryalted p38 and ERK levels were highly upregulated along with ZEB1 and fibronectin levels. The results indicate p38 and/or ERK signaling pathways may regulate EMT through a Smad-independent pathway. Pemetrexed-resistant sublines that developed EMT were correlated with downregulated E-cadherin, and upregulated Fibronectin and ZEB1. To better understand the development of EMT in pemetrexed resistance, we compared the morphologic appearances of low and high doses pemetrexed-selected stable cell lines of A549 and CL1 cells (A549/A200 and A549/A400, CL1/A50 and CL1/A200, respectively). The parental A549 and CL1 cells changed from a classical epithelial morphology to a mesenchymal, spindle-shaped morphology (Supplementary Figure 1a). Although the spindle-shaped morphology in high-dose pemetrexed-selected cells (A549/A400 and CL1/A200) was not obviously as low-dose pemetrexed-selected cells (A549/A200 and CL1/A50), the expression levels of E-cadherin and fibronectin were further confirmed through western blotting of total cell lysates (Supplementary Figure 1b). The western blots confirmed downregulation of E-cadherin and upregulation of fibronectin expression. Furthermore, the levels of pERK and ZEB1 were also upregulated in all pemetrexed-resistant sublines. Fos family member FRA1 is one of the most frequently overexpressed AP-1 proteins in solid cancers, ERK2/FRA1 induce EMT via regulation of ZEB protein expression.37 Interestingly, only FRA1 levels in A549 sublines were significantly increased (Supplementary Figure 1b).

Analysis of the pemetrexed-induced metastasis in vitro. The cells of A549 and A549/A400 were added into transwells for 4 h incubation to determine the migration ability (a) or for 16 h to measure invasion ability (b). The cells of CL1, CL1/A100 and CL1/A200 were added into transwells for 24 h incubation to determine the migration ability (c) or for 48 h to measure invasion ability (d). Western blot analyses were applied to examine the associated signaling pathways regulating EMT in A549 and CL1 sublines (e).
To verify the expressions of E-cadherin, ZEB1, pERK and fibronectin in the lung tumors, the tumor sections of A549 and A549/A400 cells were evaluated with individual antibody (Figure 2b). The IHC results clearly demonstrate a reduction in surface expression of E-cadherin and upregulation of pERK, ZEB1 and fibronectin. High-magnification pictures of IHC are provided in Supplementary Figure 5.
Regulation of EMT and MAPK signaling
The mechanisms of pemetrexed-resistant-induced EMT were further investigated by inhibition of ERK1/2 phosphorylation with U0126 and p38 phosphorylation with SB253580 followed by Western blot analysis. In the A549/A400 subline, the reduced levels of phosphorylated-ERK (pERK) by U0126 were correlated with reduced ZEB1 and fibronectin expression as well as upregulated E-cadherin expression. In contrast, inhibition of p38 by SB253580 had no effect on ZEB1, fibronectin and E-cadherin expression. Although the p-p38 levels were not significantly reduced by SB253580, we have checked the downstream p38-regulated protein Activating transcription factor 2 (ATF-2)38 and found the levels of phosphorylated ATF-2 significantly reduced by SB253580 after one hour. The inhibitor PD98059 specifically inhibits MEK-1-mediated activation of MAPK which does not directly inhibit ERK. This also caused reduced p-ERK and ZEB1 levels combined with upregulated E-cadherin expression (Figure 4a). To test the toxicities of kinase inhibitors, we treated individual subline with indicated inhibitor for 24 h and the cell viability was determined by MTT assay (Figures 4b and f). Inhibition of ERK phosphorylation resulted in A549/A400-reduced migration (Figure 4c) and invasion abilities (Figure 4d) of 50%, whereas inhibition of p38 only had a residual effect. Similar to the results obtained from A549/A400 investigation, inhibition of ERK1/2 phosphorylation with U0126 and PD98059 in CL1/A200 subline reduced p-ERK, ZEB1 and fibronectin levels in combination with upregulated E-cadherin expression (Figure 4e). Inhibition of ERK phosphorylation resulted in CL1/A200-reduced migration (Figure 4g) and invasion abilities (Figure 4h) of ~50%.

Determination of the ERK signaling pathway that regulates pemetrexed-induced EMT by kinase inhibitors. The cells of A549/A400 were treated with U0126, PD98059 and SB253580 for the indicated time and then harvested for western blot analysis. The phosphorylated ERK and p38 levels indicated the effects of kinase inhibition. The levels of ZEB1, E-cadherin and fibronectin represent the alteration of EMT upon phosphorylation inhibition (a). The possible toxicity of kinase inhibitors were measured by MTT assay (b, f). The A549/A400 cells were pretreated with the indicated kinase inhibitor for 24 h followed by migration assay (c) or invasion assay (d). The CL1/A200 cells were treated with individual kinase inhibitors for the indicated time, followed by protein detection (e). The kinase inhibitor pretreated CL1/A200 cells were analyzed by transwell migration assay (g) or invasion assay (h). U0126, PD98059 and SB253580 were purchased from Cell Signaling (Danvers, MA, USA).
Interference of ERK, ZEB1 and fibronectin expressions resulted in EMT suppression
The role of ERK1/2 in pemetrexed resistance-induced EMT was further characterized by inhibition of ERK1/2 expression with siRNA interference. In A549/A400 cells, when ERK1/2 expression was downregulated by siRNA to <50%, E-cadherin expression was significantly increased with reduced ZEB1 and fibronectin expression (Figure 5a). The migration ability of the ERK interfered A549/A400 cells was reduced to 30% (siERK#1) and 10% (siERK#2) when compared with the si-controlled cells (Figure 5b). When invasion ability was assayed, the abilities of siERK#1 and siERK#2 cells were reduced to 40% and 10%, respectively (Figure 5c). We also investigated the effect of p-38 in pemetrexed-resistant sublines by siRNA interference (Figure 5d). The results showed that the levels of ZEB1, E-cadherin and fibronectin were not significantly altered in A549/A400 cells. Furthermore, inhibition of p-38 expression by siRNA has no effect in migration and invasion abilities in vitro (Figures 5e and f). Similar approaches were also applied to CL1/A200 cells (Supplementary Figures 2a–f) and reduced ERK expression also resulted in reduced ZEB1 and fibronectin expression with E-cadherin upregulation. Interference of ERK expression also reduced CL1/A200 cells migration and invasion abilities, respectively. As shown in the data in Figure 5a and Supplementary Figure 2a, the degrees of ERK inhibition were correlated with the levels of ZEB1 and fibronectin downregulation, and were also reflected in the degrees of migration and invasion inhibition. To determine the role that ZEB1 has in pemetrexed-mediated EMT, siRNA inhibition of ZEB1 (siZEB1) was applied and the E-cadherin level was increased and the fibronectin level was decreased significantly without affecting ERK phosphorylation (Figure 5g). Inhibition of ZEB1 expression also resulted in migration (Figure 5h) and invasion (Figure 5i). Similar results were obtained from CL1/A200 cells showing that upregulated E-cadherin and downregulated fibronectin resulted in less migration and invasion under ZEB1 inhibition (Supplementary Figures 2g–l). When fibronectin was inhibited by siRNA (siFN), ZEB1 and E-cadherin levels were not altered (Figure 5j) but migration (Figure 5k) and invasion (Figure 5l) were significantly reduced to <50%. Similar data were demonstrated in CL1/A200 cells (Supplementary Figures 2j–l). We conclude that pemetrexed-mediated EMT occurs via ERK-signaling activation of ZEB1, which downregulates E-cadherin and upregulates fibronectin to promote EMT in both A549/A400 and CL1/A200 pemetrexed-resistant cancer cells.

Inhibition of ERK by siRNA technology to correlate the EMT suppression in pemetrexed-resistant sublines. The cells of A549/A400 were transfected with two different sequences of siRNA to inhibit ERK expression (siERK#1 and siERK#2). After 48 h incubation, the cells were harvested for pERK, ERK, ZEB1, E-cadherin and fibronectin protein detection (a) or seeded for migration assay (b) and invasion assay (c). Similarly, the expression of p38 was inhibited by two individual siRNAs and examined (d–f). A mixture of ZEB1 siRNAs were transfected into A549/A400 cells and the effects of inhibited ZEB1 were analyzed by western blot (g), migration (h) and invasion (i) assays. Similarly, fibronectin siRNAs were transfected into cells and analyzed (j–l), respectively. A mixed siRNA for ZEB1 (SASI_Hs02_00330526, 00330527, 00330528, 00330529) and fibronectin (SASI_Hs01_00203291, Hs02_00203292, Hs02_00203294, Hs02_00333045) with siC (universal NC control siRNA) were used. SiRNAs for p38 (#6243 and #6564) were purchased from Cell Signaling.
To answer whether overexpressed TS also contribute to ERK/ZEB1-mediated EMT, we used siRNA to inhibit TS in A549/A400 and CL1/A200 cells (Supplementary Figure 3). Inhibition of TS expression in pemetrexed-resistant A549/A400 cells by siRNA resulted in loss of cell proliferation as sub-G1 fraction significantly increased (Supplementary Figure 3d). The TS-inhibited cells further reduced migration and invasion (Supplementary Figures 3b and c), but not the ERK phosphorylation (Supplementary Figure 3a). The levels of ZEB1, E-cadherin and fibronectin were not altered in TS-inhibited cells. Inhibition of TS expression in CL1/A200 cells by siRNA has no major effects on cell proliferation, migration and invasion (Supplementary Figures 3e–h).
Vinblastine inhibits pemetrexed-resistant tumor growth in a mice xenograft model and may also inhibit EMT
According to the data listed in Table 1, pemetrexed-resistant sublines were more sensitive to vincristine than their parental cells. Therefore, we tested the efficacy of vinblastine in a mice xenograft model inoculated with A549/A400 cells. Compared with the PBS or pemetrexed treatment, vinblastine significantly inhibited the growth of A549/A400 tumors (Figure 6a). We then examined whether vincristine could also attenuate the phosphorylation of ERK1/2 and suppress EMT. When A549/A400 cells were treated with vincristine (5 nM) for 24 h, ERK and p-ERK levels were reduced to 70%. With 48 h treatment, ERK and p-ERK levels were reduced to 70% and 40% respectively, when correlated with fibronectin downregulation (Figure 6b). The mRNA levels of ZEB1, E-cadherin and fibronectin in A549/A400 cells were determined by RT–PCR (Figure 6c). The mRNA levels of ZEB1 and fibronectin were reduced, and E-cadherin levels were increased significantly. Similar results were also obtained from CL1/A200 cells treated with vincristine (Supplementary Figure 4). The results indicate that vincristine inhibits the ERK signaling pathway on ERK expression and activation. Surprisingly, E-cadherin levels were not correspondingly upregulated when ZEB1 was reduced. We speculate that vincristine induces A549/A400 cell death. This might result in E-cadherin degradation. Because pemetrexed-resistant cells are sensitive to vincristine treatment, it is difficult to recognize the possible effect on EMT inhibition; therefore, we used 5 nm of vincristine to treat A549/A400 cells for in vitro transwell assays. With 24 h of vincristine treatment, the viability of vincristine-treated cells was maintained at ~90% when compared with untreated cells (Figure 6d). Under a non-lethal dose of vincristine treatment, the migration of A549/A400 cells was reduced to 50% (Figure 6e), and the invasion ability was also reduced to 50% (Figure 6f). These results indicate that vincristine and vinblastine not only overcome pemetrexed resistance but also may suppress ERK–ZEB1 regulated metastasis in NSCLC. In summary, as shown in Figure 6g, we have demonstrated that the enhanced pemetrexed resistance of EMT is mediated by ERK and ZEB1, and vinca alkaloids may resolve resistance and reduce metastasis.

Vinblastine inhibits mice xenografted A549/A400 tumor growth and vincristine reduces phosphorylation of ERK. The A549/A400 cells were injected subcutaneously. Twelve animals were then randomly divided into three groups (PBS, VBL and pemetrexed). VBL was obtained from Teva Pharmaceutical Industries Limited (Petah Tikva, Israel). Tumor sizes were measured every 3 days following drug injection and tumor volume was calculated by the following formula: 0.5 × larger diameter (mm) × small diameter2 (mm). Due to tumor size variability, a non-parametric Mann-Whitney U test was applied for statistical analysis. (a). *P<0.05 was considered statistically significant. The effects of VCR on A549/A400 cells were investigated by western blot analysis (b). The mRNA levels of ZEB1, E-cadherin and fibronectin were analyzed by RT–PCR (c).The cell viability with VCR (5 nM) was examined by MTT assay (d). The effects of VCR on migration (e) and invasion were determined (f). A signaling pathway that is regulated by vinca alkaloids in pemetrexed-resistant lung cancer cells is proposed (g).
Discussion
Previously, we have identified pemetrexed-mediated apoptosis associated with the activation of ataxia telangiectasia mutated/TP53-dependent and -independent signaling pathways, which promote intrinsic and extrinsic apoptosis.39 However, when lung cancer cells develop pemetrexed resistance, researchers have yet to study whether TP53 status could affect the mechanism of pemetrexed resistance. Up to date, there is no clear evidence showing how to resolve resistance occurring after pemetrexed treatment. Our data suggested that the choice of docetaxel to manage first-line pemetrexed resistance could be suggested and there is no obvious bias against the status of TP53 in pemetrexed-resistant sublines of docetaxel sensitivities.
Chemoresistance is usually companied with metastasis that blocks the efficiency of cancer therapy. Our data demonstrated that wt-TP53 and mut-TP53 lung cancer cells increased their metastatic potential after pemetrexed selection. In the mice metastasis model (Figure 2), enhanced metastasis can be observed in A549/A400 cells, which developed more tumors in the lung tissue. In contrast to the highly mobile A549 cells, CL1 cells showed no metastatic abilities and did not establish any tumors in mice lungs. Therefore, when low percentage of CL1/A200 cells established tumors in vivo, it suggested that metastasis can be induced by pemetrexed resistance. We have shown that pemetrexed-induced apoptotic cell death is ERK-dependent.40 It is possible that the surviving cells using high TS expression to tolerate pemetrexed-activated ERK signaling and resulted in metastasis. Indeed, the in vitro migration and metastasis assays further support that pemetrexed-mediated EMT is associated with ERK/ZEB1 activation and E-cadherin inhibition as well as upregulation of fibronectin (Figure 4). Although p38 also activated in pemetrexed resistanct cells, this signaling pathway has no effect on pemetrexed-mediated EMT. Previously, ERK2/FRA1 induce EMT via regulation of ZEB protein expression has been shown.37 Although the si-RNAs used for ERK1/2 inhibition in our investigation cannot distinguish ERK1 or ERK2 specifically, our data further support the importance of this signaling pathway in pemetrexed-resistant-induced EMT. Activation of ZEB1 to repress E-cadherin expression has been shown,41 but the association of ZEB1 and fibronectin upregulation is a novel finding. We found that FRA1 was strongly expressed in A549/A400 but not CL1/A200 cells. We suggest that FRA1 may be a critical ERK pathway effector regulating EMT-like changes in A549/A400 cells. What could be the linking factor between ERK and ZEB1 for EMT in CL1/A200 cells? As relative low FRA1 level was detected in CL1/A200 cells, upregulated ZEB1 expression may be resulted from β-catenin signaling.42
There are four major vinca alkaloids in clinical use: vinblastine (VBL), vinorelbine (VRL), vincristine (VCR) and vindesine (VDS). VCR, VBL and VRL have been approved for use in the United States.43 However, these drugs are a substrate of the ABCB1 (P-gp) transporter, therefore overexpression of ABCB1 in cancer cells is considered the major phenotype of multidrug resistance to vinca alkaloids.44, 45 We have shown that vinca alkaloids are non-cross-resistant to pemetrexed and may be applied to reverse pemetrexed resistance of NSCLC. A retrospective study to evaluate the activity and toxicity of VRL administered to a consecutive series of pemetrexed-pretreated malignant pleural mesothelioma patients (59 patients) has been reported.46 It seems that VRL is moderately active in pemetrexed-pretreated malignant pleural mesothelioma patients, with an acceptable toxicity profile.
We have suggested that the ERK-mediated pathway is an important regulator of pemetrexed-induced apoptotic cell death.40 The surviving cancer cells with upregulated TS may enhance EMT via the ERK/ZEB1 signaling pathway and result in metastasis. Our study demonstrated that pemetrexed resistance could be relieved by vinca alkaloids and might be independent to the TP53 status.
Materials and methods
Cell culture
Human lung adenocarcinoma cells CL1 harboring a mutated TP53 gene (mut-TP53, R248W) were cultured in RPMI-1640 medium with 10% fetal bovine serum (Gibco, Life Technologies, Carlsbad, CA, USA). Human adenocarcinoma A549 cells (ATCC, Manassas, VA, USA) harboring wild-type TP53 (wt-TP53) were maintained as previously described.47
Cytotoxicity assay: MTT assay
Chemosensitivity to pemetrexed was determined using MTT assay. The detailed steps of MTT assay have been previously described.47 The cells were exposed to various concentrations of pemetrexed in fresh medium for 72 h. Mean values were calculated from three independent experiments.
Protein extraction and western blot analysis
Protein extracts were prepared as described previously.47 Transferred proteins (10–30 μg) on polyvinylidene difluoride membrane were reacted with polyclonal anti-TS (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-DHFR, FPGS, GGH (GeneTex, Hsinchu City, Taiwan), anti-β-actin (NeoMarker, Ferymont, CA, USA), anti-p38, -pERK, -pJNK, -Zeb1, -E-cadherin, -Fibronectin, -Smad2/3, -p-ATF-2 (Thr71), -FRA1 (Cell Signaling, Danvers, MA, USA) and anti-Tp53 (DAKO, Glostrup, Denmark) separately, followed by conjugation of anti-rabbit or anti-mouse IgG (Santa Cruz Biotechnology) to horseradish peroxidase.
Inhibition of ERK, ZEB1 and fibronectin by siRNA transfection
The cells (8 × 104) were seeded in six-well plates for 16 h. The siRNA (30 pmol) for ERK (Sigma, siErk#1 SASI_Hs01_00104111; siErk#2 SASI_Hs01_00104113) were transfected with Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA) for 4 h and then replaced with fresh culture medium and incubated for an additional 48 h. The cells were dissociated by trypsin/EDTA solution and reseeded for migration assay, or harvested with RIPA buffer for western blots analysis. Similar procedures were applied to inhibition of ZEB1, fibronectin and p38.
Mice metastasis and xenograft models
Cells were washed and resuspended in PBS. Subsequently, 4- to 6-week-old SCID mice (n=5 per group; BioLASCO Taiwan Co., Ltd., Taipei, Taiwan) were injected in the lateral tail vein with a single-cell suspension containing 5 × 105 cells of A549 and subline in 50 μl PBS buffer or 2 × 106 cells of CL1 and subline. Mice were killed after 55 days (A549) and 75 days (CL1). All organs were examined for metastasis formation. The lungs were removed and fixed in 10% formalin fixative. The tumor weights were measured on a microbalance. The representative lung tumors were removed, fixed and embedded in paraffin, which was then sectioned into 4 μm layers and stained with H&E for histologic analysis and IHC with the indicated antibody and photography. The current study was approved by the Chung Shan Medical University Animal Care Committee (permit number: 1542) and all efforts were made to minimize suffering. To establish A549/A400 tumor xenografts, male BALB/c mice were injected subcutaneously with 5 × 106 cells (100 μl in serum-free DMEM) plus 100 μl Matrigel (BD Biosciences, San Jose, CA, USA). Twelve animals were then randomly divided into three groups consisting of four animals each. Thirty days after cell implantation, when the tumor size reached 120 mm3, mice in each group were intraperitoneally injected with 100 μl PBS and pemetrexed (100 mg/kg per mouse per week) to serve as controls. The VBL group was injected with 4.8 mg/kg per mouse per week.
In vitro transwell migration assay
Cell migration assay was carried out in 24-well tissue culture plates filled with 1.5 ml DMEM (for A549 cells) or RPMI (for CL1 cells) containing 10% FBA with a 8-μm transwell filter membrane (Thermo Fisher, Waltham, MA, USA). The group of A549 cells (2 × 104 cells/100 μl) in DMEM containing 0.5% FBS were added to transwell plates and incubated in 5% CO2 at 37 ° for 4 h. We fixed the cells on the lower side of the insert filter quickly with ice-cold methanol for 30 min. The cells were dried at room temperature and then were stained with 20% Giemsa solution (Merck KGaA, Darmstadt, Germany) overnight. Once stained, the cells were observed by using the optical microscope and photography. Similar procedures were applied to CL1 cells (5 × 104 cells/100 μl) incubated for 24 h.
In vitro transwell matrigel invasion assay
Invasion of tumor cells was evaluated with procedures similar to those of migration assay with matrigel (BD Biosciences cat. no. 354234) added. In medium-containing plates with a membrane, 0.5 μg/150 μl of matrigel for A549 cells and 0.2 μg/150 μl of matrigel for CL1 cells were added and polymerized at 37 ° for 3 h. The groups of A549 cells (2 × 104 cells/100 μl) were incubated for 16 h and CL1 cells (5 × 104 cells/100 μl) were incubated for 48 h. After the indicated time, non-invasive cells were gently removed from the top of the matrigel with a cotton-tipped swab. Invasive cells were fixed in ice-cold methanol, stained with 20% Giemsa solution and counted under a microscope. Results were averaged from three independent experiments.
Statistical analysis
All values are presented as mean±s.d. Data were compared among groups using a t-test and *P<0.05 is considered statistically significant.
References
Rollins KD, Lindley C . Pemetrexed: a multitargeted antifolate. Clin Ther 2005; 27: 1343–1382.
Calvert H, Bunn PA Jr . Future directions in the development of pemetrexed. Semin Oncol 2002; 29: 54–61.
Schultz RM, Patel VF, Worzalla JF, Shih C . Role of thymidylate synthase in the antitumor activity of the multitargeted antifolate, LY231514. Anticancer Res 1999; 19: 437–443.
Shih C, Chen VJ, Gossett LS, Gates SB, MacKellar WC, Habeck LL et al. LY231514, a pyrrolo[2,3-d]pyrimidine-based antifolate that inhibits multiple folate-requiring enzymes. Cancer Res 1997; 57: 1116–1123.
Ozasa H, Oguri T, Uemura T, Miyazaki M, Maeno K, Sato S et al. Significance of thymidylate synthase for resistance to pemetrexed in lung cancer. Cancer Sci 2010; 101: 161–166.
Zhang D, Ochi N, Takigawa N, Tanimoto Y, Chen Y, Ichihara E et al. Establishment of pemetrexed-resistant non-small cell lung cancer cell lines. Cancer Lett 2011; 309: 228–235.
Chen CY, Chang YL, Shih JY, Lin JW, Chen KY, Yang CH et al. Thymidylate synthase and dihydrofolate reductase expression in non-small cell lung carcinoma: the association with treatment efficacy of pemetrexed. Lung Cancer 2011; 74: 132–138.
Liu Y, Yin TJ, Zhou R, Zhou S, Fan L, Zhang RG . Expression of thymidylate synthase predicts clinical outcomes of pemetrexed-containing chemotherapy for non-small-cell lung cancer: a systemic review and meta-analysis. Cancer Chemother Pharmacol 2013; 72: 1125–1132.
Wang Z, Li Y, Ahmad A, Azmi AS, Kong D, Banerjee S et al. Targeting miRNAs involved in cancer stem cell and EMT regulation: an emerging concept in overcoming drug resistance. Drug Resist Updat 2010; 13: 109–118.
Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, Gallick GE . Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol 2007; 14: 3629–3637.
Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res 2009; 69: 2400–2407.
Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II, Yang SH et al. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer 2009; 63: 219–226.
Yang AD, Fan F, Camp ER, van Buren G, Liu W, Somcio R et al. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res 2006; 12: 4147–4153.
Kajiyama H, Shibata K, Terauchi M, Yamashita M, Ino K, Nawa A et al. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol 2007; 31: 277–283.
Hiscox S, Jiang WG, Obermeier K, Taylor K, Morgan L, Burmi R et al. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of beta-catenin phosphorylation. Int J Cancer 2006; 118: 290–301.
Piek E, Moustakas A, Kurisaki A, Heldin CH, ten Dijke P . TGF-(beta) type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci 1999; 112: Pt 24 4557–4568.
Moustakas A, Heldin CH . Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci 2007; 98: 1512–1520.
Akiyoshi S, Inoue H, Hanai J, Kusanagi K, Nemoto N, Miyazono K et al. c-Ski acts as a transcriptional co-repressor in transforming growth factor-beta signaling through interaction with smads. J Biol Chem 1999; 274: 35269–35277.
Heldin CH, Miyazono K, ten Dijke P . TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997; 390: 465–471.
Nishihara A, Hanai JI, Okamoto N, Yanagisawa J, Kato S, Miyazono K et al. Role of p300, a transcriptional coactivator, in signalling of TGF-beta. Genes Cells 1998; 3: 613–623.
Massague J, Chen YG . Controlling TGF-beta signaling. Genes Dev 2000; 14: 627–644.
Feng XH, Derynck R . Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol 2005; 21: 659–693.
Wakefield LM, Hill CS . Beyond TGFbeta: roles of other TGFbeta superfamily members in cancer. Nat Rev Cancer 2013; 13: 328–341.
Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL . p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci 2002; 115: 3193–3206.
Tavares AL, Mercado-Pimentel ME, Runyan RB, Kitten GT . TGF beta-mediated RhoA expression is necessary for epithelial-mesenchymal transition in the embryonic chick heart. Dev Dyn 2006; 235: 1589–1598.
Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL . Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 2004; 6: 603–610.
Derynck R, Zhang YE . Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003; 425: 577–584.
Davies M, Robinson M, Smith E, Huntley S, Prime S, Paterson I . Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-beta1 involves MAPK, Smad and AP-1 signalling pathways. J Cell Biochem 2005; 95: 918–931.
Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A . TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell 2005; 16: 1987–2002.
Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000; 2: 84–89.
Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2000; 2: 76–83.
Takkunen M, Grenman R, Hukkanen M, Korhonen M . Garcia de Herreros A, Virtanen I. Snail-dependent and -independent epithelial-mesenchymal transition in oral squamous carcinoma cells. J Histochem Cytochem 2006; 54: 1263–1275.
Yee DS, Tang Y, Li X, Liu Z, Guo Y, Ghaffar S et al. The Wnt inhibitory factor 1 restoration in prostate cancer cells was associated with reduced tumor growth, decreased capacity of cell migration and invasion and a reversal of epithelial to mesenchymal transition. Mol Cancer 2010; 9: 162.
Ohira T, Gemmill RM, Ferguson K, Kusy S, Roche J, Brambilla E et al. WNT7a induces E-cadherin in lung cancer cells. Proc Natl Acad Sci USA 2003; 100: 10429–10434.
Buque A, Aresti U, Calvo B, Sh Muhialdin J, Munoz A, Carrera S et al. Thymidylate synthase expression determines pemetrexed targets and resistance development in tumour cells. PLoS One 2013; 8: e63338.
Chu YW, Yang PC, Yang SC, Shyu YC, Hendrix MJ, Wu R et al. Selection of invasive and metastatic subpopulations from a human lung adenocarcinoma cell line. Am J Respir Cell Mol Biol 1997; 17: 353–360.
Shin S, Dimitri CA, Yoon SO, Dowdle W, Blenis J . ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell 2010; 38: 114–127.
Ouwens DM, de Ruiter ND, van der Zon GC, Carter AP, Schouten J, van der Burgt C et al. Growth factors can activate ATF2 via a two-step mechanism: phosphorylation of Thr71 through the Ras-MEK-ERK pathway and of Thr69 through RalGDS-Src-p38. EMBO J 2002; 21: 3782–3793.
Yang TY, Chang GC, Chen KC, Hung HW, Hsu KH, Wu CH et al. Pemetrexed induces both intrinsic and extrinsic apoptosis through ataxia telangiectasia mutated/p53-dependent and -independent signaling pathways. Mol Carcinog 2013; 52: 183–194.
Yang TY, Chang GC, Chen KC, Hung HW, Hsu KH, Sheu GT et al. Sustained activation of ERK and Cdk2/cyclin-A signaling pathway by pemetrexed leading to S-phase arrest and apoptosis in human non-small cell lung cancer A549 cells. Eur J Pharmacol 2011; 663: 17–26.
Sanchez-Tillo E, Lazaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A et al. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010; 29: 3490–3500.
Dhillon AS, Tulchinsky E . FRA-1 as a driver of tumour heterogeneity: a nexus between oncogenes and embryonic signalling pathways in cancer. Oncogene 2015; 34: 4421–4428.
Moudi M, Go R, Yien CY, Nazre M . Vinca alkaloids. Int J Prev Med 2013; 4: 1231–1235.
Kartner N, Riordan JR, Ling V . Cell surface P-glycoprotein associated with multidrug resistance in mammalian cell lines. Science 1983; 221: 1285–1288.
McGrogan BT, Gilmartin B, Carney DN, McCann A . Taxanes, microtubules and chemoresistant breast cancer. Biochim Biophys Acta 2008; 1785: 96–132.
Zucali PA, Perrino M, Lorenzi E, Ceresoli GL, De Vincenzo F, Simonelli M et al. Vinorelbine in pemetrexed-pretreated patients with malignant pleural mesothelioma. Lung Cancer 2013; 84: 265–270.
Chiu LY, Ko JL, Lee YJ, Yang TY, Tee YT, Sheu GT . L-type calcium channel blockers reverse docetaxel and vincristine-induced multidrug resistance independent of ABCB1 expression in human lung cancer cell lines. Toxicol Lett 2010; 192: 408–418.
Acknowledgements
This work was supported by grants from the Ministry of Science and Technology, Taiwan (MOST-103-2320-B-040-015, MOST-104-2320-B-040-003).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Chiu, LY., Hsin, IL., Yang, TY. et al. The ERK–ZEB1 pathway mediates epithelial–mesenchymal transition in pemetrexed resistant lung cancer cells with suppression by vinca alkaloids. Oncogene 36, 242–253 (2017). https://doi.org/10.1038/onc.2016.195
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.195
This article is cited by
-
The role of the ZEB1–neuroinflammation axis in CNS disorders
Journal of Neuroinflammation (2022)
-
A reciprocal feedback between colon cancer cells and Schwann cells promotes the proliferation and metastasis of colon cancer
Journal of Experimental & Clinical Cancer Research (2022)
-
Histone deacetylase 3 promotes alveolar epithelial–mesenchymal transition and fibroblast migration under hypoxic conditions
Experimental & Molecular Medicine (2022)
-
Slug and Vimentin downregulation at the metastatic site is associated with Skip-N2 metastasis of lung adenocarcinoma
Discover Oncology (2022)
-
Transcriptional and post-transcriptional control of epithelial-mesenchymal plasticity: why so many regulators?
Cellular and Molecular Life Sciences (2022)
