
Abstract
Tyrosinase is responsible for the two initial enzymatic steps in the conversion of tyrosine to melanin. Many tyrosinase mutations are the leading cause of albinism in humans, and it is a prominent biotechnology and pharmaceutical industry target. Here we present crystal structures that show that both monophenol hydroxylation and diphenol oxidation occur at the same site. It is suggested that concurrent presence of a phenylalanine above the active site and a restricting thioether bond on the histidine coordinating CuA prevent hydroxylation of monophenols by catechol oxidases. Furthermore, a conserved water molecule activated by E195 and N205 is proposed to mediate deprotonation of the monophenol at the active site. Overall, the structures reveal precise steps in the enzymatic catalytic cycle as well as differences between tyrosinases and other type-3 copper enzymes.
Similar content being viewed by others

Introduction
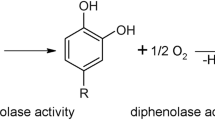
Tyrosinase is a di-copper oxidase responsible for the production of melanin in many organisms. It is involved in wound healing, UV protection, detoxification of phenols and more1. Mutations in tyrosinase cause albinism in mammals and are associated with melanoma2. The active site of all type-3 copper proteins comprises six histidine residues that coordinate the two copper ions CuA and CuB. It is identical among tyrosinases, catechol oxidases (COx) and haemocyanins, yet each member of the family has different function(s). Tyrosinase performs two successive reactions in the presence of molecular oxygen; the hydroxylation of monophenols to form ortho-diphenols (monophenolase activity) and the oxidation of o-diphenols to o-quinones (diphenolase activity). COx are strict diphenolases while haemocyanins bind oxygen and normally do not exhibit catalytic activity, however, some activity was observed in the presence of SDS3.
The catalytic mechanism of type-3 copper proteins has been studied extensively for the past two decades, recently assisted by the availability of three-dimensional structures4,5,6,7,8. Based on these structures, it is evident that the differences in function are due to variations in the residues surrounding the substrate-binding pocket. A number of mechanistic studies have also contributed to the current understanding of active site states, substrate inhibition and copper involvement9,10. However, to date, both mechanistic and structural studies have not provided direct evidence of the catalytic cycle, as visualization of substrates in the active site has not been obtained. Thus, the subtle mechanistic differences between members of this protein family are still undetermined10.
In this study, we present structural evidence for the identical binding mode of substrates in the active site, as opposed to previous assumptions. This led us to the identification of a more likely mechanism of catalysis, and to an explanation for the difference between tyrosinase and COx.
Results
Structures with substrates elucidate reaction mechanism
We have determined the crystal structure of tyrosinase from Bacillus megaterium (TyrBm) with substrates bound to the active site. This enzyme’s activity in crystal7 hampered initial attempts, and the resulting electron density maps lacked evidence of bound molecules. We previously showed that replacing the Cu2+ with Zn2+ ions can be performed in TyrBm without structural consequences7, while the presence of Zn2+ ions inhibited the activity of tyrosinase on both monophenols and diphenols7,11. Based on these observations, we soaked the crystals in ZnCl2 before soaking them in the substrate solution in order to trap the substrates within the active site of TyrBm in the crystal. This approach was successful, and for the first time high-resolution structures with clear electron density for L-tyrosine or L-Dopa (3,4-dihydroxyphenylalanine) in the active site of tyrosinase were obtained at 2.2 Å resolution (Fig. 1 and Table 1).

(a) Tyrosine (green) is observed in the active site. Its carboxyl side chain forms hydrogen bonds with R209. Zinc ions are presented as grey spheres. (b) L-Dopa (pink) is observed in the active site in the same position as tyrosine. (c) p-Tyrosol (teal) with its 2Fo-Fc electron density map (grey wire) contoured at 1.3σ. Copper ions are shown as brown spheres and a water molecule as a red sphere. A green arrow points at the electron density that we propose shows hydroxylation has occurred in crystal. (d) Superposition with the apo-TyrBm structure (light green, PDB code 3NQ0) shows CuA and residues V218 and H60 that have moved, whereas H208 has not.
The TyrBm structure with bound L-tyrosine (TyrBm:T) is presumed to be in the enzymes’ met form1,7,10. As expected12,13, the tyrosine hydroxyl group is directed towards ZnA (at the equivalent position of CuA) at a distance of 1.9 Å and its benzyl ring is oriented through hydrophobic π–π interactions with H208 (Fig. 1a). In this orientation, the ortho position is electrophilic and directed towards the proposed oxygen species binding site located between CuA and CuB. Previous studies have suggested that diphenols are oriented towards CuB9,10, thus relating each of the two activities to a different Cu ion. This suggestion is also based on the structure of COx in which CuA is shielded by a bulky phenylalanine residue (F261) and only the diphenolase activity is observed. Previous results from our laboratory, using directed evolution on TyrBm, showed that when access to CuB is hindered by mutation R209H, the diphenolase activity decreases while the monophenolase activity increases14. However, the crystal structure at 2.2 Å resolution of TyrBm with L-Dopa in the active site (TyrBm:D; Table 1) unequivocally shows that the diphenol binds to ZnA in the same orientation as tyrosine, assisted by interactions with H208 (Fig. 1b and Supplementary Fig. 1). This result indicates that both the hydroxylation and oxidation activities occur without significant binding site reorganization. The question that arises, therefore, is what differentiates the activity of tyrosinases and the homologous COx? Superposition of TyrBm:T and TyrBm:D, with sweet potato COx4 (PDB code 1BUG), indicates that both tyrosine and L-Dopa can be positioned in the active site, considering the location of F261 of COx (Fig. 2). However, the flexibility of F261 is restricted owing to the thioether bond fixing one of the histidines4 (which is flexible in bacterial tyrosinases6,15), and therefore it cannot allow for any substrate rotation during the reaction. During the monophenolase reaction, a rotation of the tyrosine is suggested to occur, allowing the electrophilic attack that leads to hydroxylation12,16 (Fig. 3). This was suggested by Decker and colleagues16, and was further confirmed by using molecular dynamics by Deeth and Dietrich13. These studies, however, have not dealt with the molecular mechanism of reaction in the case where the diphenol enters the active site. We suggest that since the reaction on L-Dopa does not involve an electrophilic attack but only the oxidation of the two hydroxyl groups, the rotation in this case is not necessary and does not occur. Thus, the diphenolase reaction in COx can be performed despite the partial blocking of the active site by the perturbing phenylalanine.

Tyrosine (in green) and L-Dopa (in pink) can be positioned in the active site of COx considering the distance shown from F261 (in teal). H244, of COx (in teal), is positioned similarly to H208 of TyrBm (in grey). H109 of COx is restricted by the thioether bond with C92.

During the monophenolase reaction, a rotation of tyrosine is suggested to occur, allowing the electrophilic attack that leads to hydroxylation.
Substrate deprotonation scenario
The newly obtained TyrBm:T and TyrBm:D structures reveal an additional important element of the reaction mechanism. It has been suggested that substrates bind to the active site as a phenolate, therefore a mechanism of deprotonation16 by the enzyme is expected. This proton needs to then be returned to the active site for the generation of water and the release of the quinone product. A number of options have been previously suggested for the identity of the deprotonating base6,16. The highly conserved E195 has been suggested as the potential base; however, its distance from the active site (7 Å) is too great for such activity. Examination of the vicinity of this residue in TyrBm:T and TyrBm:D (as well as over 30 other TyrBm structures we have determined) showed that almost invariably, a water molecule is located bound equidistant between the E195 carboxyl group and the conserved N205 carbonyl group (Fig. 4) previously shown to be vital for activity and copper uptake17. A similar water molecule is also observed in the structures of sweet potato COx (1BT3; ref. 4), Streptomyces tyrosinase (1WX2; ref. 6), Agaricus bisporus tyrosinase (2Y9W; ref. 8) and Manduca sexta tyrosinase (3HHS; ref. 5). We propose that this conserved water molecule is activated by E195 and N205, and serves as the deprotonating base. The involvement of water molecules or hydroxide ions in enzyme catalysis has been previously shown in several studies with other enzymes18,19,20. A recent QM calculation study performed by Siegbahn and Borowski21 examined the reaction mechanism and suggested a similar deprotonation scenario. They showed that it is energetically feasible to move the proton from tyrosine through the water molecule to the glutamate residue (E195). They also considered the involvement of three additional residues in this scenario that are also highly conserved, R191, N192 and D178, and which stabilize the water molecule through E195 (Fig. 4). Attempts to mutate E195 to arginine or leucine resulted in insoluble protein further strengthening the importance of this conserved residue.

Stereoview of structure TyrBm:T in which a conserved water molecule is shown oriented by E195 and N205 (orange) with their 2Fo-Fc electron density map (grey wire) contoured at 2.2σ. Conserved residues R191 N192 and D178 (orange) are also assumed to be involved in the deprotonation scenario. All dashed lines represent a distance of ~3.0 Å.
TyrBm active structure presents product formation
In order to obtain a substrate in the active site in the presence of Cu2+, we soaked crystals with p-tyrosol, which TyrBm hydroxylates relatively slowly in comparison with tyrosine. The TyrBm p-tyrosol structure (TyrBm:P) with Cu2+ in the active site was determined at a resolution of 2.5 Å (Fig. 1c and Table 1). Tyrosol is observed in both subunits in the asymmetric unit, directed towards CuA and oriented by His208, as was the case for tyrosine. In one of the subunits, clear electron density at the ortho position of the p-tyrosol is observed (Fig. 1c), which we interpret as a snapshot of hydroxytyrosol (the diphenol), or perhaps even the final quinone product. Two major differences in the active site are seen in the TyrBm:P structure: (i) V218, the residue in the loop above the active site, moves 1.4 Å to allow substrate entrance, confirming its role as recently suggested15; and (ii) CuA moves 1.0 Å towards the hydroxyl of the bound substrate (Fig. 1d). Our results support findings by Kleifeld et al.22 that showed that during different steps along catalytic cycles, metal ions may accommodate alternative locations.
Discussion
We present here for the first time a detailed structure-based mechanism of tyrosinase. The structures of TyrBm with bound substrates presented here confirm that monophenols and diphenols bind identically at the active site towards CuA. Based on these structures and on a number of recent studies13,15,16, we suggest that although both mono- and diphenols orient within the active site in the same way, only monophenols rotate during the reaction. This fact enables to differentiate between enzymes with both mono- and diphenolase activities and those that are strict diphenolases. Thus, in sweet potato COx and similar enzymes, two constraints need to coexist in order to prevent monophenolase activity: (i) a bulky residue above the active site (such as F261) and (ii) a restricting thioether bond on the active site histidine coordinating CuA. The presence of both constraints prevents substrate axial to equatorial rearrangement needed for monophenolase hydroxylation, but allows for the oxidation of a diphenol. Consequently, TyrBm variant V218F can hydroxylate tyrosine owing to the absence of a thioether bond15, and tyrosinase from A. bisporus performs hydroxylation despite the thioether bond because of the presence of V283 above the active site8. This provides a compelling and structurally founded explanation for the long-lasting debate on the different activities of type-3 copper proteins.
Another major issue discussed in the literature concerning the catalytic cycle of tyrosinases has been the deprotonation of the entering substrates16, which is in fact the initial step of the reaction. We suggest here that the base that deprotonates the substrate is a conserved hydroxide or water molecule oriented and activated by two highly conserved residues, a glutamic acid and an asparagine. Importantly, substrate deprotonation has been suggested to be involved in diphenol substrate inhibition or suicide inactivation9,23. The suicide inactivation pathway of diphenols is apparently facilitated by the incorrect proton transfer to the active site peroxide instead of to an adjacent base. In light of the new evidence showing that both substrates bind to CuA, the existence of such an inactivation mechanism also explains the ability to influence the monophenolase/diphenolase activity ratio, as in TyrBm mutants14,15,17.
Finally, the formation of product within the TyrBm structure has been presented. The use of p-tyrosol as a substrate, which is hydroxylated at a slower rate than tyrosine, enabled us not only to determine the crystal structure of the TyrBm:substrate complex in the presence of Cu2+ in the active site, but also to observe product formation in the crystal. The fact that tyrosine in the TyrBm:T structure is observed in a very similar orientation as p-tyrosol in the TyrBm:P structure, supports our claim that Zn2+ in the active site may only prevent protein activity but not substrate binding.
Methods
Purification and crystallization of tyrosinase from TyrBm
A tyrosinase producing TyrBm was isolated from soil samples and the gene encoding for the tyrosinase was cloned into Escherichia coli BL21. The enzyme was expressed, purified and crystalized as previously described7,24,25. Briefly, E. coli cells were grown overnight at 37 °C in 0.5 l of TB medium (tryptone 1.2%, yeast extract 2.4%, glycerol 0.4% (w/v) and potassium phosphate buffer 89 mM) and then harvested by centrifugation (8,000 g for 10 min at room temperature). Cells were treated twice in a pressure cell (French Press, Spectronic Instruments Inc., Rochester, NY, USA) and cell debris was removed by centrifugation (16,000 g for 20 min at 15 °C). The supernatant containing TyrBm was applied to a Ni(II)-bound affinity column and eluted with an appropriate buffer (20 mM Tris–HCl, pH 7.5, 500 mM NaCl and 500 mM imidazole). Crystallization was performed using the hanging-drop vapour diffusion method at 293 K. Drops consisting of 2 μl protein solution (2 mg ml−1) and 2 μl of the mother liquor were equilibrated against a 600-μl reservoir containing 18% PEG 8000 and 0.1 M cacodylic acid, pH 6.5.
Substrate binding in crystal
In order to trap ligands in the active site, mature crystals were soaked overnight in 1 mM of either CuSO4 or ZnCl2 and subsequently in 1 mM of the appropriate ligand (L-tyrosine, p-tyrosol and L-Dopa). To ensure maximal occupancy, an additional soaking of the ligand for 1–5 min was performed before crystal freezing.
Data collection and structure determination
X-ray diffraction data was collected at the European Synchrotron Radiation Facility, Grenoble, France, at beamlines ID14-1 and ID 23-1. All data were indexed, integrated, scaled and merged using Mosflm and Scala26. The initial structure of TyrBm was solved by molecular replacement using Phaser27 and the coordinates of earlier determined TyrBm structure (PDB code 3NM8). A single solution was obtained for two monomers in the asymmetric unit. Refinement was performed using Phenix28 coupled with rounds of manual model building, real-space refinement and structure validation performed using COOT29. Data collection, phasing and refinement statistics are presented in Table 1.
Additional information
Accession codes: Coordinates and structure factors of TyrBm have been deposited in the RCSB Protein Data Bank under accession code 4P6R for TyrBm:T, 4P6S for TyrBm:D and 4P6T for TyrBm:P.
How to cite this article: Goldfeder, M. et al. Determination of tyrosinase substrate-binding modes reveals mechanistic differences between type-3 copper proteins. Nat. Commun. 5:4505 doi: 10.1038/ncomms5505 (2014).
References
Fairhead, M. & Thöny-Meyer, L. Bacterial tyrosinases: old enzymes with new relevance to biotechnology. N. Biotechnol. 29, 183–191 (2012).
Olivares, C. & Solano, F. New insights into the active site structure and catalytic mechanism of tyrosinase and its related proteins. Pigm. Cell Melanoma Res. 22, 750–760 (2009).
Cong, Y. et al. Structural mechanism of SDS-induced enzyme activity of scorpion hemocyanin revealed by electron cryomicroscopy. Structure 17, 749–758 (2009).
Klabunde, T., Eicken, C., Sacchettini, J. C. & Krebs, B. Crystal structure of a plant catechol oxidase containing a dicopper center. Nat. Struct. Mol. Biol. 5, 1084–1090 (1998).
Li, Y., Wang, Y., Jiang, H. & Deng, J. Crystal structure of Manduca sexta prophenoloxidase provides insights into the mechanism of type 3 copper enzymes. Proc. Natl Acad. Sci. USA 106, 17002–17006 (2009).
Matoba, Y., Kumagai, T., Yamamoto, A., Yoshitsu, H. & Sugiyama, M. Crystallographic evidence that the dinuclear copper center of tyrosinase is flexible during catalysis. J. Biol. Chem. 281, 8981–8990 (2006).
Sendovski, M., Kanteev, M., Shuster Ben-Yosef, V., Adir, N. & Fishman, A. First structures of an active bacterial tyrosinase reveal copper plasticity. J. Mol. Biol. 405, 227–237 (2011).
Ismaya, W. T. et al. Crystal structure of Agaricus bisporus mushroom tyrosinase: Identity of the tetramer subunits and interaction with tropolone. Biochemistry 50, 5477–5486 (2011).
Muñoz-Muñoz, J. L. et al. Unravelling the suicide inactivation of tyrosinase: a discrimination between mechanisms. J. Mol. Catal. B Enzym. 75, 11–19 (2012).
Ramsden, C. A. & Riley, P. A. Tyrosinase: the four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Biorg. Med. Chem. 22, 2388–2395 (2014).
Han, H. Y. et al. The inhibition kinetics and thermodynamic changes of tyrosinase via the zinc ion. Biochim. Biophys. Acta 1774, 822–827 (2007).
Decker, H., Schweikardt, T. & Tuczek, F. The first crystal structure of tyrosinase: all questions answered? Angew. Chem. Int. Ed. 45, 4546–4550 (2006).
Deeth, R. & Diedrich, C. Structural and mechanistic insights into the oxy form of tyrosinase from molecular dynamics simulations. J. Biol. Inorg. Chem. 15, 117–129 (2010).
Shuster Ben-Yosef, V., Sendovski, M. & Fishman, A. Directed evolution of tyrosinase for enhanced monophenolase/diphenolase activity ratio. Enzyme Microb. Technol. 47, 372–376 (2010).
Goldfeder, M., Kanteev, M., Adir, N. & Fishman, A. Influencing the monophenolase/diphenolase activity ratio in tyrosinase. Biochim. Biophys. Acta 1834, 629–633 (2013).
Rolff, M., Schottenheim, J., Decker, H. & Tuczek, F. Copper-O2 reactivity of tyrosinase models towards external monophenolic substrates: molecular mechanism and comparison with the enzyme. Chem. Soc. Rev. 40, 4077–4098 (2011).
Kanteev, M., Goldfeder, M., Chojnacki, M., Adir, N. & Fishman, A. The mechanism of copper uptake by tyrosinase from Bacillus megaterium. J. Biol. Inorg. Chem. 18, 895–903 (2013).
Bailey, L. J., McCoy, J. G., Phillips, G. N. & Fox, B. G. Structural consequences of effector protein complex formation in a diiron hydroxylase. Proc. Natl Acad. Sci. USA. 105, 19194–19198 (2008).
Lavie, A., Allen, K. N., Petsko, G. A. & Ringe, D. X-ray crystallographic structures of D-xylose isomerase-substrate complexes position the substrate and provide evidence for metal movement during catalysis. Biochemistry 33, 5469–5480 (1994).
Walter, N. G. Ribozyme catalysis revisited: Is water involved? Mol. Cell 28, 923–929 (2007).
Siegbahn, P. E. M. & Borowski, T. Comparison of QM-only and QM/MM models for the mechanism of tyrosinase. Faraday Discuss. 148, 109–117 (2011).
Kleifeld, O., Frenkel, A., Martin, J. M. L. & Sagi, I. Active site electronic structure and dynamics during metalloenzyme catalysis. Nat. Struct. Mol. Biol. 10, 98–103 (2003).
Muñoz-Muñoz, J. L. et al. Catalysis and inactivation of tyrosinase in its action on o-diphenols, o-aminophenols and o-phenylendiamines: potential use in industrial applications. J. Mol. Catal. B Enzym. 91, 17–24 (2013).
Sendovski, M., Kanteev, M., Shuster Ben-Yosef, V., Adir, N. & Fishman, A. Crystallization and preliminary X-ray crystallographic analysis of a bacterial tyrosinase from Bacillus megaterium. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 1101–1103 (2010).
Shuster, V. & Fishman, A. Isolation, cloning and characterization of a tyrosinase with improved activity in organic solvents from Bacillus megaterium. J. Mol. Microbiol. Biotechnol. 17, 188–200 (2009).
Leslie, A. G. W. Joint CCP4+ESF-EAMCB Newsletter on Protein Crystallography, no. 26 (1992).
McCoy, A. J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. Sect. D Biol. Crystallogr. 63, 32–41 (2007).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132 (2004).
Acknowledgements
This work was supported by the Israel Science Foundation founded by the Israel Academy of Sciences and Humanities, grant number 193/11. We gratefully thank the staff of the European Synchrotron Radiation Facility (beamlines ID23-1 and 14-4) for provision of synchrotron radiation facilities and assistance. Furthermore, we would like to thank Dr Raz Zarivach for fruitful discussions.
Author information
Authors and Affiliations
Contributions
M.G. and M.K. performed and analysed the experiments, and wrote the manuscript; S.I.-O. assisted in protein purification and crystallization; N.A. and A.F. conceived and designed the study, analysed the results and wrote the manuscript; and all authors discussed and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figure 1 (PDF 113 kb)
Rights and permissions
About this article

Cite this article
Goldfeder, M., Kanteev, M., Isaschar-Ovdat, S. et al. Determination of tyrosinase substrate-binding modes reveals mechanistic differences between type-3 copper proteins. Nat Commun 5, 4505 (2014). https://doi.org/10.1038/ncomms5505
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms5505
This article is cited by
-
Pigmentation and TYRP1 expression are mediated by zinc through the early secretory pathway-resident ZNT proteins
Communications Biology (2023)
-
Thioquinoline derivatives conjugated to thiosemicarbazide as potent tyrosinase inhibitors with anti-melanogenesis properties
Scientific Reports (2023)
-
Biochemical characterization of Dimocarpus longan polyphenol oxidase provides insights into its catalytic efficiency
Scientific Reports (2022)
-
Enzymatic browning and genome-wide polyphenol oxidase gene identification in three contrasting avocado accessions
Plant Biotechnology Reports (2022)
-
Whole Cell-mediated Biocatalytic Synthesis of Helicid Cinnamylate and Its Biological Evaluation as a Novel Tyrosinase Inhibitor
Biotechnology and Bioprocess Engineering (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
