
Abstract
Von Hippel–Lindau (VHL) disease is an autosomal dominant familial cancer syndrome caused by germline mutations in VHL tumor suppressor gene. It is characterized by hemangioblastoma in central nervous system and retina, renal cell carcinoma or cyst, pheochromocytoma, pancreatic cyst and tumor, endolymphatic-sac tumor, and papillary cystadenoma in epididymis and broad ligament. Here, we used PCR-direct sequencing and universal primer quantitative fluorescent multiplex PCR (UPQFM-PCR) to detect VHL mutations in 16 patients clinically diagnosed with VHL disease. PCR-direct sequencing detected 12 germline mutations (75%, 12/16), in which a novel mutation of c.451A>T/p.Ile151Phe found in one proband had not been reported previously. UPQFM-PCR found two large deletions (12.5%, 2/16). The two remaining patients carried non-typical disease-causing mutations, including one silent mutation (c.481C>A/p.Arg161Arg) and one mutation in 3′-UTR (c.642+70C>A). Remarkably, 56.3% (9/16) probands did not have family history of VHL disease, suggesting the higher frequency of de novo mutations in Chinese patients. We also summarized Chinese VHL disease patients with VHL mutation findings published in the literature to provide information about the spectrum of VHL mutations in Chinese VHL disease patients.
Similar content being viewed by others
Introduction
Von Hippel–Lindau (VHL) disease is an autosomal dominant familial cancer syndrome caused by germline mutations in VHL tumor suppressor gene. The incidence of this disease is roughly 1/36 000 live births, and its penetrance is estimated to be more than 90% by 65 years of age.1, 2 It is characterized by hemangioblastoma in central nervous system (CNS) and retina, renal cell carcinoma and cyst, pheochromocytoma, pancreatic cyst and tumor, endolymphatic sac tumor, and papillary cystadenoma in epididymis and broad ligament.3, 4, 5
VHL gene locates in chromosome 3p25-26 and contains three coding exons that encode VHL protein (pVHL). pVHL combines with elongin B, elongin C and Cullin 2 (CUL2) to form VCB–CUL2 complex, which mediates the hypoxia-inducible factor.6 Abnormal pVHL may disrupt its tumor-suppression function directly or indirectly though hypoxia-inducible factor-mediated effects. Through transcriptional regulation, hypoxia-inducible factor promotes glucose uptake and expression of angiogenic, growth and mitogenic factors, including vascular endothelial growth factor, platelet derived growth factor, erythropoietin and transforming growth factor α, known to be involved in tumorigenesis.7, 8, 9, 10
Clinical criteria for the diagnosis of VHL disease include one of the following items: (1) more than one hemangioblastoma in CNS or retina; (2) a single hemangioblastoma in CNS or retina plus a visceral complication (such as multiple renal, pancreatic or hepatic cysts, pheochromocytoma or renal cancer) with the exception of epididymal and renal cysts; (3) any one of above manifestations with a family history.11 There is also a classification of VHL disease based on its clinical manifestations. VHL disease families without a family member affected with pheochromocytoma are classified as VHL disease type 1; VHL disease families with a family member affected with pheochromocytoma are classified as type 2, and type 2 is further subdivided into type 2A (without renal cell carcinoma or pancreatic cysts), 2B (with renal cell carcinoma or pancreatic cysts), and 2C (only pheochromocytoma).11
Genetic testing is the golden standard for VHL disease diagnosis. PCR-direct sequencing is accurate and relatively inexpensive, but is unsuitable for identification of large partial and complete VHL deletions.12 Southern blot or multiplex ligation-dependent probe associated with PCR-direct sequencing can achieve 100% mutation detection,13, 14, 15 but they are time-consuming and inconvenient for clinical application. Recently, the relative simple and practical method of universal primer quantitative fluorescent multiplex PCR (UPQFM-PCR) has been used to detect large deletions.16, 17 Approximately 20% of VHL disease patients do not have a family history and carry a ‘de novo mutation’.18, 19 Actually, these ‘de novo mutations’ may be inherited from their germline mosaic parents who are phenotypically normal and may have no mutations in their somatic cells.4
Here, we used the methods of PCR-direct sequencing and UPQFM-PCR to investigate germline mutations in VHL in 16 probands diagnosed with VHL disease by clinical diagnosis criteria. We found VHL mutations in 14 probands, including 11 point mutations (7 missense mutations and 4 nonsense mutations), 1 micro-deletion and 2 large deletions, in which a novel mutation of c.451A>T/p.Ile151Phe found in one proband had not been reported previously. The other two probands had non-typical disease-causing mutations including one silent mutation (c.481C>A/p.Arg161Arg) and one mutation in 3′-UTR (c.642+70C>A). Notably, no family history was found in 9 of the 16 patients (56.3%), suggesting that de novo mutations in VHL gene may be relatively prevalent in Chinese patients. We also analyzed our VHL disease families in association with the VHL mutation data in Chinese VHL disease patients reported in the literature to provide the spectrum of VHL gene mutations in Chinese population.
Patients and methods
Patients
A total of 16 unrelated patients diagnosed with VHL disease based on the clinical diagnosis criteria mentioned above as well as their family members were tested for germline mutations in VHL gene. This project was approved by the Medical Ethics Committee of Peking University First Hospital. Informed consent was obtained from all patients and their relatives.
DNA sequencing
Genomic DNA was isolated from peripheral blood samples by Qiagen DNA Blood mini kit (Qiagen, Hilden, Germany) following the manufacturer’s instruction. Genomic DNA (50–100 ng) was amplified by PCR in a standard PCR buffer containing 200 μM per each dNTPs, 1.5 mM MgCl2, 0.25 units of Taq polymerase and 0.2 μM per each primers with the thermal cycling program of 95 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s for 30 cycles. PCR products were purified and sequenced in an ABI 3730 automated sequencer (ABI, Foster City, CA, USA). The primer pairs used for the amplification of the three exons and their flanking introns of VHL gene were 1F: 5′-ggtggtctggatcgcgga-3′ and 1R: 5′-ggcttcagaccgtgctatcg-3′ (product size 418bp), 2F: 5′-gtggctctttaacaacctttgc-3′ and 2R: 5′-cctgtacttaccacaacaaccttatc-3′ (product size 208 bp), and 3F: 5′-gcaaagcctcttgttcgttc-3′ and 3R: 5′-caaaaatgccaccaccttct-3′ (product size 504 bp).
UPQFM-PCR to detect large deletions and insertions
UPQFM-PCR for the detection of large deletions and insertions in VHL gene was first described by Cybulski et al.16 Three exons of VHL gene and a fragment of β-globin gene (internal control) were amplified in two steps of PCR. The first PCR (PCR1) consisted of 100 ng genomic DNA and primer pairs tagged with designed universal sequences (in Table 1) for the amplification of the three exons of VHL gene and a fragment of β-globin gene (internal control) in 25 μl, with the thermal cycling program of 94 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s for 9 cycles. The second PCR (PCR 2) contained 2 μl of PCR 1 product and the universal primer pair of which the reverse primer was conjugated with FAM fluorescence. PCR 2 was run at 94 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s for 20 cycles. The PCR product of PCR 2 was separated by capillary electrophoresis in ABI 310 capillary sequencer (ABI), and the peaks were analyzed by Peak Scanner v.1.0 software (ABI). Detection of large deletions and insertions relied on the comparison between the peak areas of VHL gene exons and the area of β-globin. We first detected 20 normal individuals to calculate the area ratio of the three exons to β-globin for 95% confidence interval. The ratio of exon 1 was 1.100∼1.912, that of exon 2 0.287∼0.611 and that of exon 3 0.912∼1.328. If the ratio exceeded the 95% CI and reduced by nearly 50%, exon deletion was defined.
Results
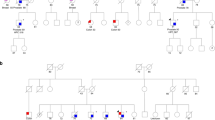
All of the 16 probands fulfilled the VHL disease diagnostic criteria. Nine of the probands (including one proband with familial lipoma history) had no family history. In all, 15 families were classified as type 1 (without pheochromocytoma) and one family belonged to type 2 (with pheochromocytoma) VHL disease (Table 2). Direct sequencing detected nucleotide substitutions or small deletions in 12 probands (12/16, 75%), including 7 missense mutations, 4 nonsense mutations and 1 micro-deletion. Proband 5 with novel mutation (c.451A>T/p.Ile151Phe) had not been reported previously. UPQFM-PCR revealed exon 3 deletion (Figure 1) in two probands (12.5%, 2/16). The remaining two probands were found to have non-typical disease-causing mutations, including one silent mutation (c.481C>A/p.Arg161Arg) and one mutation in 3′-UTR (c.642+70C>A), but the pathogenesis of these two mutations is unclear at present.

A large deletion detected by universal primer quantitative fluorescent multiplex PCR. (a) Normal control showing the peak areas and locations of the three von Hippel-Lindau (VHL) exons, and a β-globin fragment; (b) Exon 3 deletion, the arrow points to exon 3 deletion. A full color version of this figure is available at the Journal of Human Genetics journal online.
In this series of patients, the most usual manifestations were CNS hemangioblastoma, clear cell renal cell carcinoma, pancreatic cysts and tumors (Table 2). Pheochromocytoma, endolymphatic sac tumors and epididymal or ovarian cyst adenomas were rarely found. Proband 5 with a novel mutation (c.451A>T/p.Ile151Phe) had no VHL disease family history, and dysacusis (caused by endolymphatic sac tumors) was the initial symptom at 18 years of age, and ovarian cystadenoma, multiple renal cell carcinoma and pancreatic tumors were identified 20 years later. Probands 8 and 9 had the same mutation (c.263G>A/p.Trp88X), but their clinical symptoms were different. The mother of proband 9 had CNS hemangioblastoma at 34 years of age, but she is now 81 years old without any other VHL disease related lesions. Proband 15 showed multiple CNS hemangioblastomas, multiple renal cell carcinomas and pancreatic neuroendocrine tumors and cysts but without family history, and carried a silent mutation (c.481C>A/p.Arg161Arg) (Supplementary Figure S1). Proband 16 had retinal angiomas as the initial symptom, and then developed multiple CNS hemangioblastomas, multiple renal cell carcinomas and pancreatic neuroendocrine tumors but without family history, and carried a mutation in 3′-UTR (Supplementary Figure S2).
Several families also had some unusual clinical symptoms. Proband 4 with familial lipoma history had syringomyelia associated with Chiari type I malformation as the first presentation. His father and five brothers and sisters all had lipoma but no VHL disease lesions. Proband 7 and his younger brother were found to have mesh-like hemangioblastomas in CNS.
Discussion
In this study, we found an interesting phenomenon that more than 56.3% Chinese VHL disease patients had no family history of VHL disease but they were typical to be diagnosed with VHL disease by the clinical diagnosis criteria. Recently, Siu et al.20 reported that 55.6% (5/9) Chinese patients with VHL syndrome had no family history. Therefore, de novo mutations in VHL gene may be frequently seen in Chinese patients. Report from NIH demonstrated that 23% of the VHL syndrome kindreds had no family history, and thus the mutations may derived from sporadic (de novo) germline VHL mutations or an asymptomatic germline mosaic parent. Nordstrom-O’Brien et al.11 stated that only 3% VHL disease families had an established or ‘most likely’ de novo VHL mutation. It is still unknown whether the higher prevalence of sporadic VHL germline mutations relates to the increasing environmental pollution in China.
We also found two patients with nonpathogenic mutations, one silent mutation and one mutation in 3′-UTR. According to the basic knowledge of genetics, silent mutation has no effect on the protein structure. However, proband 15 presented clinical features of typical VHL disease. We had to consider the relationship between the silent mutation and VHL disease. Proband 16 with typical VHL disease manifestations carried a mutation in 3′-UTR. Some researchers also detected VHL mutations in 3′-UTR without any other mutations in VHL gene.21 Subsequently, we need to further investigate the pathogenesis of these two unusual mutations.
The prevalence of large deletions was 11∼40% in VHL disease patients.13, 14, 15, 16, 17, 20 When the data from PCR-direct sequencing are negative, we should first detect the large deletions by the method that is relatively inexpensive and uncomplicated. In this study, we found large deletions in 12.5% families by using UPQFM-PCR. So we recommend to use PCR-direct sequencing combined with UPQFM-PCR to search for VHL mutations for the patients with typical VHL disease manifestations as well as their family members. VHL mutation carriers should be treated based on the recommendations in the VHL handbook (http://www.vhl.org/).
We combined our findings from the 16 probands with the data from previous reports of Chinese VHL disease patients to summarize the clinical characteristics and genotype/phenotype relationships in Chinese patients.17, 20, 22, 23, 24, 25 There were totally 70 families that were clinically diagnosed as VHL disease and had the data of VHL mutations (Table 3). The spectrum of VHL mutations in Chinese patients is comparable to the findings of large scale investigations from other countries,11, 21 but with some unique characteristics (Table 3). The prevalence of germline mutations was 92.9% (65/70) including three non-typical disease-causing mutations. The prevalence of intragenic mutations detected by direct sequencing was 80% (56/70), and that of large deletions was 17.3% (9/52).
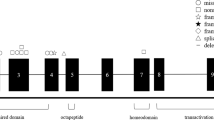
There were 18 mutations (found in 26 families) in exon 1, 7 mutations (found in 7 families) in exon 2 and 11 mutations (detected in 18 families) in exon 3, 3 mutations located in intron 1, 1 mutation in intron 2 and 1 mutation in 3′-UTR (Figure 2). Obviously, exon 1 was the hot mutation exon, accounting for 51.0% (26/51) of the families. The region of VHL germline mutations located between codons 65–178 (Figure 2). For nucleotide substitutions in the coding exons, the frequent VHL germline mutation regions were codons 65–98 (24 families) and codons 161–168 (15 families), inconsistent with the data from western countries and Japan. However, the prevalence of codon 161 mutations seemed to be higher than that of frequent mutations codon 167 in China.

Schematically demonstrating the point mutations and small insertions/deletions in von Hippel-Lindau (VHL) gene in 70 Chinese families with VHL disease reported in the literature. We searched the literature for Chinese VHL disease patients with the results of VHL gene mutation study. We summarized the 54 Chinese patient families reported in six papers in association with the 16 families presented here. A total of 56 families were found to have point mutations or small deletions, 9 families to have gross deletions and VHL mutations were not detected in 5 families. Only the 56 families with point mutations and small deletions are depicted in this figure. Location and nucleotide mutation/consequence are shown. Clinical type and number of the family are in brackets. ‘*’ Denotes new mutation not described previously, including three non-typical disease-causing mutations: c.481C>A/p.Arg161Arg, 3′-UTR mutation (c.642+70C>A) and IVSI-38C>T/Unknown. Filled areas beside exon 1 and 3 represent 5′- and 3′-untranslated region, respectively.
In the previous reports, the genotype/phenotype relationship had been discussed, especially that of VHL germline mutations and pheochromocytoma.16, 21, 26, 27 Pheochromocytoma appeared in patients with missense mutations, and rarely happened in those with nonsense mutations and large deletions.11, 21 All of the 13 Chinese families with type 2 VHL disease carried missense mutations (Table 3). Interestingly, all of the four families with the c.482G>A/p.Arg161Gln mutation demonstrated type 2C VHL disease (Figure 2), similar to the report by Zbar et al.21 in which all of the three VHL disease families with this mutation exhibited pheochromocytoma. It seems that p.Arg161Gln is a specific mutation for pheochromocytoma. The amino-acid residue 161 located in the Elongin C/P53-binding domain (residues 154–164) may be a key element for the function of this domain, and mutations in this region may thus develop pheochromocytoma.28, 29, 30
In this study, we used PCR-direct sequencing and UPQFM-PCR methods to detect nucleotide substitutions and deletions in VHL gene in probands clinically diagnosed with VHL disease. Up to 56.3% probands did not have the family history of VHL disease, suggesting that de novo mutations in VHL gene may be relatively common in Chinese VHL disease patients. We also summarized the Chinese VHL disease patients with the data of VHL mutations published in the literature, and compared the data with those from other countries. The clinical and molecular genetic characteristics were basically similar between VHL disease of Chinese patients and the patients from other countries. Pheochromocytoma was only found in patients with missense mutations and not in those with other types of VHL mutations. The incidence of large deletions in VHL gene was 17.3%, suggesting the requirement of methods for the detection of large VHL deletions to be included in routine VHL mutation screening.
References
Neumann, H. P. & Wiestler, O. D. Clustering of features of von Hippel-Lindau syndrome: evidence for a complex genetic locus. Lancet 337, 1052–1054 (1991).
Maher, E. R., Yates, J. R., Harries, R., Benjamin, C., Harris, R., Moore, A. T. et al. Clinical features and natural history of von Hippel-Lindau disease. Q. J. Med. 77, 1151–1163 (1990).
Singh, A. D., Shields, C. L. & Shields, J. A. von Hippel–Lindau Disease. Surv. Ophthalmol. 46, 117–142 (2001).
Lonser, R. R., Glenn, G. M., Walther, M., Chew, E. Y., Libutti, S. K., Linehan, W. M. et al. von Hippel-Lindau disease. Lancet 361, 2059–2067 (2003).
Maher, E. R., Neumann, H. P. & Richard, S. von Hippel-Lindau disease: a clinical and scientific review. Eur. J. Hum. Genet. 19, 617–623 (2011).
Latif, F., Tory, K., Gnarra, J., Yao, M., Duh, F. M., Orcutt, M. L. et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 260, 1317–1320 (1993).
Levy, A. P., Levy, N. S., Iliopoulos, O., Jiang, C., Kaplin, W. G. Jr. & Goldberg, M. A. Regulation of vascular endothelial growth factor by hypoxia and its modulation by the von Hippel-Lindau tumor suppressor gene. Kidney Int. 51, 575–578 (1997).
Kourembanas, S., Hannan, R. L. & Faller, D. V. Oxygen tension regulates the expression of the platelet-derived growth factor-B chain gene in human endothelial cells. J. Clin. Invest. 86, 670–674 (1990).
Krieg, M., Haas, R., Brauch, H., Acker, T., Flamme, I. & Plate, K H Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 19, 5435–5443 (2000).
Kaelin, W. G. The von Hippel-Lindau Gene, Kidney Cancer, and Oxygen Sensing. J. Am. Soc. Nephrol. 14, 2703–2711 (2003).
Nordstrom-O’Brien, M., van der Luijt, R. B., van Rooijen, E., van den Ouweland, A. M., Majoor-Krakauer, D. F., Lolkema, M. P. et al. Genetic analysis of von Hippel-Lindau disease. Hum. Mutat. 31, 521–537 (2010).
Klein, B., Weirich, G. & Brauch, H. DHPLC-based germline mutation screening in the analysis of the VHL tumor suppressor gene: usefulness and limitations. Hum. Genet. 108, 376–384 (2001).
Stolle, C., Glenn, G., Zbar, B., Humphrey, J. S., Choyke, P., Walther, M. et al. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum. Mutat. 12, 417–423 (1998).
Cho, H. J., Ki, C. S. & Kim, J. W. Improved detection of germline mutations in Korean VHL patients by multiple ligation-dependent probe amplification analysis. J. Korean. Med. Sci. 24, 77–83 (2009).
Rocha, J. C., Silva, R. L., Mendonca, B. B., Marui, S., Simpson, A. J. & Camargo, A. A. High frequency of novel germline mutations in the VHL gene in the heterogeneous population of Brazil. J. Med. Genet. 40, e31 (2003).
Cybulski, C., Krzystolik, K., Murgia, A., Gorski, B., Debniak, T., Jakubowska, A. et al. Germline mutations in the von Hippel-Lindau (VHL) gene in patients from Poland: disease presentation in patients with deletions of the entire VHL gene. J. Med. Genet. 39, E38 (2002).
Zhang, J., Huang, Y., Pan, J., Liu, D., Zhou, L., Xue, W. et al. Germline mutations in the von Hippel-Lindau disease (VHL) gene in mainland Chinese families. J. Cancer. Res. Clin. Oncol. 134, 1211–1218 (2008).
Richards, F. M., Payne, S. J., Zbar, B., Affara, N. A., Ferguson-Smith, M. A. & Maher, E. R. Molecular analysis of de novo germline mutations in the von Hippel-Lindau disease gene. Hum. Mol. Genet. 4, 2139–2143 (1995).
Sgambati, M. T., Stolle, C., Choyke, P. L., Walther, M. M., Zbar, B., Linehan, W. M. et al. Mosaicism in von Hippel-Lindau disease: lessons from kindreds with germline mutations identified in offspring with mosaic parents. Am. J. Hum. Genet. 66, 84–91 (2000).
Siu, W. K., Ma, R. C., Lam, C. W., Mak, C. M., Yuen, Y. P., Lo, F. M. et al. Molecular basis of von Hippel-Lindau syndrome in Chinese patients. Chin. Med. J. (Engl). 124, 237–241 (2011).
Zbar, B., Kishida, T., Chen, F., Schmidt, L., Maher, E. R., Richards, F. M. et al. Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum. Mutat. 8, 348–357 (1996).
Tong, A. L., Zeng, Z. P., Li, H. Z., Yang, D., Lu, L., Li, M. et al. von Hippel-Lindau gene mutation in non-syndromic familial pheochromocytomas. Ann. N. Y. Acad. Sci. 1073, 203–207 (2006).
Mao, X. C., Su, Z. P., Yu, W. Q., Zheng, W. M. & Zeng, Y. J. Familial and genetic researches on three Chinese families with von Hippel-Lindau disease. Neurol. Res. 31, 743–747 (2009).
Zhou, D. H., Wang, Y. M., Lan, T., Dong, Y. C., Zhang, B., Li, W. P. et al. [Mutation screening of VHL gene in a Chinese family with nonsyndromic pheochromocytoma]. Zhonghua. Yi. Xue. Yi. Chuan. Xue. Za. Zhi. 24, 365–368 (2007).
Zhou, J., Wang, J., Li, N., Zhang, X., Zhou, H., Zhang, R. et al. Molecularly genetic analysis of von Hippel-Lindau associated central nervous system hemangioblastoma. Pathol. Int. 60, 452–458 (2010).
Crossey, P. A., Richards, F. M., Foster, K., Green, J. S., Prowse, A., Latif, F. et al. Identification of intragenic mutations in the von Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype. Hum. Mol. Genet. 3, 1303–1308 (1994).
Ong, K. R., Woodward, E. R., Killick, P., Lim, C., Macdonald, F. & Maher, E. R. Genotype-phenotype correlations in von Hippel-Lindau disease. Hum. Mutat. 28, 143–149 (2007).
Forman, J. R., Worth, C. L., Bickerton, G. R., Eisen, T. G. & Blundell, T. L. Structural bioinformatics mutation analysis reveals genotype-phenotype correlations in von Hippel-Lindau disease and suggests molecular mechanisms of tumorigenesis. Proteins 77, 84–96 (2009).
Lee, S., Nakamura, E., Yang, H., Wei, W., Linggi, M. S., Sajan, M. P. et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell 8, 155–167 (2005).
Roe, J. S., Kim, H., Lee, S. M., Kim, S. T., Cho, E. J. & Youn, H. D. p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol. Cell 22, 395–405 (2006).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant number: 30872560 and 81172418). We are very grateful to all members of the families who participated in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Wu, P., Zhang, N., Wang, X. et al. Family history of von Hippel–Lindau disease was uncommon in Chinese patients: suggesting the higher frequency of de novo mutations in VHL gene in these patients. J Hum Genet 57, 238–243 (2012). https://doi.org/10.1038/jhg.2012.10
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2012.10
Keywords
This article is cited by
-
A meta-analysis of different von Hippel Lindau mutations: are they related to retinal capillary hemangioblastoma?
Molecular Genetics and Genomics (2022)
-
Intronic mutation of the VHL gene associated with central nervous system hemangioblastomas in two Chinese families with Von Hippel–Lindau disease: case report
BMC Medical Genetics (2020)
-
Biological and clinical impact of central nervous system hemangioblastomas in Chinese patients with von Hippel-Lindau disease: implications for treatment
Hereditary Cancer in Clinical Practice (2020)
-
A novel mutation in the VHL gene in a Chinese family with von Hippel-Lindau disease
BMC Medical Genetics (2018)
-
Genotype phenotype correlation in Asian Indian von Hippel–Lindau (VHL) syndrome patients with pheochromocytoma/paraganglioma
Familial Cancer (2018)

{kind=link}
{kind=link}