
Abstract
We describe a new early-onset neuromuscular disorder due to a homozygous loss-of-function variant in the kyphoscoliosis peptidase gene (KY). A 7.5-year-old girl with walking difficulties from 2 years of age presented with generalized muscle weakness; mild contractures in the shoulders, hips and feet; cavus feet; and lordosis but no scoliosis. She had previously been operated with Achilles tendon elongation. Whole-body MRI showed atrophy and fatty infiltration in the calf muscles. Biopsy of the vastus lateralis muscle showed variability in fiber size, with some internalized nuclei and numerous very small fibers with variable expression of developmental myosin heavy chain isoforms. Some small fibers showed abnormal sarcomeres with thickened Z-discs and small nemaline rods. Whole-exome sequencing revealed a homozygous one-base deletion (c.1071delG, p.(Thr358Leufs*3)) in KY, predicted to result in a truncated protein. Analysis of an RNA panel showed that KY is predominantly expressed in skeletal muscle in humans. A recessive variant in the murine ortholog Ky was previously described in a spontaneously generated mouse mutant with kyphoscoliosis, which developed postnatally and was caused by dystrophy of postural muscles. The abnormal distribution of Xin and Ky-binding partner filamin C in the muscle fibers of our patient was highly similar to their altered localization in ky/ky mouse muscle fibers. We describe the first human case of disease associated with KY inactivation. As in the mouse model, the affected child showed a neuromuscular disorder – but in contrast, no kyphoscoliosis.
Similar content being viewed by others


Introduction
A neuromuscular disorder caused by a homozygous variant in the kyphoscoliosis peptidase gene (Ky) was first described in a mouse model, which was a spontaneous mutant of the BDL strain.1 In this model, kyphoscoliosis develops due to atrophy of postural muscles during postnatal growth.2, 3 The muscle phenotype of the ky/ky was morphologically characterized by muscle fiber regeneration, necrosis, and internally located nuclei in the soleus, gracilis, paraspinal, and back muscles.3 The Ky locus was mapped to a small region of the distal part of the mouse chromosome 9 that is homologous to human chromosome 3q21.4 The causative variant, a GC deletion in Ky resulting in frameshift and a premature stop codon, was identified by a positional cloning strategy.5 The ky transcript codes for a muscle-specific protein and is only expressed in skeletal and cardiac muscle in mice.5 The KY protein belongs to a family of transglutaminase-like proteins.5, 6 However, recent data indicate that KY should be considered a Z-disc-associated protein that provides structural support.7
Here we report the first human case of a neuromuscular disease associated with a variant in KY.
Patients and methods
Muscle pathology
Open muscle biopsy of the quadriceps femoris muscle, vastus lateralis, was performed at the age of 7.5 years.
Specimens were frozen in propane chilled with liquid nitrogen for histochemical analysis or fixed in glutaraldehyde for electron microscopy. Standard techniques were used for histochemical staining of cryostat sections and for electron microscopy. Immunohistochemical staining of embryonic and fetal myosin heavy chains (DSHB F1.652 and Leica NCL MHCn) was performed as previously described.8
For immunofluorescence microscopy, cryostat sections were fixed, stained and mounted mainly as described.9 Antibodies used in this study are (1) mouse IgG1 monoclonal antibody XR1 against Xin isoforms A and B,10 dilution 1:10; (2) anti-filamin C mouse IgA monoclonal antibody RR90,11 dilution 1:10; and (3) rabbit polyclonal antiserum specific for desmin, dilution 1:100 (Cat. No. 10570; Progen, Heidelberg, Germany). FITC (fluorescein isothiocyanate)-, Cy3- and Cy5-conjugated secondary antibodies were obtained from Southern Biotech (Birmingham, AL, USA) and Jackson Immunoresearch Laboratories (West Grove, PA, USA). Images were acquired using an Axio Imager M1 microscope equipped with epifluorescent illumination or a LSM710 confocal laser scanning microscope (both Carl Zeiss GmbH, Oberkochen, Germany).
Genetic analysis
For whole-exome sequencing, target enrichment was performed with 3 μg of genomic DNA using the Sure SelectXT Human All Exon kit version 5 (Agilent Technologies, Santa Clara, CA, USA) and sequenced on the HiSeq2500 platform (Illumina, San Diego, CA, USA) as paired-end 100-bp reads with 60 × coverage. Base calling was performed with the Illumina pipeline, with default parameters. Sequence reads were aligned to the reference genome (hg19) with the DNAnexus software (DNAnexus Inc., Mountain View, CA, USA; www.dnanexus.com). Filtering of called variants was performed in several steps using Ingenuity Variant Analysis including genes listed in the Neuromuscular Disorder (NMD) Gene Table (http://www.musclegenetable.fr/). Further filtering was carried out to predict variants that affect function using SIFT and PolyPhen2, the level of conservation and how common the variants are in the population (using 1000 Genome (http://www.1000genomes.org/); NHLBI Exome Sequencing Project (ESP) (http://evs.gs.washington.edu/EVS/) and The Exome Aggregation Consortium (ExAC)) were investigated to reduce the number of variants. Confirmation of variants and analysis of parental and siblings samples were done by Sanger sequencing.
The tissue expression patterns of KY (NM_178554.4) and the β-actin gene (NM_001101.3) as a control were performed with reverse transcriptase polymerase chain reaction (RT-PCR) using cDNA from the mRNA extracted from the patients skeletal muscle and a human RNA panel with 20 normal tissues (Human Total RNA Master Panel II; Clontech, Mountain View, CA, USA).
Results
Clinical features
This girl was the second of three children with healthy, related Kurdish parents. Her siblings were healthy (Figure 1a). Pregnancy was normal and she was born at term, after an uneventful delivery with a birth weight of around 3.5 kg. Early developmental milestones were normal. She sat unsupported at 8 months of age and walked unsupported at around 1 year of age. Walking became increasingly difficult from 3 years of age, because of progressive equinus contractures of the feet, and when the girl came to Sweden as a refugee at 5.5 years of age she was wheelchair-dependent. At 6.5 years of age, bilateral Achilles tendon elongation surgery was performed with improvement in her walking abilities. There has been no progression of symptoms from 6.5 to 7.5 years of age.

Molecular genetics. (a) Pedigree of the family. Dots indicates confirmed carriers. (b) Chromatogram demonstrating the c.1071delG variant in KY in one of the heterozygous parents. The patient was homozygous. (c) Tissue expression of KY using a human RNA panel screened by RT-PCR, demonstrating that KY is mainly expressed in skeletal muscle.
Clinical examination at 7.5 years of age (Figure 2a) revealed normal eye movements and facial expressions. She walked unsupported with slightly increased lumbar lordosis and somewhat increased knee lifting bilaterally. She was not able to walk on her heels but could walk on her toes and run. The gait and run were slightly asymmetric due to a more pronounced involvement of the lower leg on the left side compared with the right side. She could skip single steps on one leg, lift her head while lying on her back, sit up without trick movements and rise to standing without the Gower’s sign.

Patient image and whole-body magnetic resonance imaging (MRI) at 3.0T at 7.5 years of age. (a) Demonstrating visible muscle atrophy in the lower legs. (b–f) Demonstrating that visible muscle atrophy and fat replacement was restricted to the calf muscles with predominant changes on the left side involving the gastrocnemius and soleus muscles, whereas the tibialis anterior and peroneus longus muscles were relatively spared. On the right side, the medial head of the gastrocnemius muscle was predominantly affected. A mild involvement of the vastus intermedius (c, d) and vastus lateralis (d) was also seen. Pasted image, T1, Fast-spin echo (TR/TE=500/8.4 ms). (c–f) LAVA-FLEX, fat-only phase, (TR/TE=8.4/1.1 and 2.3 ms). Panels c and d are from different levels of the thighs, while e and f are from different levels of the lower legs.
There were associated contractures, most pronounced in the lateral rotation of the shoulders (−45o/−45o), elbow supination (−10o/−5o), hip extension (−10o/−10o), plantar flexion (−20o/−20o) and dorsiflexion (−15o/−15o) of the feet. She also had cavus positioning of especially the left foot but no scoliosis. The lumbar lordosis was flexible and compensatory to a decreased hip extension. Muscle strength measurements showed a generalized mild weakness, between 3 and 4+ according to the MRC scale in axial, proximal and distal muscles – except for the lower legs, where she scored 2 and 3− for dorsal flexion in the right and left foot, respectively. Especially on the left side, the peroneal muscles seemed more affected than the anterior tibial muscle with varus positioning of the foot. The reflexes were weak in the brachioradialis and patellar tendons, and absent in the Achilles, biceps and triceps tendons. According to the Hammersmith functional motor scale, she scored 34 out of 40 points with deduction for standing on the heels (0), standing on one leg (1) and skipping on the left leg (1). Cognitive and fine motor skills were normal as well as respiratory and heart function. She was incontinent for urine and stool and needed diaper until her orthopedic operation. The incontinence for stool then disappeared while she still has occasional nocturnal enuresis. Whole-body MRI showed muscle atrophy and fat replacement of the calf muscles with predominant changes on the left side involving the gastrocnemius and soleus muscles, whereas the tibialis anterior and peroneus longus muscles appeared to be spared (Figures 2b–f). MRI of the brain and spine was normal. Serum creatine kinase levels and metabolic screening investigations were normal. Nerve conduction velocities were normal in the medianus, peroneus and suralis nerves. Needle electromyography (EMG) was normal in the biceps brachii and tibialis anterior muscles whereas there was an increased frequency of large motor unit potentials suggestive of neurogenic changes in the right gastrocnemius muscle but there was no spontaneous activity at rest.
Muscle pathology
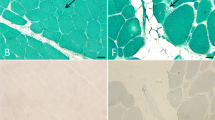
Biopsy of the vastus lateralis muscle at the age of 7.5 years revealed mild but distinctive, generalized changes. In addition to increased variability of fiber size affecting type 1 and type 2 fibers and internalized nuclei, there were numerous extremely small fibers (Figure 3a) that expressed variable myofibrillar ATPase activity, indicating expression of different myosin isoforms (Figure 3b). Immunohistochemical staining revealed that the majority of these small fibers expressed fetal (perinatal) myosin heavy chain (Figure 3c). Some (but not all) expressed embryonic (developmental) myosin heavy chain, indicating that some of these very small fibers were regenerating (Figure 3d). No inclusions or rods were identified in modified Gomori trichrome staining and the intermyofibrillar network appeared regular with NADH-TR staining. Fiber type grouping or type 1 fiber predominance was not present. There was a very mild focal increase in interstitial connective tissue. Electron microscopy showed abnormal sarcomeres in some of the small fibers with Z-line thickening and formation of small nemaline rods (Figures 3e and f). Large fibers showed regular sarcomers without apparent Z-line alterations.

Pathology of left vastus lateralis of the quadriceps femoris muscle. (a) Variability in muscle fiber size, occasional internalized nuclei and numerous very small fibers (arrows) (hematoxylin and eosin staining). (b) The numerous small fibers showed variable myofibrillar ATPase expression (arrows), ATPase, pH 4.3. (c) Immunohistochemistry revealed that most small fibers expressed fetal (perinatal) myosin heavy chain. (d) Immunohistochemistry revealed that some of the small fibers expressed embryonic (developmental) myosin heavy chain. (e, f) Some of the small fibers showed abnormal sarcomeres with thickened Z-discs (arrowheads) and rod structures (arrows).
Since the mutation in the Ky gene in the mouse is associated with an abnormal distribution of its binding partner FLNC and Xin,12, 13 which is a FLNC-binding partner10 and general muscle damage marker,14 we stained skeletal muscle sections of our patient for both proteins. Even though some parts of the sections appeared largely unaffected, in other areas inclusions containing the proteins were seen inside the fibers, with an incidence ranging from mild (fibers with mostly small inclusions) to severe (numerous fibers affected and very large inclusions) (Supplementary Figure S1). Detailed examination of strongly affected fibers using confocal microscopy showed that in these areas the two proteins colocalized whereas desmin was not enriched (Figure 4, left panel). Furthermore, examination of longitudinal sections showed that these areas were not amorphous protein aggregates as observed in the muscle fibers of myofibrillar myopathy patients (which would be desmin-positive), but their structure resembled sarcomeric remodeling associated with post-exercise muscle weakness.15, 16 As observed for myotilin and α-actinin in the ky/ky mice,12, 13 desmin was normally distributed in a cross-striated pattern.

Skeletal muscles of the patient show sarcomeric abnormalities similar to those found in ky/ky mice. Transverse (left panel) and longitudinal (right panel) sections through a severely affected area of the biopsy stained for FLNC, Xin and desmin. Note colocalization of FLNC and Xin but the absence of desmin enrichment in these areas. The longitudinal section clearly shows that these areas are not amorphous protein aggregates, but filamentous sarcomeric lesions. Scale bar: 10 μm.
Genetic analysis
Whole-exome sequencing showed that the patient had a homozygous one-base deletion in KY (c.1071delG, p.(Thr358Leufs*3)) (NM_178554.4), resulting in frameshift and premature termination (Figure 1b). The variant was not present in the Single Nucleotide Polymorphism Database (dbSNP), in the NHLBI Exome Variant Server, in the Exome Aggregation Consortium (ExAC) or in 1000 Genomes. The variant has been submitted to the Leiden Muscular Dystrophy pages (http://www.LOVD.nl/KY). Most called variants identified in genes listed in the NMD Gene Table after filtering were predicted to be likely benign or with uncertain significance according to computed ACMG Guidelines classification. No other variants that likely affects function were detected. Genetic analysis of the parents as well as the older sister revealed that they were heterozygous for the variant, whereas the younger brother had normal sequence compared with reference sequence (Figure 1b).
To examine the tissue expression of KY transcripts, we performed RT-PCR on a human tissue RNA panel. KY was most highly expressed in skeletal muscle and to a moderate level in cardiac and uterine muscles and brain. It was expressed at low or undetectable levels in other tissues (Figure 1c). The KY transcript with the c.1071delC variant was expressed in skeletal muscle, indicating that no alternative splicing and no nonsense-mediated mRNA decay had occurred. However, the variant induces a premature stop codon and a truncated non-functional protein can be predicted.
Discussion
We have described the first human case of a disease associated with inactivation of the kyphoscoliosis peptidase gene, KY. There are several lines of evidence that the homozygous c.1071delG, p.(Thr358Leufs*3) variant causes the disease by introducing a premature stop codon. First, the variant is clearly deleterious. Second, the gene is expressed in the affected tissue, skeletal muscle. Third, the variant segregates with the disease in the family. Fourth, the inactivation of the gene in a mouse model causes a similar myopathy. Fifth, an abnormal distribution of the Ky-binding protein FLNC, similar to what has been described in the ky/ky mouse, was shown.
The mouse Ky gene variant was a spontaneous event in the BDL mouse strain, resulting in kyphosis and rotational deformity of the spine, which was apparent from about 100 days of age.1 It was inherited as a recessive trait. The mutants have been extensively investigated as a possible model of idiopathic kyphoscoliosis in humans, but there was no evidence of a primary disease of the spine.2, 17 A neuromuscular cause for the deformities was found by systematic histological investigation of mutant mouse muscles at various ages. Necrosis, regeneration, and subsequent fibrosis and atrophy of muscles were identified from about 1 week of age, indicating a primary muscular dystrophy.3 However, different muscles were affected to different degrees and at various time points during the course. Early and severe involvement was evident in the soleus, gracilis, paraspinal and back muscles. The occurrence of large group atrophy and axonal sprouting was suggestive of a neurogenic disorder although there were no changes in peripheral nerves or in the spinal cord.3
The Ky gene was identified in 2001 by positional cloning, and a novel muscle-specific protein was identified.5 The KY protein harbors a transglutaminase/protease domain, which is homologous to other proteins in the transglutaminase family, and it has been proposed to be a protease.5, 6 However, multiple alignment with proteins belonging to the transglutaminase superfamily showed that a group of transglutaminase proteins including KY lack the enzymatic active site triad of amino acids, and the protease function has been questioned.13, 18 Futher studies indicates that KY should be considered to be a Z-disc-associated structural protein that interacts with sarcomeric partners such as filamin C (FLNC), myosin binding protein C, slow type (MYBPC1), KY interacting protein 1 (KYIP1, IGFN1) and titin (TTN).12, 13 More recent data have demonstrated that KY is part of a Z-band associated protein complex together with FLNC and IGFN1 likely to provide structural support to the sarcomeres in the skeletal muscle.7
Our immunolocalization studies showed an abnormal distribution of FLNC and Xin highly reminiscent of the localization patterns of both proteins in the muscle fibers of ky/ky mice.12, 13 This is a further indication that the KY variant is indeed causative for the disease phenotype of our patient. Both proteins were localized in sarcomeric lesions, which were suggested to contribute to (post-exercise) muscle weakness.15, 16, 19
Our patient had an autosomal recessive neuromuscular disease caused by a deleterious variant in the human ortholog of the mouse Ky gene. The patient developed early-onset walking difficulties because of foot drop and progressive equinus deformities of the feet. This was operated by Achilles tendon elongation, which allowed the patient to become ambulatory. Clinical examination revealed predominantly distal and asymmetric muscle weakness in the lower legs, affecting the left side more than the right one and there was also cavus feet. Weakness was most severe in the peroneal (MRC 2/2) and tibialis anterior (MRC 2/3−) muscles. In contrast, MRI investigations demonstrated atrophy and fat infiltration of the gastrocnemius and soleus muscles, whereas the anterior compartment was relatively spared. We do not know the reason for this peculiar distribution. While EMG did not demonstrate any changes in most examined muscles there was neurogenic pattern in the gastrocnemius muscle, which may indicate that there is a combined neurogenic and myogenic pathogenesis as was suggested in the ky/ky mouse.3 A partly neurogenic pathogenesis of the disease may be of importance for the development of cavus feet and a KY-associated neuromuscular disease would be important to consider in similar cases.
Clinical and muscle MRI investigations demonstrated predominantly distal leg involvement, but biopsy was performed in the more mildly affected vastus lateralis muscle. Unspecific myopathic pathological changes, without typical neurogenic alterations such as large group atrophy or fiber type grouping, were identified, and also an unusual pattern with large numbers of very small fibers with characteristics of various fiber types. Many were apparently early-regenerating fibers, but some fibers were probably atrophic or hypotrophic. Notably, some small fibers showed Z-disc alterations that were a characteristic finding in the ky/ky mouse.13
More than 20 different myopathies with predominantly distal involvement have been reported, most of which show onset in adulthood.20 Distal myopathies with childhood onset are rare. Laing early-onset myopathy (MPD1), caused by dominant variants in the slow/beta cardiac myosin heavy chain gene (MYH7), frequently presents as walking difficulties due to problems with ankle dorsiflexion in early childhood.21, 22 A dominantly inherited myopathy caused by a variant of the Kelch-like homolog 9 gene (KLHL9) was reported by Cirak et al.23 In this family, the muscle weakness started between the age of 8 and 16 years with weakness of ankle extensors and foot drop.
Our patient also had initial urine and stool incontinence which improved considerably after her orthopedic surgery. Her younger brother also sometimes has nocturnal enuresis so we believe that there could be other causes for her incontinence than her KY deficiency even though this is also possible considering that the KY protein is also expressed in smooth muscle.
No kyphosis or scoliosis had developed in our patient by the age of 7.5 years and, unlike the mouse model, there did not appear to be any major involvement of the paraspinal muscles. Finding of other individuals affected by KY inactivation and following them for a longer period will help to clarify the natural course of the disease and the phenotypic variability.
References
Dickinson AG, Meikle VM : Genetic kyphoscoliosis in mice. Lancet 1973; 1: 1186.
Mason RM, Palfrey AJ : Intervertebral disc degeneration in adult mice with hereditary kyphoscoliosis. J Orthop Res 1984; 2: 333–338.
Bridges LR, Coulton GR, Howard G, Moss J, Mason RM : The neuromuscular basis of hereditary kyphoscoliosis in the mouse. Muscle Nerve 1992; 15: 172–179.
Skynner MJ, Gangadharan U, Coulton GR et al: Genetic mapping of the mouse neuromuscular mutation kyphoscoliosis. Genomics 1995; 25: 207–213.
Blanco G, Coulton GR, Biggin A et al: The kyphoscoliosis (ky) mouse is deficient in hypertrophic responses and is caused by a mutation in a novel muscle-specific protein. Hum Mol Genet 2001; 10: 9–16.
Makarova KS, Aravind L, Koonin EV : A superfamily of archaeal, bacterial, and eukaryotic proteins homologous to animal transglutaminases. Protein Sci 1999; 8: 1714–1719.
Baker J, Riley G, Romero MR et al: Identification of a Z-band associated protein complex involving KY, FLNC and IGFN1. Exp Cell Res 2010; 316: 1856–1870.
Tajsharghi H, Kimber E, Kroksmark AK, Jerre R, Tulinius M, Oldfors A : Embryonic myosin heavy-chain mutations cause distal arthrogryposis and developmental myosin myopathy that persists postnatally. Arch Neurol 2008; 65: 1083–1090.
Chevessier F, Schuld J, Orfanos Z et al: Myofibrillar instability exacerbated by acute exercise in filaminopathy. Hum Mol Genet 2015; 24: 7207–7220.
van der Ven PF, Ehler E, Vakeel P et al: Unusual splicing events result in distinct Xin isoforms that associate differentially with filamin c and Mena/VASP. Exp Cell Res 2006; 312: 2154–2167.
van der Ven PFM, Obermann WMJ, Lemke B, Gautel M, Weber K, Fürst DO : Characterization of muscle filamin isoforms suggests a possible role of gamma-filamin/ABP-L in sarcomeric Z-disc formation. Cell Motil Cytoskeleton 2000; 45: 149–162.
Beatham J, Gehmlich K, van der Ven PFM et al: Constitutive upregulations of titin-based signalling proteins in KY deficient muscles. Neuromuscul Disord 2006; 16: 437–445.
Beatham J, Romero R, Townsend SK, Hacker T, van der Ven PFM, Blanco G : Filamin C interacts with the muscular dystrophy KY protein and is abnormally distributed in mouse KY deficient muscle fibres. Hum Mol Genet 2004; 13: 2863–2874.
Nilsson MI, Nissar AA, Al-Sajee D et al: Xin is a marker of skeletal muscle damage severity in myopathies. Am J Pathol 2013; 183: 1703–1709.
Carlsson L, Yu JG, Moza M, Carpén O, Thornell LE : Myotilin: a prominent marker of myofibrillar remodelling. Neuromuscul Disord 2007; 17: 61–68.
Fridén J : Muscle soreness after exercise: implications of morphological changes. Int J Sports Med 1984; 5: 57–66.
Venn G, Mason RM : Changes in mouse intervertebral-disc proteoglycan synthesis with age. Hereditary kyphoscoliosis is associated with elevated synthesis. Biochem J 1986; 234: 475–479.
Anantharaman V, Koonin EV, Aravind L : Peptide-N-glycanases and DNA repair proteins, Xp-C/Rad4, are, respectively, active and inactivated enzymes sharing a common transglutaminase fold. Hum Mol Genet 2001; 10: 1627–1630.
Orfanos Z, Gödderz MP, Soroka E et al: Breaking sarcomeres by in vitro exercise. Sci Rep 2016; 6: 19614.
Udd B : Distal myopathies – new genetic entities expand diagnostic challenge. Neuromuscul Disord 2012; 22: 5–12.
Darin N, Tajsharghi H, Ostman-Smith I, Gilljam T, Oldfors A : New skeletal myopathy and cardiomyopathy associated with a missense mutation in MYH7. Neurology 2007; 68: 2041–2042.
Lamont PJ, Wallefeld W, Hilton-Jones D et al: Novel mutations widen the phenotypic spectrum of slow skeletal/beta-cardiac myosin (MYH7) distal myopathy. Hum Mutat 2014; 35: 868–879.
Cirak S, von Deimling F, Sachdev S et al: Kelch-like homologue 9 mutation is associated with an early onset autosomal dominant distal myopathy. Brain 2010; 133: 2123–2135.
Acknowledgements
We thank Brith Leidvik and Janna Lundgren for technical assistance, Professor Rolf Schröder for valuable advice, and the patient and the family for their support. This study was supported by the Nilsson-Ehle Foundation, by the Research Fund for Neuromuscular Disorders in West Sweden and by the Swedish Research Council (project no. 07122).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Hedberg-Oldfors, C., Darin, N., Olsson Engman, M. et al. A new early-onset neuromuscular disorder associated with kyphoscoliosis peptidase (KY) deficiency. Eur J Hum Genet 24, 1771–1777 (2016). https://doi.org/10.1038/ejhg.2016.98
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.98
This article is cited by
-
Progressive hereditary spastic paraplegia caused by a homozygous KY mutation
European Journal of Human Genetics (2017)

{kind=link}