
Abstract
Hypokalaemic periodic paralysis (HypoPP) is an autosomal dominant disorder, which is characterized by periodic attacks of muscle weakness associated with a decrease in the serum potassium level. A major disease-causing gene for HypoPP has been identified as CACNA1S, which encodes the skeletal muscle calcium channel α-subunit with four transmembrane domains (I–IV), each with six transmembrane segments (S1–S6). To date, all CACNA1S mutations identified in HypoPP patients are located within the voltage-sensor S4 segment. In this study we report a novel CACNA1S mutation in a new region of the protein, the S3 segment of domain III. We characterized a four-generation South American family with HypoPP. Genetic analysis identified a novel V876E mutation in all HypoPP patients in the family, but not in normal family members or 160 control people. Clinical analysis indicates that mutation V876E is associated with a severe outcome as characterized by a very early age of onset, complete penetrance and a severe prognosis including death. These results identify a new mutation in CACNA1S and expand the spectrum of CACNA1S mutations associated with HypoPP.
Similar content being viewed by others


Introduction
Primary periodic paralyses are autosomal dominant disorders characterized by episodic flaccid muscle weakness and intermittent myotonia, and are classified as hypokalemic (hypokalaemic periodic paralysis, HypoPP) or hyperkalemic (HyperPP) on the basis of the serum potassium level during the attack of weakness. HypoPP can be provoked by a low concentration of serum potassium, whereas in the case of HyperPP serum potassium is elevated or normal.1
Hypokalaemic periodic paralysis is the most common form of periodic paralysis in humans, but it is still considered a rare disease because its prevalence rate is about 1 in 100 000 livebirth.2 Molecular genetic analyses showed that HypoPP was caused by mutations in the calcium channel gene CACNA1S or the sodium channel gene SCN4A.3, 4 A voltage-gated potassium channel β-subunit gene KCNE3 was claimed as a HypoPP gene with the identification of one mutation (Arg83His) in two families, but recent studies suggest that Arg83His may be a benign polymorphic variant.5, 6
The CACNA1S gene encodes the voltage-gated calcium channel α-subunit Cav1.1, maps to chromosomes 1q31–q32, spans 90 kb, and is organized into 44 exons. CACNA1S has an important role in Ca2+-mediated excitation-contraction coupling. CACNA1S contains four homologous transmembrane domains (DI–DIV), each of which contains six α-helical segments spanning the membrane (S1–S6). The six transmembrane segments consist of two distinct modules: the pore domain (S5–S6) and the voltage-sensor domain (S1–S4). To date, mutations in CACNA1S account for approximately 70% of HypoPP cases, but only several missense mutations in the CACNA1S have been identified in the highly conserved S4 segment. These mutations include R528H, R528G, R1239H, R1239G and R897S.3, 7, 8, 9, 10 In addition, the R1086H mutation has been linked to malignant hyperthermia.11
In this study, we identified and characterized a South American family with HypoPP. Genetic studies identified a novel c.2627T>A (p.V876E) mutation in CACNA1S that is responsible for the disease. This mutation is located in a new region of the CACNA1S protein, the S3 segment of domain III, whereas all other mutations identified previously are within the voltage-sensing S4 segment of CACNA1S.
Materials and methods
Study subjects and isolation of genomic DNA
The study participants were identified and enrolled in Unidad de Genética, Universidad del Rosario in Colombia. This study was approved by local Institutional Review Boards on Human Subject Research, and informed written consent was obtained from each participant. Detailed records on family medical history, physical examinations and clinical data were obtained. The diagnosis of the HypoPP was carried out on the basis of symptoms, physical signs and the blood potassium level.
Human genomic DNA was extracted from peripheral blood using standard protocols.
Linkage analysis
Linkage analysis was carried out using microsatellite markers at the CACNA1S and SCN4A loci as described previously by us.12, 13
Mutation screening
To identify the molecular basis of HypoPP in this family, we carried out direct DNA sequence analysis of the CACNA1S gene. All 44 coding exons and exon–intron boundaries of the CACNA1S gene were PCR amplified and sequenced (see Table 1 for information on primers). DNA sequence analysis was performed using the BigDye Terminator Cycle Sequencing v1.1 kit on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). All mutant PCR fragments were sequenced on both strands using both forward and reverse primers.
Results
Clinical features of a South American family with HypoPP
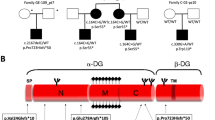
We identified and characterized a South American family with 12 living individuals in four generations (Figure 1). In the family, six members (I-2, I-5, II-2, II-4, III-2 and IV-1) were clinically diagnosed as being affected with HypoPP. The proband, individual II-2, is a 37-years old female patient whose symptoms began at the age of 4 years and were diagnosed at the age of 14. The frequency of HypoPP attacks is six times per year. The symptoms include muscle weakness and flaccid paralysis triggered by the rest after the exercise, the intake of foods rich in carbohydrates or cold. The paralysis affects extremities. The patient was from the sixth pregnancy, and birth weight and size were normal and without perinatal complications. Physical examinations showed that the lower limbs had decreased strength and tone of dominance, but sensitivity was preserved. Her treatment started with oral intake of potassium (K+) diclofenamida, which improved symptoms. She is currently on acetazolamide, which showed partial improvement of symptoms. The proband does not have any other disease.

Pedigree structure of a South American family affected with hypokalaemic periodic paralysis (HypoPP) and co-segregation of mutation V876E in CACNA1S in this family. Affected males and females are indicated by filled squares and circles, respectively. Normal individuals are shown as empty symbols. Deceased individuals are indicated by slashes (/). The proband is indicated by an arrow. V/V, wild-type allele; V/E, mutation V876E.
Patient I-2 was a male patient with the onset of HypoPP as early as in the first year of life. He had frequent episodes of muscle weakness and flaccid paralysis with low potassium. He died at the age of 47 years, apparently from the disease because of respiratory stress.
Patient I-5 is a female patient with symptoms of muscular weakness and flaccid paralysis of limbs.
Patient II-4 was a male HypoPP with the same symptoms as I-2. His symptoms started at the age of 9 years. He never received treatment for HypoPP and died at the age of 16 years.
Patient III-2 is a 19-year-old female patient with HypoPP started at the age of 9 years. She developed muscle weakness and flaccid paralysis because of low levels of K (2.8 mEq l−1). The frequency of HypoPP attacks is five times per year. Her treatment with acetazolamide showed irregular, partial improvement of symptoms with recovery of the serum potassium (3.5 mEq l−1).
Patient IV-1 is a 3-year-old female patient who presents symptoms of muscular weakness. She is not on any medication.
Identification of a novel CACNA1S mutation associated with HypoPP
For genetic analysis of the South American family with HypoPP, we first carried out linkage analysis for two candidate genes identified for HypoPP: CACNA1S and SCN4A. A genetic recombination was found with marker D17S1816 at the SCN4A locus (data not shown). We further sequenced the S4 segments of the SCN4A gene, but did not detect any mutation. These results suggest that SCN4A is unlikely to be a candidate gene for HypoPP in the family.
Linkage results suggest that two markers D1S413 and D1S249 at the CACNA1S locus are positively linked to HypoPP in the family (data not shown), indicating that CACNA1S is a strong candidate gene for HypoPP in the family. We then carried out direct DNA sequence analysis of the whole CACNA1S gene, all exons and exon–inton boundaries of CACNA1S were PCR-amplified and sequenced. This analysis identified a heterozygous T to A transition at nucleotide 2627 of CACNA1S, which results in substitution of a highly conserved valine residue with a glutamic acid residue at codon 876 (c.2627T>A (p.V876E)) (Figure 2a). The V876E mutation is located in the transmembrane segment S3 of domain III. The mutated V876 residue is an evolutionarily, highly conserved residue in the S3 segment of domains I, II, III and IV (Figure 2b). Furthermore, V876 is not only evolutionally conserved in the CACNA1S proteins from Caenorhabditis elegans to Homo sapiens (Figure 2c), but also in various calcium channel α-subunits (Figure 2d).

Identification of a novel mutation 2627T → A (V876E) in the CACNA1S gene in the South American family with hypokalaemic periodic paralysis (HypoPP). (a) DNA sequences for a normal family member (normal) and the proband (affected) are shown. The nucleotide residue where the mutation occurs is indicated by an arrow. The T to A change at codon 876 results in the substitution of a valine residue by a glutamine acid residue in DIIIS3. (b) Alignment of amino-acid sequences of the S3 segments of domains I, II, III and IV. The site of the mutation is indicated by asterisk. (c) Alignment of amino-acid residues around V876E for proteins orthologous to CACNA1S. (d) Alignment of amino-acid sequences around V876E from various calcium channels α-subunits.
Further sequence analysis showed that all affected members in this family carried the V876E mutation (Figure 1). All normal individuals in this family do not carry this mutation. The mutation was not present in 160 normal controls.
Discussion
In this study, we have identified a novel mutation in the CACNA1S gene that causes autosomal dominant muscle disorder HypoPP in a South American family. Mutation V876E is highly likely to be the disease-causing mutation in the HypoPP family on the basis of the following reasons. First, the V876E mutation co-segregates with the disease in this family; second, the mutated V876 residue is evolutionarily, highly conserved not only among CACNA1S proteins from various species, but also among different calcium channel α-subunits; third, the V876E mutation is not present in 160 normal controls. Together with the fact that CACNA1S is the major disease-causing gene for HypoPP, we conclude that the V876E mutation in CACNA1S is the pathogenic mutation in the South American HypoPP family.
To date, several specific missense mutations have been identified in the CACNA1S gene in families with HypoPP, including R528H and R528G located in DIIS4, R1239H and R1239G in DIVS4.3, 7, 8, 9 In addition, a de novo mutation R897S in DIIIS4 has been reported recently.10 All these mutations identified previously are located in the critical voltage-sensor S4 segment, and occur at an arginine residue. Interestingly, the V876E mutation identified in our study occurs at a different valine residue located in the S3 transmembrane segment of domain III, a segment that had never been associated with HypoPP. These results suggest that HypoPP mutations in CACNA1S can occur in domains other than voltage-sensor S4.
The V876E mutation is associated with an unusual early age of onset, complete penetrance and severe prognosis of HypoPP. In the family with mutation V876E, the mean age at onset of HypoPP attacks is 5.2±3.6 years, which is earlier than those with the R528H mutation (14±5 years) or patients with the R1239H mutation (7±4 years).14 It is of note that one patient in the family with mutation V876E developed the symptom even within 1 year after birth. The infantile presentation is rarely reported in HypoPP cases, although it was reported in HyperPP cases.10, 14 Incomplete penetrance, particularly in females, has been reported for several HypoPP mutations, such as CACNA1S R528H and SCN4A R672C mutations,15, 16 but the penetrance of HypoPP in both male and female patients in the family under this study is complete. Two male patients (I-2 and II-4) developed HypoPP attacks involving respiratory muscles and died.
The functional effect of the V876E mutation on the skeletal calcium channel is not known. The S4 segment of each transmembrane domain acts as a voltage sensor for the calcium channel. Other segments, such as S3, are indispensable for regulating the gating properties of the channel. Future electrophysiological characterization of the V876E mutation will provide insights into the mechanism underlying the pathogenesis of HypoPP in this South American family.
Conflict of interest
The authors declare no conflict of interest.
References
Stedwell, R. E., Allen, K. M. & Binder, L. S. Hypokalemic paralyses: a review of the etiologies, pathophysiology, presentation, and therapy. Am. J. Emerg. Med. 10, 143–148 (1992).
Fontaine, B., Vale-Santos, J., Jurkat-Rott, K., Reboul, J., Plassart, E., Rime, C. S. et al. Mapping of the hypokalaemic periodic paralysis (HypoPP) locus to chromosome 1q31-32 in three European families. Nat. Genet. 6, 267–272 (1994).
Ptácek, L. J., Tawil, R., Griggs, R. C., Engel, A. G., Layzer, R. B., Kwieciński, H. et al. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell 77, 863–868 (1994).
Bulman, D. E., Scoggan, K. A., Bulman, D. E., Scoggan, K. A., Van Oene, M. D., Nicolle, M. W. et al. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology 53, 1932–1936 (1999).
Abbott, G. W., Butler, M. H., Bendahhou, S., Dalakas, M. C., Ptacek, L. J. & Goldstein, S. A. MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell 104, 217–231 (2001).
Sternberg, D., Tabti, N., Fournier, E., Hainque, B. & Fontaine, B. Lack of association of the potassium channel-associated peptide MiRP2-R83H variant with periodic paralysis. Neurology 61, 857–859 (2003).
Fouad, G., Dalakas, M., Servidei, S., Mendell, J. R., Van den Bergh, P., Angelini, C. et al. Genotype-phenotype correlations of DHP receptor alpha 1-subunit gene mutations causing hypokalemic periodic paralysis. Neuromuscul. Disord. 7, 33–38 (1997).
Jurkat-Rott, K., Lehmann-Horn, F., Elbaz, A., Heine, R., Gregg, R. G., Hogan, K. et al. A calcium channel mutation causing hypokalemic periodic paralysis. Hum. Molec. Genet. 3, 1415–1419 (1994).
Wang, Q., Liu, M., Xu, C., Tang, Z., Liao, Y., Du, R. et al. Novel CACNA1S mutation causes autosomal dominant hypokalemic periodic paralysis in a Chinese family. J. Mol. Med. 83, 203–208 (2005).
Chabrier, S., Monnier, N. & Lunardi, J. Early onset of hypokalaemic periodic paralysis caused by a novel mutation of the CACNA1S gene. J. Med. Genet. 45, 686–688 (2008).
Monnier, N., Procaccio, V., Stieglitz, P. & Lunardi, J. Malignant- hyperthermia susceptibility is associated with a mutation of the alpha-1-subunit of the human dihydropyridine-sensitive L-type voltage-dependent calcium-channel receptor in skeletal muscle. Am. J. Hum. Genet. 60, 1316–1325 (1997).
Du, W., Bautista, J. F., Yang, H., Diez-Sampedro, A., You, S. A, Wang, L. et al. Calcium-sensitive potassiumchannelopathy in human epilepsy and paroxysmal movement disorder. Nat. Genet. 37, 733–738 (2005).
Zhang, X., Chen, S., Yoo, S., Chakrabarti, S., Zhang, T., Ke, T. et al. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell. 135, 1017–1027 (2008).
Miller, T. M., Dias da Silva, M. R., Miller, H. A., Kwiecinski, H., Mendell, J. R., Tawil, R. et al. Correlating phenotype and genotype in the periodic paralyses. Neurology 63, 1647–1655 (2004).
Elbaz, A., Vale-Santos, J., Jurkat-Rott, K., Lapie, P., Ophoff, R. A., Bady, B. et al. Hypokalemic periodic paralysis and the dihydropyridine receptor (CACNL1A3): genotype/phenotype correlations for two predominant mutations and evidence for the absence of a founder effect in 16 Caucasian families. Am. J. Hum. Genet. 56, 374–380 (1995).
Kim, M. K., Lee, S. H., Park, M. S., Kim, B. C., Cho, K. H., Lee, M. C. et al. Mutation screening in Korean hypokalemic periodic paralysis patients: a novel SCN4A Arg672Cys mutation. Neuromuscul. Disord. 14, 727–731 (2004).
Acknowledgements
This study was supported by an Established Investigator Award from the American Heart Association (0440157N), an NHLBI Grant (R01 HL66251), a State of Ohio Wright Center of Innovation grant and Biomedical Research and Technology Transfer Partnership Award (BRTT, Ohio's Third Frontier Project), the National Basic Research Program of China (973 Grant 2007CB512000) and the National Natural Science Foundation of China (30670857 and 30700455).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ke, T., Gomez, C., Mateus, H. et al. Novel CACNA1S mutation causes autosomal dominant hypokalemic periodic paralysis in a South American family. J Hum Genet 54, 660–664 (2009). https://doi.org/10.1038/jhg.2009.92
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2009.92
Keywords
This article is cited by
-
Whole-exome sequencing in eccrine porocarcinoma indicates promising therapeutic strategies
Cancer Gene Therapy (2022)
-
Mutations associated with hypokalemic periodic paralysis: from hotspot regions to complete analysis of CACNA1S and SCN4A genes
neurogenetics (2022)
-
Skeletal muscle CaV1.1 channelopathies
Pflügers Archiv - European Journal of Physiology (2020)
-
A Novel Mutation in CACNA1S Gene Associated with Hypokalemic Periodic Paralysis Which has a Gender Difference in the Penetrance
Journal of Molecular Neuroscience (2012)
-
Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels
Pflügers Archiv - European Journal of Physiology (2010)
