
Abstract
Fast-moving nanoscale systems offer the tantalizing possibility for rapid processing of materials, energy or information1. Frictional forces can easily dominate the motion of these systems, yet whereas nanomechanical techniques, such as atomic force microscopy, are widely used to measure static nanoscale friction2, they are too slow to measure the kinetic friction crucial for short-timescale motion. Here, we report measurements of frictional damping for a prototypical nanoscale system: benzene on a graphite surface, driven by thermal motion. Spin-echo spectroscopy is used to measure the picosecond time dependence of the motion of single benzene molecules, indicating a type of atomic-scale continuous Brownian motion not previously observed. Quantifying the frictional coupling between moving molecules and the surface, as achieved in these measurements, is important for the characterization of phononically driven nanomechanical tools. The data also provide a benchmark for simulations of nanoscale kinetic friction and demonstrate the applicability of the spin-echo technique.
Similar content being viewed by others

Main
Recent nanoscale linear motors have shown that phonon flow, from hot to cold regions, can drive a section of nanotube along a coaxial ‘track’1,3, in a reversal of the normal frictional process. Speeds are potentially ∼108 μm s−1, enabling atomic-scale motion on picosecond timescales1. Measurements of nanoscale friction have been carried out with friction force microscopy (FFM), using the remarkable ability of scanning probe techniques to manipulate objects and measure small forces4. FFM is, however, limited by the instrument’s inertia to ∼10 μm s−1 (ref. 5), and there is a need for much faster probes to study the kinetic friction associated with nanoelectromechanical systems. As the moving parts are not loaded laterally or normally in the conventional sense, one approach is to study the dynamics of molecules directly as they move over a surface. For example, Krim et al. 6 used a quartz-crystal microbalance to study the friction of layers of molecules and fluorescence correlation spectroscopy7 provides diffusion information in the time domain. However, neither of these techniques, nor any other, offers spatial information on an atomic scale together with picosecond time resolution, as in the present work.
Here, we demonstrate an alternative approach, carrying out in effect a nanotribological measurement of kinetic friction in the single-molecule limit. Friction and the coupling to surface phonons is deduced for a model nanoscale system from the way individual molecules move on the atomic scale as they are pushed around on picosecond timescales by thermal excitation. Our work is also driven by the knowledge that macroscale friction is fundamentally a microscopic phenomenon8, because real surfaces contact only at microscopic asperities, and that energy dissipation, just as in nanoscale systems, is often dominated by the creation of phonons4. Simple theoretical models of lateral motion of surface species are well established9. In general, two factors control the motion: barriers in the lateral variation of the adsorbate potential energy and the rate at which the energy required to move and overcome these barriers is exchanged with the substrate10. With benzene/graphite, we find the role of the substrate potential to be insignificant, with the observed motion corresponding to an unprecedented regime dominated by kinetic dissipation.
To follow the motion over both atomic length scales and picosecond timescales, we use the established techniques of helium11 and neutron12 spin-echo (HeSE and NSE respectively). Helium atoms or neutrons scatter from mobile benzene molecules on the graphite surface and the spin-echo technique reveals the motion directly through the time dependence of structural correlations on the surface. An experiment provides measurements of the time decay of the correlation function, I(ΔK,t), for different momentum transfers, ΔK, of the scattered particles (see the Methods section for full details). The dynamics then follow from the form of the correlation function and its dependence on ΔK. Specifically, the continuous Brownian motion that we observe here is characterized by a decay in correlation with a pure exponential time dependence,

as demonstrated in Fig. 1. Also the dephasing rate, α, the rate at which spatial correlations decay, has a simple quadratic dependence on ΔK, such that α=DΔK2 (ref. 13), as we show in Fig. 2a. The diffusion coefficient, D, relates to friction through the Einstein theory of Brownian motion, where D=kBT/(m η). The friction coefficient, η, quantifies the energy exchange rate between the adsorbate, of mass m, and the substrate at a temperature T, through fluctuation and dissipation10. Thus, η, which is identical to the friction coefficient used in, for example, the Langevin equation of motion9, can be obtained directly from the experiment, as we demonstrate below.

The single exponential decay, solid line, that is the characteristic of random motion: HeSE data (circles) and NSE data (triangles) (at 0.5 ML, 140 K, ΔK=0.32 Å−1). Inset: Data for several momentum transfer values. The decay constants of the exponential curves give the values of the dephasing rate, α, which are plotted in Fig. 2a.

a, The dark blue line shows a quadratic fit to the momentum transfer data, characteristic of Brownian motion. Experimental data (at 0.5 ML, 140 K) is shown for HeSE (circles) and for NSE (triangles), with Langevin molecular dynamics (MD) simulations as squares. The marked points correspond to the measurements in Fig. 1. b, An Arrhenius plot (ln α versus 1/T) of α (for 0.5 ML, ΔK=0.4 Å−1). The lack of any significant activated component in the motion is evident from the shallow gradient of the line of best fit (see text).
Measurements of the normalized time-dependent correlation function, for 0.5 monolayers (ML) benzene/graphite, are shown in Fig. 1. The lin–log plot illustrates a large time range, over which the single exponential decay in correlation is indicated by a line of best fit. The excellent agreement of the fit demonstrates a system showing random motion14,15. The data also show good agreement between the neutron and helium data. The inset shows examples of the correlation measurement at three momentum transfers, each resulting in different values of the dephasing rate, α, obtained from the exponential decay rate. Figure 2a shows results for the variation of α between 0<ΔK<1.5 Å−1 at 140 K. The solid line is a quadratic fit to the data, which, together with the single exponential in Fig. 1, indicates that the motion is continuous, atomic-scale Brownian motion, not previously observed16. The fit gives the diffusion coefficient directly as D=5.39±0.13×10−9 m2 s−1. To confirm the assignment of Brownian motion, and rule out any significant hopping, we have demonstrated that a pure quadratic gives the best fit to the data, because the most probable coefficient of the next highest term (quartic) is zero (see the Methods section). We have also measured the temperature dependence of α, shown as an Arrhenius plot in Fig. 2b. For thermally activated diffusion by a jump mechanism, an Arrhenius plot (ln(α) versus 1/T) gives a straight line with activation energy, Ea, proportional to the gradient. The plot shows an activation energy of 17±12 meV, indicating that any corrugation can safely be neglected because it would be easily overcome by thermal motion (kBT=12 meV at 140 K). As a final demonstration that Brownian motion predominates, we have carried out a quantitative molecular dynamics study within the Langevin framework9,17 (see the Methods section). The squares in Fig. 2a show results from a simulation (carried out for 50 molecules, following the trajectories for 5 ns using 2 fs time steps), reproducing the magnitude and curvature of the experimental data. A potential corrugation of <20 meV must be used, consistent with the small activation energy derived from Fig. 2b. The results are insensitive to the shape and the magnitude of the potential, provided it is small. The best description of the experiment is achieved with η=2.2±0.2 ps−1. This value of the benzene/graphite friction agrees well with a simple estimate from the diffusion constant, D. The measured value of D, when corrected for a collisional contribution 0.55 ps−1(ref. 18) gives a value of η=2.2±0.1 ps−1, which corresponds to a thermal mean-free-path of only 0.7 Å. These values confirm our interpretation, and our measurement of unusually strong kinetic friction for this prototypical carbon system.
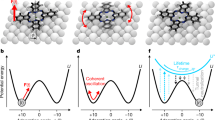
Our observation of continuous Brownian motion is strikingly different from the hopping diffusion encountered in almost all previous surface studies (Fig. 3), where the usual situation of diffusion dominated by the corrugation of the energy landscape (Fig. 3d,e) gives a typical trajectory dominated by vibration with occasional hops (Fig. 3f). In contrast, the present Brownian regime (Fig. 3a,b) has a flat landscape and the motion (Fig. 3c) is a continuous, random walk. The measured dissipation corresponds directly to the velocity-dependent drag term used to describe kinetic friction using linear response theory, where F=−η m v. As graphite has a negligible electronic density of states at the Fermi level, the electronic contribution will be minimal8, and therefore the phonon contribution must dominate, as we have confirmed with force-field molecular dynamics simulations (see the Methods section). Although atomic force microscopy and FFM-related techniques are ideal for measuring static friction, they are less well suited to submicrosecond measurements of kinetic behaviour19. The complementary picosecond technique demonstrated here can be applied widely to characterizing frictional dissipation in freely moving nanoscale systems. HeSE and NSE can be readily applied to single-molecule dynamics on either well-characterized surfaces or in porous matter and nanotubes.

a–f, Illustration of surface motion in the newly measured high-friction, viscous regime (a–c), alongside typical hopping motion (d–f). Comparison of the trajectories of a molecule on the surface resulting from the molecular dynamics simulations described in the text shows a clear difference between motion where the molecule vibrates about energetically favourable adsorption sites, which appear as dense areas, with occasional hops between them (f), and the friction-dominated regime where the molecule appears to carry out a random walk across the surface in a flat energy landscape (c).
The identification and characterization of a truly Brownian regime of continuous fluctuation and dissipation on the atomic scale suggests a range of new technological opportunities in graphitic systems. Phonon-driven nanomechanics, as recently demonstrated1,3, provide a promising avenue for fast-acting nanosystems. In such systems, two essential characteristics are a minimum in potential corrugation, to minimize the static friction and allow the components to move, plus well-defined frictional coupling to enable the systems to be driven by phononic excitations, for example between two thermal reservoirs. The benzene/graphite system fits this scenario exactly and might be extended to a more general system composed of polycyclic hydrocarbons or graphene on graphite. The low corrugation in the energy means there is negligible barrier to motion and the high friction gives sensitivity to small thermal gradients and the certainty of static behaviour in the absence of a gradient, suggesting a phononically controlled flow of benzene molecules could perhaps be used as an active medium to drive larger nanoscale constructions20. More generally, the experimental quantities we derive provide a quantitative picture of motion, and the necessary information to validate simulations of nanoscale dynamics and friction.
Methods
NSE and HeSE yield the correlation function, I(ΔK,t), known as the intermediate scattering function. The intermediate scattering function is the spatial Fourier transform of the van Hove pair correlation function, G(R,t), which represents the probability of a particle at (R,t)given there was a particle at (0,0) and gives a complete statistical description of adsorbate motion14,21.
We present data from the Cambridge helium-3 spin-echo spectrometer22, with complementary NSE measurements from ‘IN11C’ (ref. 12) at the Institut Laue-Langevin. Spin-echo is an established technique, originally using neutrons to measure a sample’s correlation function in real time21. Here, we scatter a beam of helium-3 atoms or neutrons from mobile benzene atoms on the graphite surface. The probe particles are spin-polarized at the entrance of the spectrometer, then exposed to a magnetic field before scattering from the sample (Fig. 4). The probe’s wavefunctions are split by the magnetic field into two parts, of which one is accelerated and the other decelerated with respect to the particles’ mean speed, so that the part wavefunctions arrive at the sample with a time separation t. In the second spectrometer arm, the probe particles are exposed to a second magnetic field, equal and opposite to the first, recombining the two part wavefunctions before passing through a polarization analyser and into the detector. Structural changes in the sample over a length scale given by the scattering momentum transfer ℏΔK, within the time interval t lead to losses in coherence and spin-polarization. Thus, we measure the correlation of the configuration of the molecules on the surface with time, that is, the motion of the benzene atoms. The main difference between NSE and HeSE is that neutrons interact with the nuclei of the sample, whereas helium atoms interact with the outer valence electrons at the surface, giving a much greater surface sensitivity. HeSE also benefits from a higher intensity and increased momentum transfer range, allowing ready access to a variety of surface systems. Our comparative measurements show that the nature of the probe–sample interaction does not affect the results of the dynamics measurement, particularly, that multiple scattering effects do not influence the HeSE data.

Magnetic fields are used to separate spin-polarized wave packets, which subsequently arrive at the sample with a time delay, leading to a loss in coherence, and consequently in spin-polarization on recombination. The decay in spin-polarization with time gives a measurement of the time-dependent surface correlation and hence the surface motion.
The HeSE measurements are from a highly oriented pyrolytic graphite surface (grade SPI-1, Structure Probe), cleaved, quickly installed in the ultrahigh-vacuum sample chamber (base pressure 5×10−11 mbar) and cleaned by heating to >800 K. An exfoliated graphite sample (Papyex, Le Carbone-Lorraine), outgased in vacuum for four days at 973 K, is used for the NSE experiments, giving a large surface area (23 m2 g−1) and related signal. For both spectrometers, benzene is deposited from the gas phase to a coverage of 0.5 ML, with monolayer coverage defined by the  overlayer structure from ref. 23. For the HeSE measurements, coverage is established from changes in specular signal on adsorption. In the NSE experiments, the adsorbed mass is measured directly. Hydrogenated benzene (>99.9%, Sigma-Aldrich) is used for the HeSE experiments and fully deuterated benzene (99.6% D, CEN Saclay) in NSE, giving coherent scattering in both experiments.
overlayer structure from ref. 23. For the HeSE measurements, coverage is established from changes in specular signal on adsorption. In the NSE experiments, the adsorbed mass is measured directly. Hydrogenated benzene (>99.9%, Sigma-Aldrich) is used for the HeSE experiments and fully deuterated benzene (99.6% D, CEN Saclay) in NSE, giving coherent scattering in both experiments.
To identify the underlying microscopic behaviour of the system, we must determine whether the data in Fig. 2a follow a quadratic form (indicating continuous diffusion) or sin2 form (indicating hopping)15. Visual inspection strongly suggests continuous motion; however, as the measured range represents only the first fraction of the longest possible period of oscillation, we use Bayesian statistics to determine the better fit without prejudice. Fitting a x2+b x4, an expansion of sin2 about zero, enables us to compare Brownian motion (b=0) with hopping motion ( ). The most probable value of the ratio b/a is 0±0.01 Å2, indicating a negligible quartic term and favouring the quadratic fit. The shortest possible jump length for benzene on graphite would correspond to the C–C bond length, giving a minimum value of b/a=0.35 Å2. As this is much larger than the experimental value, the motion of the benzene molecules must be predominately continuous.
). The most probable value of the ratio b/a is 0±0.01 Å2, indicating a negligible quartic term and favouring the quadratic fit. The shortest possible jump length for benzene on graphite would correspond to the C–C bond length, giving a minimum value of b/a=0.35 Å2. As this is much larger than the experimental value, the motion of the benzene molecules must be predominately continuous.
To confirm the contributions to the dynamics from the benzene–graphite friction and benzene–benzene interactions, we can apply approximate stochastic models18 or carry out molecular dynamics simulations of the system17. We chose the latter as it is most widely applicable. The simulations trace adsorbate trajectories using the Langevin equation:

where m is the mass of a benzene molecule and vi is the velocity of the ith benzene molecule. The parameters adjusted to fit the data are the surface–adsorbate potential energy surface, U, the kinetic friction coefficient, η, and the pairwise adsorbate–adsorbate force Fi,j. U is a simple sinusoidal potential. The energy flow from the surface to the adsorbate is simulated through random impulses ζ(t) derived from a Gaussian distribution, the width of which is determined from η using fluctuation dissipation theory and so is not separately adjustable10. As there are also no sharp features in the data corresponding to correlated motion induced by long-range repulsions22, we model the benzene–benzene interaction as a simple hard-wall potential of adjustable width. The form of the momentum transfer dependency also rules out molecules that are rotating rather than translating in the measured range as this motion would give extended plateaux rather than the quadratic functional form seen24. The benzene–benzene hard-wall interaction distance (the size of one molecule as seen by the others) is 5–5.3 Å. Larger molecules give a peaked structure factor, not seen in the experiment, as confinement leads to correlated motion. Smaller molecules are excluded by the sum of internal bond lengths. Other data, not shown, indicate a weak coverage dependency, with the diffusion coefficient increasing by <10% as the coverage is halved, but with no evidence for a change in diffusion mechanism. The Langevin molecular dynamics simulations also correctly reproduce the coverage dependence of the data over the measured range.
In addition, force-field molecular dynamics simulations were carried out using the second-generation force-field COMPASS within the package CERIUS, following molecular trajectories over 1 fs steps for 50 ps, using a coverage of 0.5 ML on 7×8×2 graphite unit cells at 140 K. The simulations show evidence of Brownian diffusion, in good agreement with the experimental data, and giving a diffusion coefficient of D=6.11±0.52×10−9 m2 s−1, which compares favourably with the experimental value. These results confirm the dominant phononic contribution to the kinetic friction measured in this system as electronic excitations are not included in the simulations.
References
Barreiro, A. et al. Subnanometer motion of cargoes driven by thermal gradients along carbon nanotubes. Science 320, 775–778 (2008).
Albers, B. J. et al. Three-dimensional imaging of short-range chemical forces with picometre resolution. Nature Nanotech. 4, 307–310 (2009).
Somada, H., Hirahara, K., Akita, S. & Nakayama, Y. A molecular linear motor consisting of carbon nanotubes. Nano Lett. 9, 62–65 (2009).
Meyer, E., Overney, R. M., Dransfield, K. & Gyalog, T. Nanoscience: Friction and Rheology on the Nanoscale (World Scientific, 1998).
Dienwiebel, M. et al. Superlubricity of graphite. Phys. Rev. Lett. 92, 126101 (2004).
Krim, J., Solina, D. H. & Chiarello, R. Nanotribology of a Kr monolayer: A quartz-crystal microbalance study of atomic-scale friction. Phys. Rev. Lett. 66, 181–184 (1991).
Zhao, J. & Granick, S. How polymer surface diffusion depends on surface coverage. Macromolecules 40, 1243–1247 (2007).
Persson, B. N. J. Sliding Friction (Springer, 1998).
Ala-Nissila, T., Ferrando, R. & Ying, S. C. Collective and single particle diffusion on surfaces. Adv. Phys. 51, 949–1078 (2002).
Kubo, R. The fluctuation-dissipation theorem. Rep. Prog. Phys. 29, 255–284 (1966).
Alexandrowicz, G. & Jardine, A. P. Helium spin-echo spectroscopy: Studying surface dynamics with ultra-high-energy resolution. J. Phys. Condens. Matter 19, 305001 (2007).
Farago, B. Recent neutron spin-echo developments at the ILL (IN11 and IN15). Physica B 267–268, 270–276 (1999).
Frenken, J. W. M., Hinch, B. J., Toennies, J. P. & Wöll, Ch. Anisotropic diffusion at a melting surface studied with He-atom scattering. Phys. Rev. B 41, 938–946 (1990).
van Hove, L. Correlations in space and time and born approximation scattering in systems of interacting particles. Phys. Rev. 95, 249–262 (1954).
Hulpke, E. Helium Atom Scattering from Surfaces Ch. 12 (Springer, 1992).
Barth, J. V. Transport of adsorbates at metal surfaces: From thermal migration to hot precursors. Surf. Sci. Rep. 40, 75–149 (2000).
Ellis, J. & Graham, A. P. The use of quasielastic helium atom scattering to study correlated motion in adsorbate overlayers. Surf. Sci. 377–379, 833–842 (1997).
Martinez-Casado, R., Vega, J. L., Sanz, A. S. & Miret-Artes, S. Line shape broadening in surface diffusion of interacting adsorbates with quasielastic He atom scattering. Phys. Rev. Lett. 98, 216102 (2007).
Maier, S. et al. Fluctuations and jump dynamics in atomic friction experiments. Phys. Rev. B 72, 245418 (2005).
Browne, W. R. & Feringa, B. L. Making molecular machines work. Nature Nanotech. 1, 25–35 (2006).
Mezei, F. Neutron Spin-Echo, Lecture Notes in Physics Vol. 128 (Springer, 1979).
Jardine, A. P., Alexandrowicz, G., Hedgeland, H., Allison, W. & Ellis, J. Studying the microscopic nature of diffusion with helium-3 spin-echo. Phys. Chem. Chem. Phys. 11, 3355–3374 (2009).
Meehan, P., Rayment, T., Thomas, R. K., Bomchil, G. & White, J. M. Neutron diffraction from benzene adsorbed on graphite. J. Chem. Soc. Faraday Trans. I 76, 2011–2016 (1980).
Bee, M. J. Quasielastic Neutron Scattering (Adam Hilger, 1988).
Acknowledgements
The authors would like to thank E. Tosatti, S. Clarke, M. R. Johnson and H. Lauter for helpful discussions.
Author information
Authors and Affiliations
Contributions
Experimental work: H.H., P.F., A.P.J., G.A.; data analysis: H.H., P.F., A.P.J., J.E.; simulations: H.H., P.F., A.P.J.; manuscript preparation: H.H., P.F., A.P.J., G.A., W.A., J.E.; project planning: P.F., W.A., J.E.
Corresponding authors
Rights and permissions
About this article
Cite this article
Hedgeland, H., Fouquet, P., Jardine, A. et al. Measurement of single-molecule frictional dissipation in a prototypical nanoscale system. Nature Phys 5, 561–564 (2009). https://doi.org/10.1038/nphys1335
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nphys1335
This article is cited by
-
Molecular motion of a nanoscopic moonlander via translations and rotations of triphenylphosphine on graphite
Communications Chemistry (2024)
-
Capillary grip-induced stick-slip motion
Nano Research (2022)
-
Deconvolution of dissipative pathways for the interpretation of tapping-mode atomic force microscopy from phase-contrast
Communications Physics (2021)
-
Nanoscopic diffusion of water on a topological insulator
Nature Communications (2020)
-
Macroscopic self-reorientation of interacting two-dimensional crystals
Nature Communications (2016)
