
Abstract
Pelizaeus-Merzbacher disease (PMD) is an X-linked recessive hypomyelination disorder caused by mutations in the proteolipid protein 1 gene (PLP1) located on chromosome Xq22. A male patient showed severe developmental delay, pendular nystagmus and laryngeal wheezing. The auditory brain stem response showed only the first wave and brain magnetic resonance imaging showed white matter hypomyelination, suggesting typical PMD. A novel PLP1 mutation, F240L, which was inherited from his mother, was identified.
Similar content being viewed by others

Pelizaeus-Merzbacher disease (PMD; MIM #312080) is a rare X-linked recessive hypomyelination disorder caused by mutations in the proteolipid protein 1 gene (PLP1) located on chromosome Xq22.1 PLP1 is highly conserved among vertebrates and encodes two major compact myelin proteins, PLP1 and its splicing variant DM20.2 More than 100 mutations have been reported in PMD, including duplications, point mutations, insertions and deletions. Chromosomal Xq22 microduplications involving PLP1 are the most frequent mutations, which have been detected in ~60–70% of PMD patients. Point mutations, including missense, nonsense and splicing, have been detected in 10–25% of patients and deleterious mutations are rare.3
Most patients with typical PMD manifest pendular nystagmus in early infancy as well as generalized hypotonia that later changes into spasticity, tremor and motor developmental delay.4 According to the severity of clinical manifestations, PMD is classified into subgroups, including connatal, classical, X-linked spastic paraplegia and a PLP1 null syndrome. These subgroups correspond to the different types of PLP1 mutations, namely missense mutations, chromosomal Xq22 microduplications, missense mutations in the specific region in exon 4 and whole-gene deletions, respectively.2,5 Here we report the case of a patient with PMD with a novel PLP1 mutation.
A 3-year-old boy was born at 38 weeks of gestation with a birth weight of 2774 g. This boy is the only child of healthy and non-consanguineous parents. Family history is unremarkable. Soon after birth, automated auditory brainstem response hearing screening detected abnormalities with no detection of waves after the first wave. At that time, pendular nystagmus was also noted. Because he had feeding difficulties due to laryngeal wheezing, tube feeding was initiated. Brain magnetic resonance imaging at 6 months of age revealed a hypomyelination pattern (Figure 1). The patient exhibited severe developmental delay with no head control or meaningful language development. From these findings, connatal type PMD was suggested as a candidate diagnosis.

Brain magnetic resonance imaging at 6 months of age. (a) T1-weighted sagittal image. There are no abnormalities in the corpus callosum or cerebellum. (b) T2-weighted axial image shows high intensity in the white matter, indicating hypomyelination.
This study was performed in accordance with the Declaration of Helsinki and was approved by the ethics committee of Tokyo Women’s Medical University. After receiving written informed consent, we obtained blood samples from the patient and his mother for genomic DNA extraction. Because PLP1 duplications are predominantly observed in PMD patients, chromosomal microarray testing was first performed as described previously,1 but PLP1 duplication was not observed. Sanger sequencing for all PLP1 exons was then performed as described in a previous report.6
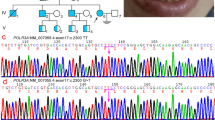
Sanger sequencing of the patient’s sample revealed a nucleotide alteration (c.718T>C) in exon 6 of PLP1, resulting in a protein change, p.Phe240Leu (F240L). The mother was heterozygous for this mutation, indicating that this mutation was maternally inherited (Figure 2). The prediction scores of this mutation were evaluated using wANNOVAR (http://wannovar.usc.edu/); the results were: SIFT_score=0 (Damaging), Polyphen2_HDIV_score=0.979 (Damaging), Polyphen2_HVAR_score=0.982 (Damaging), MutationTaster_score=1 (Damaging) and CADD_phred=34. From these findings, c.718T>C (p.Phe240Leu) was considered to be the cause of disease.

Sanger sequencing results. (a) Hemizygous and heterozygous mutations of c.718T>C (p.Phe240Leu [F240L]) are shown in the patient and his mother, respectively. (b) The amino-acid sequences of the different species are shown, indicating conservation among species.
Previously reported nucleotide alterations in coding regions are summarized in Supplementary Table S1. In total, 112 mutations had been reported. Since the c.718T>C (p.Phe240Leu) mutation identified in this study has not been reported previously, we determined it to be a novel mutation.
Previous studies have shown that PLP1 is important for the central nervous system because it maintains and stabilizes the lamellar structure of oligodendrocytes.7,8 PLP1 is a membrane protein with four alpha-helix transmembrane (TM) domains that can mediate its conformational structure through sequence-specific interactions between the TM alpha-helices of the protein monomers.9
As mentioned above, 112 nucleotide alterations have been reported. Among them, 48 mutations are located in the TM domains. As previously suggested, most of the missense mutations in the TM domains were related to the very severe connatal type PMD.9,10 The mutation identified in this study altered the amino acid phenylalanine (Phe) to leucine (Leu) in TM4. A similar Phe-to-Leu mutation in TM1 (p.Phe32Leu) was previously reported,5 and the patient exhibited a very severe connatal type similar to the patient reported in this study.
Amino acid changes in the TM domain may lead to changes in protein conformational structure or result in an unfolded protein, affecting TM transport of the protein and resulting in accumulation of PLP1 in the endoplasmic reticulum.2 Previously, we identified the following four missense substitutions located within the TM domain: p.Gly83Arg, p.Leu85Pro, p.Ala243Val and p.Ala247Thr.6,11,12 All four mutations were related to connatal type PMD.
In conclusion, we identified a novel PLP1 mutation, c.718T>C (p.Phe240Leu), in a patient with connatal type PMD. This novel mutation on the fourth alpha-helix in the TM domain supports the genotype–phenotype correlation in PMD that relates missense mutations in TM domains to severe clinical manifestations.
References
References
Yamamoto T, Shimojima K . Pelizaeus-Merzbacher disease as a chromosomal disorder. Congenit Anom 2013; 53: 3–8.
Inoue K . PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics 2005; 6: 1–16.
Woodward KJ . The molecular and cellular defects underlying Pelizaeus-Merzbacher disease. Expert Rev Mol Med 2008; 10: e14.
Koeppen AH, Robitaille Y . Pelizaeus-Merzbacher disease. J Neuropathol Exp Neurol 2002; 61: 747–759.
Cailloux F, Gauthier-Barichard F, Mimault C, Isabelle V, Courtois V, Giraud G et al. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Clinical European Network on Brain Dysmyelinating Disease. Eur J Hum Genet 2000; 8: 837–845.
Shimojima K, Inoue T, Hoshino A, Kakiuchi S, Watanabe Y, Sasaki M et al. Comprehensive genetic analyses of PLP1 in patients with Pelizaeus-Merzbacher disease applied by array-CGH and fiber-FISH analyses identified new mutations and variable sizes of duplications. Brain Dev 2010; 32: 171–179.
Gudz TI, Schneider TE, Haas TA, Macklin WB . Myelin proteolipid protein forms a complex with integrins and may participate in integrin receptor signaling in oligodendrocytes. J Neurosci 2002; 22: 7398–7407.
Harlow DE, Saul KE, Komuro H, Macklin WB . Myelin proteolipid protein complexes with alphav integrin and AMPA receptors in vivo and regulates AMPA-dependent oligodendrocyte progenitor cell migration through the modulation of cell-surface GluR2 expression. J Neurosci 2015; 35: 12018–12032.
Dhaunchak AS, Colman DR, Nave KA . Misalignment of PLP/DM20 transmembrane domains determines protein misfolding in Pelizaeus-Merzbacher disease. J Neurosci 2011; 31: 14961–14971.
Garbern JY . Pelizaeus-Merzbacher disease: genetic and cellular pathogenesis. Cell Mol Life Sci 2007; 64: 50–65.
Yamamoto T, Nanba E . A novel mutation (A246T) in exon 6 of the proteolipid protein gene associated with connatal Pelizaeus-Merzbacher disease. Hum Mutat 1999; 14: 182.
Yamamoto T, Nanba E, Zhang H, Sasaki M, Komaki H, Takeshita K . Jimpy(msd) mouse mutation and connatal Pelizaeus-Merzbacher disease. Am J Med Genet 1998; 75: 439–440.
Data Citations
Yamamoto, Toshiyuki HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.920
Acknowledgements
We express our gratitude to the patient and his family for their cooperation. This work was supported by Japan China Sasakawa Medical Fellowship (YL). This study was partially supported by the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development (AMED), a Grant-in-Aid for Scientific Research from Health Labor Sciences Research Grants from the Ministry of Health, Labor and Welfare, Japan, and JSPS KAKENHI Grant Number 15K09631 (TY).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Lu, Y., Shimojima, K., Sakuma, T. et al. A novel PLP1 mutation F240L identified in a patient with connatal type Pelizaeus-Merzbacher disease. Hum Genome Var 4, 16044 (2017). https://doi.org/10.1038/hgv.2016.44
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2016.44
This article is cited by
-
Novel mutations in the GJC2 gene associated with Pelizaeus–Merzbacher-like disease
Molecular Biology Reports (2019)
