
Abstract
Three new γ-pyrones named marinactinones A–C (1–3) were isolated from marine-derived actinomycete Marinactinospora thermotolerans SCSIO 00606. These structures were elucidated by extensive spectroscopic methods. All three new compounds were evaluated for cytotoxic effects on six cancer cell lines and inhibitory activities of DNA topoisomerase II. Compounds 1–3 exhibited moderate cytotoxicities against SW1990, HepG2 and SMCC-7721 cell lines, and compound 2 showed weak DNA topoisomerase II inhibition activity. This is the first report on the chemical constituents and their biological activities from Marinactinospora, a novel genus of marine actinomycetes.
Similar content being viewed by others
Introduction
Numerous secondary metabolites with novel structures and distinct biological activities have been discovered from marine actinomycetes, which represent new resources having great potential for novel drug exploration.1, 2, 3, 4 In our efforts to search for new antitumor compounds, organic extracts of more than 2000 actinomycete strains isolated from sediments of the northern South China Sea were screened by brine shrimp lethality assays.5, 6 Among them, the ethyl acetate extracts of a strain Marinactinospora thermotolerans SCSIO 00606 exhibited good cytotoxic activity. The subsequent chemical investigation on this strain resulted in the isolation of three new γ-pyrone derivates, namely, marinactinones A–C (1–3).
γ-Pyrone natural products constitute a large class of biologically active compounds, and most of them were derived from marine organisms.7 To the best of our knowledge, marinactinones A–C (1–3) were the first three γ-pyrone natural products with a methylhexyl side chain. Previously, we described M. thermotolerans as a novel marine actinobacterial genus.8 Herein, we report the isolation, structure elucidation of the three new γ-pyrones (1–3) from the strain M. thermotolerans SCSIO 00606, and we also evaluated the new compounds for their cytotoxicities against six human cancer cell lines and their potential to inhibit DNA topoisomerase II. This is the first report on the chemical constituents from this new group of marine microbes and their biological activities.
Results and Discussion
Structure determination
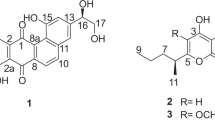
Marinactinone A (1) was obtained as pale yellow oil. Its HR–ESI–MS gave a [M+H]+ ion peak at m/z 253.1812 (calcd for [M+H]+ 253.1804), in combination with 1H and 13C NMR data (Table 1), suggesting the molecular formula to be C15H24NO3. The 1D NMR data for 1 indicated one carbonyl, five quarternary carbons, one methine, four methylenes and five methyls. Heteronuclear single quantum coherence experiments revealed all the connective relationship between the proton and carbon signals in 1. The IR absorptions at 1670 and 1602 cm−1, the absorption at 253.5 nm in the UV spectrum, together with the signals at δ 99.3 (C-3, s), 118.2 (C-5, s), 158.4 (C-6, s) and 162.1 (C-2, s) p.p.m. in the 13C NMR spectrum showed the presence of γ-pyrone moiety.9 Furthermore, the HMBC correlations from H-14 (δ 1.84, s, 3H) to C-2, C-3 and C-4, from H-15 (δ 1.93, s, 3H) to C-4, C-5 and C-6 and from 2-OMe (δ 3.95, s, 3H) to C-2 (Figure 2) confirmed that the skeleton of 1 is 6-substituted 2-methoxy-3,5-dimethyl-4H-pyrane-4-one. Detailed NMR analyses including 1D NMR, 1H–1H double-quantum-filtered correlation spectroscopy and HMBC, showed the presence of one methine, four methylenes and two methyls, indicating that the substitution at C-6 positions was a 5-methylhexyl group. The HMBC correlations from H-7 (δ 2.58, t, J=7.5 Hz, 2H) to C-6, C-7 (δ 30.7, t) and C-8 (δ 27.3, t) confirmed the link between γ-pyrone moiety and the 5-methylhexyl group (Figure 2). Taken together, compound 1 was identified as 2-methoxy-3,5-dimethyl-6-(5-methylhexyl)-4H-pyran-4-one (Figure 1).

Selected 1H–1H double-quantum-filtered correlation spectroscopy and HMBC correlations for compounds 1–3.

Structures of marinactinones A–C (1–3).
Marinactinone B (2) was isolated as pale yellow oil. The HR–ESI–MS showed the [M+H]+ ion peak at m/z 267.1978 (calcd for [M+H]+ 267.1960), establishing the molecular formula as C16H26O3. The UV, IR and 1D NMR showed it probably had a similar structure of compound 1. Detailed comparison of the 1D NMR spectra of 2 and 1 revealed that the presence of an ethyl group at C-5 in 2, instead of a methyl group at C-5 in 1 (Table 1). Consistently, the 1H NMR spectrum of 2 showed the two proton signals at δ 2.43 p.p.m. (H-15, q, J=7.5 Hz), and the 13C NMR spectrum showed the presence of one methylene carbon at δ 18.0 p.p.m. (C-15, t) in 2. Furthermore, the HMBC correlations from H-16 (δ 1.06, t, J=7.5 Hz) to C-5 (δ 123.9, s) and C-15, from H-15 to C-4 (δ 180.6, s), C-5, C-6 (δ 158.6, s) and C-16 (δ 13.7, q) in 2 (Figure 2) confirmed the slight difference between 2 and 1. Conclusively, the structure of 2 was elucidated as 2-methoxy-3-methyl-5-ethyl-6-(5-methylhexyl)-4H-pyran-4-one (Figure 1).
Marinactinone C (3) was isolated as pale yellow oil. The molecular formula of 3 was determined as C16H26O3 by HR–ESI–MS at m/z 267.1973 (calcd for [M+H]+ 267.1960). The structure of 3 was established by comparing its spectroscopic data with those of 1 and 2. Compound 3 was first confirmed to have the same 6-substituted 2-methoxy-3-methyl-5-ethyl-4H-pyran-4-one skeleton as of 2; however, a 4-methylhexyl group at C6, instead of the 5-methylhexyl group at C-6 in 2, was present in compound 3. The conclusion could be drawn from the detailed comparisons of the NMR data of 3 and 2. The carbon signals of C-9 (δ 27.1, t), C-10 (δ 38.7, t), C-11 (δ 27.9, d) and C-12 (δ 22.6, q) in 2 were replaced by two methylene (δ 29.0, t, C-9; δ 29.2, t, C-11), one methine (δ 31.7, d, C-10) and one methyl (δ 14.1, q, C-12) in 3, consistent with the changes of proton signals in the 1H NMR spectrum (Table 1). Furthermore, the heteronuclear single quantum coherence and HMBC correlations also supported the corresponding changes (Figure 2). Thus, compound 3 was established as 2-methoxy-3-methyl-5-ethyl-6-(4-methylhexyl)-4H-pyran-4-one. The C-10 was only one chiral center of compound 3, but we could not confirm its configuration for sample limitation.
Compounds 1–3 were evaluated for their cytotoxicities against MCF-7, SW1990, HepG2, NCI-H460, SMCC-7721 and HeLa cancer cell lines with 5-fluorouracil as a positive control. From our observations, compounds 1–3 exhibited moderate cytotoxicities to SW1990, HepG2 and SMCC-7721, but no significant cytotoxicities to other cell lines (Table 2). Subsequently, compounds 1–3 were tested for inhibitory effects on in vitro DNA topoisomerase II enzymatic activity. Only compound 2 exhibited dose-dependent weak inhibition activity on topoisomerase II, with IC50 value of 607 μM (Figure 3).

Assay for DNA topoisomerase II activity and inhibitory activity. (a) DNA topoisomerase II activity, lane 1–6, plasmid DNA (pBV220) 0.5 μg + topoisomerase II 0, 0.5, 1, 1.5, 2 and 4 μl; lane 7, maker; lane 8, pBV220 0.5 μg; (b) DNA topoisomerase II inhibitory activity by marinactinone B, lane 1, pBV220 0.5 μg; lane 2–8, same as lane 1 + topoisomerase II (1 U, 2 μl) + 0, 12.5, 25, 50, 100, 200 and 400 μg ml−1 marinactinone B (2).
Methods
General
Optical rotation was recorded on PerkinElmer Model 341 polarimeter (PerkinElmer Inc., Waltham, MA, USA). UV spectra were recorded on Beckman DU 640 spectrophotometer (Beckman Coulter Inc., Brea, CA, USA). IR spectra were taken on Bruker VECTOR22 IR spectrophotometer (Bruker Corporation, Ettlingen, Germany) in KBr disks. 1H-, 13C-NMR and DEPT spectra and 2D NMR were recorded on a Bruker AVANCE DRX-500 spectrometer using TMS as internal standard, and chemical shifts were recorded as δ (p.p.m.) values. ESI–MS were measured with an API 2000 LC/MS/MS Mass Spectrometer (AB SCIEX, Foster City, CA, USA) HR–ESI–MS spectra were measured on a Waters Q-Tof Micro mass spectrometer (Waters Corporation, Milford, MA, USA). Semi-preparative HPLC was performed using an ODS column (YMC-Pack ODS-5-A, 250 × 10 mm, 5 μm, 3 ml min−1) and monitored by UV detector.
Actinomycete material
M. thermotolerans SCSIO 00606 was isolated and identified by Dr Xinpeng Tian from a sea sediment sample collected from site E103 (21°30.658′N 115°25.580′E; black soft mud at 114 m depth) in the northern South China Sea in September 2006. It was preserved in Key Laboratory of Marine Bio-resources Sustainable Utilization, South China Sea Institute of Oceanology, Chinese Academy of Sciences. Working stocks were prepared on ISP 2 agar medium slants (Yeast extract 4.0g, Malt extract 5.0g, Dextrose 4.0g, Agar 20.0g, Seawater 1.0l) modified with seawater instead of distilled water, stored at 4 °C.
Fermentation, extraction and isolation
The strain SCSIO 00606 was inoculated into 500 ml Erlenmeyer flasks containing150 ml liquid medium (soluble starch 10.0 g, yeast extract 10.0 g, KH2PO4 0.5 g, cornmeal 3.0 g, glucose 20.0 g, MgSO4·7H2O 0.5 g, beef extract 3.0 g, CaCO3 2.0 g and 30.0 g sea salt dissolved in 1.0 l distill water, pH 7.0), followed by shaking incubation at 180 r.p.m. for 9 days at 28 °C. The fermented whole broth (45.0 l) was filtered through cheesecloth to separate into supernatant and mycelia. The former was concentrated under reduced pressure to about a quarter of the original volume and then extracted three times with ethyl acetate to give an ethyl acetate solution (crude extract 15.0 g), whereas the latter was extracted three times with acetone/H2O (80:20). The acetone solution was concentrated under reduced pressure to afford an aqueous solution. The aqueous solution was extracted three times with ethyl acetate to give another ethyl acetate solution (crude extract 5.0 g). Both ethyl acetate solutions were combined and concentrated under reduced pressure to give a crude extract (20.0 g).
The crude extract (20.0 g) was separated into 13 fractions on a silica gel column using a step gradient elution of petroleum ether/CHCl3/MeOH. The fraction 5 (753.2 mg) was subjected on Sephadex LH-20 eluting with CHCl3/MeOH (1:1), and thereafter followed by silica gel column chromatography using a stepwise gradient elution of petroleum ether–acetone, to yield subfraction 5-3-2 (117.0 mg). The subfraction 5-3-2 was further purified by semi-preparative HPLC (75% MeOH, 3.0 ml min−1), to give compound 1 (5.0 mg, tR 16.9 min), 2 (4.5 mg, tR 21.8 min) and 3 (2.0 mg, tR 23.4 min).
Physico–chemical properties
Marinactinone A (1): obtained as pale yellow oil (CHCl3); UV (MeOH) λmax (log ɛ) 208.9 (4.02), 253.5 (3.94) nm; IR (KBr) νmax cm−1 2925, 1670, 1602, 1467, 1415, 1320, 1250, 1158; HR–ESI–MS m/z 253.1812 [M+H]+ (calcd for C15H25O3, 253.1804); 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz; see Table 1).
Marinactinone B (2): obtained as pale yellow oil (CHCl3); UV (MeOH) λmax (log ɛ) 212.3 (3.99), 254.6 (3.89) nm; IR (KBr) νmax cm−1 2930, 1668, 1600, 1458, 1402, 1247, 1170; HR–ESI–MS m/z 267.1978 [M+H]+ (calcd for C16H27O3, 267.1960); 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz; see Table 1).
Marinactinone C (3): obtained as pale yellow oil (CHCl3); [α]26D+3.0 (c 0.1, CHCl3); UV (MeOH) λmax (log ɛ) 213.5 (4.00), 253.5 (3.91) nm; IR (KBr) νmax cm−1 2929, 1670, 1600, 1465, 1410, 1258, 1172; HR–ESI–MS m/z 267.1973 [M+H]+ (calcd for C16H27O3, 267.1960); 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz; see Table 1).
Cytotoxicity assays
Cytotoxicity was evaluated by the MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) method10 using MCF-7, SW1990, HepG2, NCI-H460, SMCC-7721 and HeLa cell lines. The cell lines were grown in RPMI-1640 supplemented with 10% fetal bovine serum under a humidified atmosphere of 5% CO2 and 95% air at 37°C. A volume of 200 μl of those cell suspensions at a density of 1 × 105 cells per ml was plated in 96-well microtiter plates and incubated for 24 h at the above condition. Then 2 μl of the test compound solutions (in DMSO) at different concentrations was added to each well and further incubated for 72 h in the same condition. A volume of 20 μl of the MTT solution (5 mg ml−1 in IPMI-1640 medium) was added to each well and incubated for 4 h. A volume of 150 μl of an old medium containing MTT was then gently replaced by DMSO and pipetted to dissolve any formazan crystals formed. Absorbance was then determined on a SpectraMax Plus plate reader (Molecular Devices Inc., Sunnyvale, CA, USA) at 570 nm.
Assay for topoisomerase II inhibition in vitro
Compounds 1–3 were also screened for their ability to inhibit topoisomerase II enzymatic activity,11, 12, 13 with etoposide as positive control. The plasmid pBV220 was purified with the AxyPrep Plasmid midiPrep Kit (Axygen Biosciences, Union City, CA, USA) from ampicillin-resistant Escherichia coli. Active DNA topoisomerase II was obtained from SD (Sprague Dawley) rat liver cells. The reaction volumes were 20 μl in an assay buffer (200 mM Tris-HCl, pH 7.5, 340 mM KCl, 40 mM MgCl2, 20 M dithiothreitol, 120 μg ml−1 bovine serum albumin and 4 mM ATP) containing 1 unit of topoisomerase II (2 μl) and 5 μg plasmid pBV220 (4 μl). Samples were solubilized using DMSO and tested for inhibitory activity at the final concentrations of 0, 12.5, 25, 50, 100, 200 and 400 μg ml−1. The reaction mixtures were incubated at 37 °C for 30 min and terminated by adding 10 volume of loading buffer (5% sarkosyl, 0.0025% bromophenol blue, 25% glycerol). Reaction products were electrophoresed on a 0.7% agarose gel in Tris-acetate-EDTA running buffer containing ethidium bromide (0.5 μg ml−1) at 80 V for 30 min. For the visualizing and quantitative analyses of topoisomerase II activity, the gels were directly scanned with Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA) and the area representing supercoiled DNA was calculated. Concentrations for 50% inhibition (IC50) were determined by interpolation from plots of topoisomerase II activity versus inhibitor concentration.
References
Jensen, P. R., Mincer, T. J., Williams, P. G. & Fenical, W. Marine actinomycete diversity and natural product discovery. Anton. Leeuw. Int. J. G. 87, 43–48 (2005).
Lam, K. S Discovery of novel metabolites from marine actinomycetes. Curr. Opin. Microbiol. 9, 245–251 (2006).
Molinski, T. F., Dalisay, D. S., Lievens, S. L. & Saludes, J. P. Drug development from marine natural products. Nat. Rev. Drug. Discov. 8, 69–85 (2009).
Olano, C., Mendez, C. & Salas, J. A. Antitumor compounds from marine actinomycetes. Mar. Drugs 7, 210–248 (2009).
Solis, P. N., Wright, C. W., Anderson, M. M., Gupta, M. P. & Phillipson, J. D. A microwell cytotoxicity assay using artemia-salina (Brine Shrimp). Planta. Med. 59, 250–252 (1993).
Mongelli, E., Martino, V., Coussio, J. & Ciccia, G. Screening of argentine medicinal plants using the brine shrimp microwell cytotoxicity assay. Int. J. Pharm. 34, 249–254 (1996).
Wilk, W., Waldmann, H. & Kaiser, M. γ-Pyrone natural products—a privileged compound class provided by nature. Bioorg. Med. Chem. 17, 2304–2309 (2009).
Tian, X. P. et al. Marinactinospora thermotolerans gen. nov., sp nov., a marine actinomycete isolated from a sediment in the northern South China Sea. Int. J. Syst. Evol. Microbiol. 59, 948–952 (2009).
Kanazawa, T. et al. Gamma-pyrones from Gonystylus keithii, as new inhibitors of parathyroid hormone (PTH)-induced Ca release from neonatal mouse calvaria. Chem. Pharm. Bull. (Tokyo) 45, 1046–1051 (1997).
Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63 (1983).
Miller, K. G., Liu, L. F. & Englund, P. T. A homogeneous type-II DNA topoisomerase from hela-cell nuclei. J. Biol. Chem. 256, 9334–9339 (1981).
Liu, M. H. et al. Study on the antitumor activity of Albaconol and its effect on DNA topoisomerase II. Chinese Pharmacological Bull. 20, 1224–1227 (2004).
Polycarpou-Schwarz, M. et al. Thanatop: a novel 5-nitrofuran that is a highly active, cell-permeable inhibitor of topoisomerase II. Cancer Res. 67, 4451–4458 (2007).
Acknowledgements
This work was financially supported by the National Basic Research Program of China (No. 2010CB833800), the Chinese National Natural Science Fund (No. 40906076, 40906075) and the Innovative Program of The Chinese Academy of Sciences (No. KSCX2-YW-G-065 and KSCX2-YW-G-073). The NMR experiments were performed by NMR laboratory of South China Sea Institute of Oceanology, Chinese Academy of Sciences. The antitumor assays were performed at the Department of Biochemistry and Molecular Biology, College of Basic Medical Sciences, Second Military Medical University, Shanghai, PRC.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, F., Tian, X., Huang, C. et al. Marinactinones A–C, new γ-pyrones from marine actinomycete Marinactinospora thermotolerans SCSIO 00606. J Antibiot 64, 189–192 (2011). https://doi.org/10.1038/ja.2010.153
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2010.153
Keywords
This article is cited by
-
Metabolites derived from fungi and bacteria suppress in vitro growth of Gnomoniopsis smithogilvyi, a major threat to the global chestnut industry
Metabolomics (2022)
-
Lobophorins E and F, new spirotetronate antibiotics from a South China Sea-derived Streptomyces sp. SCSIO 01127
The Journal of Antibiotics (2011)
