
Abstract
Structural brain malformations associated with Tuberous Sclerosis Complex (TSC) are related to the severity of the clinical symptoms and can be visualized by magnetic resonance imaging (MRI). Tuberous Sclerosis Complex is caused by inactivating TSC1 or TSC2 mutations. We investigated associations between TSC brain pathology and different inactivating TSC1 and TSC2 variants, and examined the potential prognostic value of subdivision of TSC2 variants based on their predicted effects on TSC2 expression. We performed genotype-phenotype associations of TSC-related brain pathology on a cohort of 64 children aged 1.4–17.9 years. Brain abnormalities were assessed using MRI. Individuals were grouped into those with an inactivating TSC1 variant and those with an inactivating TSC2 variant. The TSC2 group was subdivided into changes predicted to result in TSC2 protein expression (TSC2p) and changes predicted to prevent expression (TSC2x). The TSC2 group was associated with more and larger tubers, more radial migration lines, and more subependymal nodules than the TSC1 group. Subependymal nodules were also more likely to be calcified. Subdivision of the TSC2 group did not reveal additional, substantial differences, except for a larger number of tubers in the temporal lobe and a larger fraction of cystic tubers in the TSC2x subgroup. The severity of TSC-related brain pathology was related to the presence of an inactivating TSC2 variant. Although larger studies might find specific TSC2 variants that have prognostic value, in our cohort, subdivision of the TSC2 group did not lead to better prediction.
Similar content being viewed by others


Introduction
Tuberous Sclerosis Complex (TSC) is an autosomal dominant disorder caused by inactivating TSC1 or TSC2 variants.1, 2 Most TSC-associated lesions are thought to arise due to somatic second-hit mutations that inactivate the remaining wild-type TSC1 or TSC2 allele. The protein products of TSC1 and TSC2 form the TSC complex, that inhibits the mammalian Target of Rapamycin Complex 1 (mTORC1).3 Loss or inactivation of the TSC complex results in constitutive activation of mTORC1, and mTORC1 inhibitors have been shown to be useful for treating hamartoma-related complications of TSC.4, 5
Our aim was to investigate genotype-phenotype associations in a well-characterized cohort of TSC individuals, focusing on the relationships between specific TSC1 and TSC2 variants and macrostructural brain lesions detected by magnetic resonance imaging (MRI), including cortical tubers, radial migration lines (RMLs), subependymal nodules (SENs) and subependymal giant cell astrocytomas (SEGAs). In most studies, inactivating TSC2 variants are associated with increased numbers of cortical tubers and a higher prevalence of SEGAs.6, 7, 8, 9, 10, 11, 12, 13, 14 We investigated whether there was additional clinical value for subdivision of TSC2 variants, as has been described recently for cognitive function in TSC.15 We compared TSC-related brain pathology as assessed by MRI, in individuals with an inactivating TSC1 variant to brain pathology in individuals with an inactivating TSC2 variant. In addition, we compared the TSC1 group to individuals with a TSC2 variant predicted to prevent TSC2 mRNA expression (TSC2x) and to individuals with a TSC2 variant predicted to either alter the TSC2 amino acid sequence or result in reduced TSC2 expression (TSC2p).
Methods
Patients
Children treated at the ENCORE-TSC Expertise Center of the Erasmus MC-Sophia Children’s Hospital, Rotterdam, the Netherlands with a genetically confirmed TSC diagnosis and at least one brain MRI were eligible for inclusion. Inactivating TSC1 or TSC2 variants were identified in 108 individuals, of whom 101 had at least one MRI available. In 64 cases the quality of the MRI was suitable for analysis, based on the criteria described below.
Genetic analysis and functional assessment
Molecular testing was performed at the Department of Clinical Genetics of the Erasmus MC. All identified variants were assessed with ALAMUT mutation prediction software (version 2.6.1 (January 2015); Interactive Biosoftware, Rouen, France). Exons were numbered according to genomic reference sequences NG_012386.1 (TSC1) and NG_005895.1 (TSC2); cDNA notation was according to transcript reference sequences NM_000368.4 (TSC1) and NM_000548.3 (TSC2).
Functional assessment was performed as described.16 For the analysis of TSC2 variants, HEK 293T cells in which exons 2–38 of TSC2 had been deleted by CRISPR/Cas9 genome editing17 were used. Briefly, guide oligos 5′-caccgacggagtttatcatcaccg-3′ and 5′-aaaccggtgatgataaactccgtc-3′ (exon 2), and 5′-caccggttatcgccacgcaccact-3′ and 5′-aaacagtggtgcgtggcgataacc-3′(intron 38) were cloned into the pX458 and pX459 vectors,18 and transfected into HEK 293T cells. Following puromycin selection, GFP-positive cells were single-cell sorted and grown in 96-well plates. The resultant colonies were trypsinised, expanded and validated by PCR, sequencing and immunoblotting. A single subclone, 3H9, was used for subsequent functional assessments.
For the detection of mosaic individuals, targeted Next Generation Sequencing of the TSC1 and TSC2 loci was performed, as described previously.19 Clinical, genetic and functional data from this study have been submitted to the TSC1 and TSC2 Leiden Open Variant Databases (LOVD) (http://www.lovd.nl/TSC2; http://www.lovd.nl/TSC1).
Magnetic resonance imaging
Brain MRIs were made at the Erasmus MC-Sophia Children’s Hospital on a 1.5 Tesla General Electric scanner using a standard protocol of axial and coronal T1, T2 and fluid-attenuated inversion recovery sequences. To achieve an as uniform as possible sample, the MRI made closest to 8 years of age was selected. MRIs from individuals less than 12 months of age were not used because, at that age, myelination has not progressed enough to be able to measure tuber size and detect RML reliably. MRIs were excluded if there were movement artifacts, if axial images were absent, or when secondary structural abnormalities not directly related to TSC were present.
All MRIs were assessed by two trained medical students, and re-assessed by a pediatric neuroradiologist and a pediatric neurologist, who were blinded to the genotype and clinical characteristics of the patient. Picture Archiving and Communication System software was used for all assessments.
The numbers and locations of all TSC-related brain abnormalities were assessed and verified in all available MRI sequences. For each tuber, the largest axes parallel and perpendicular to the gyrus were measured on axial slices, and multiplied to obtain an estimate of the maximum cross-sectional area. All lesions were inspected for cystic changes or calcifications on T2, fluid-attenuated inversion recovery and, if available, susceptibility weighted angiography sequences.
Statistical analysis
Univariate regression analysis was used to compare continuous outcomes. A Student’s t-test was used for comparing two groups of continuous data, and a chi-square test was used for categorical data. For comparisons between multiple groups, an analysis of variance test with a Bonferroni post-hoc correction was used for continuous data, and a chi-square test was used for categorical data. To correct for multiple testing, a false discovery rate test was used. All the outcomes of the statistical testing are included in Supplementary Table 3; q values are given in the text where the corresponding P value was no longer significant after correction for multiple testing.
Results
Patient population and genetic variant subdivision
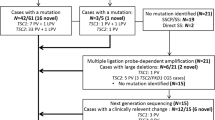
In total, 64 patients aged 1.4–17.9 years were included (Table 1); 21 (33%) had an inactivating TSC1 variant and 43 (67%) had an inactivating TSC2 variant (Figure 1, Table 1 and Supplementary Tables 1 and 2). We defined inactivating variants as those that were predicted to either prevent mRNA expression, truncate the open reading frame prematurely, or affect TSC complex function. We divided the variants into three groups: TSC1, TSC2x and TSC2p. The TSC1 group consisted of 21 individuals with 20 different TSC1 variants, including eight predicted frameshift variants, eight predicted nonsense variants, two large deletions, one predicted missense variant and one substitution predicted to affect splicing. The TSC2x group consisted of 26 variants that were predicted to either prevent TSC2 mRNA expression, or render the TSC2 mRNA subject to nonsense mediated decay (NMD). This group included seven frameshift and eight nonsense variants, four large deletions and seven variants predicted to affect splicing. The TSC2p group consisted of variants that were predicted to alter the TSC2 amino acid sequence or to result in reduced levels of functional TSC2 mRNA. We defined functional mRNA as not subject to NMD and encoding the TSC2 GAP domain (amino acids 1616–1654).1 Nonsense and frameshift variants in the last exon and the last 18 codons of the penultimate exon were presumed to escape NMD.20 The TSC2p group consisted of 13 different variants in 16 individuals, and included 7 missense variants (1 variant in 2 individuals), an in-frame deletion, a nonsense and a frameshift variant both predicted to escape NMD, and 2 variants (1 variant in 3 individuals) that were predicted to affect splicing, but might still result in expression of functional TSC2 mRNA.

Schematic overview of TSC1 and TSC2 variants. Exon numbering is according to genomic reference sequences NG_012386.1 (TSC1) and NG_005895.1 (TSC2) (build GRCh37 (hg19) of the human reference sequence); cDNA numbering is according to reference transcripts NM_000368.4 (TSC1) and NM_000548.3 (TSC2). (a) TSC1. Approximate positions of the TSC1 variants identified in our TSC cohort are indicated relative to exons 1–23. Large deletions are boxed with the approximate position of the distal extent of the deletion, relative to the exons, indicated. Non-coding 5′ and 3′ untranslated regions (UTR) are shaded in gray; the 3′ UTR in exon 23 is not drawn to scale. (b) TSC2. Approximate positions of the TSC2 variants identified in our TSC patient cohort are indicated relative to exons 2–42. Large deletions are boxed and the approximate positions of the distal extent of the deletions are indicated. The region encoding the TSC2 GAP domain (amino acids 1616–1654) is shaded gray. All variants predicted to result in the absence of TSC2 (TSC2x) are shown above the exons; variants for which expression of a mutant form of TSC2 (TSC2p) was considered possible are shown below the exons (see Supplementary Tables and text for details). TSC, Tuberous Sclerosis Complex.
To investigate the effects of TSC1 and TSC2 variants on the TSC complex-dependent inhibition of mTORC1, we expressed the variant proteins together with an S6K reporter construct and determined the T389 phosphorylation status of the S6K reporter (Figures 2 and 3). First, we compared the effect of the TSC1 c.562T>G p.(F188V) substitution to the inactivating TSC1 c.350T>C p.(L117P) variant16 (Figure 2). Compared to wild-type TSC1, expression of the p.F188V and p.L117P variants resulted in reduced TSC1 signals and increased S6K-T389 phosphorylation. Next, we assessed the effects of 10 TSC2 variants on TSC complex function (Figure 3). In nine cases, expression of the variant failed to inhibit S6K-T389 phosphorylation. We did not observe significant differences in S6K-T389 phosphorylation between cells completely lacking TSC2, and those expressing the TSC2 variants, indicating that in our in vitro assay, the variants resulted in complete inactivation of the TSC complex. The TSC2 p.L160V variant had the same effect on S6K-T389 phosphorylation as wild-type TSC2. We did not obtain evidence that the p.L160V substitution affected TSC complex function. However, splice site prediction analysis indicated that the TSC2 c.478C>G, p.(L160V) substitution created a new 5′ splice donor site 4 nucleotides upstream of the normal splice site (TSC2 c.478C>G, p.(A161Tfs*20)). The splicing defect was confirmed by RT-PCR and sequence analysis of RNA from cultured skin fibroblasts (Supplementary Figure 2). There was no evidence that the original splice site was utilized in mRNA expressed from the variant (G) allele, indicating that the predicted TSC2 p.L160V protein was unlikely to be expressed. Therefore, we classified the TSC2 c.478C>G (p.A161Tfs*20) variant as TSC2x.

Functional assessment of the TSC1 c.562T>G p.(F188V) variant. We compared the effects of expression of the TSC1 p.F188V variant with wild-type TSC1 and the TSC1 p.L117P variant using a transfection-based immunoblot assay. Immunoblots are shown in (a); please note that for simplicity some lanes have been removed from the blot. The original, complete blots are shown in Supplementary Figure 1. Signals for TSC2, TSC1, total S6K (S6K) and T389-phosphorylated S6K (T389) were determined per variant, relative to the wild-type control (TSC1) in four transfection experiments. The mean TSC2 (b), TSC1 (c) and S6K (e) signals and mean T389/S6K ratio (d) are shown for each variant. The dotted lines indicate the signal obtained upon expressing wild-type TSC1 (=1.0). Error bars represent the standard error of the mean; variants that were significantly different from the wild-type are indicated with an asterisk (P<0.05; Student’s t-test). In TSC2/S6K no TSC1 protein is present. Amino acid changes are given according to the TSC1 reference transcript NM_000368.4. TSC, Tuberous Sclerosis Complex. Variants that were significantly different from the wild-type are indicated with an asterisk.

Functional assessment of TSC2 variants. We compared the effects of expression of wild-type TSC2 with 10 different TSC2 variants in HEK 293T (TSC2−/−; 3H9) cells using a transfection-based immunoblot assay. All the variants were identified in our patient cohort with the exception of the p.L1750fs variant. This variant is similar to the TSC2 c.5252_5259+19del27, p.(R1751Hfs*41) variant identified in our cohort. In both cases, the variant mRNA transcript is predicted to escape NMD, and the C-terminal epitope used for TSC2 protein detection is absent. Immunoblots are shown in (a). The signals for TSC2, TSC1, total S6K (S6K) and T389-phosphorylated S6K (T389) were determined per variant, relative to the wild-type control (TSC2) in four transfection experiments. The mean TSC2 (b), TSC1 (c) and S6K (d), signals and mean T389/S6K ratio (e) are shown for each variant. The dotted lines indicate the signal/ratio for TSC2 (=1.0). Error bars represent the standard error of the mean; variants that were significantly different from TSC2 are indicated with an asterisk (P<0.05; Student’s t-test). Cells were cotransfected with TSC1 and S6K expression constructs, except for the mock transfected cells (pcDNA3 only). TSC1/S6K refers to cells transfected with the TSC1 and S6K expression constructs only; RHEB refers to cells transfected with an RHEB expression construct. Amino acid changes are given according to the TSC2 reference transcript NM_000548.3. Variants that were significantly different from the wild-type are indicated with an asterisk.
Cortical tubers
Cortical tubers were detected in 62 patients (97%); 19/21 (90%) from the TSC1 group, and all individuals from the TSC2 group (Table 1 and Supplementary Figure 3). Tubers were most often found in the frontal lobe, consistent with this lobe having the largest volume (Table 1). Cystic tubers were present in 23 patients (36%; range 1–18 cystic tubers per patient; median: 6). Calcified tubers were present in 11 patients (17%; range: 1–17; median: 3). Representative MRIs of cystic and calcified tubers are shown in Figures 4a and b.

Examples of TSC-specific brain abnormalities assessed in this study. (a) T1 sequence showing cystic cortical tubers (arrows). (b) T2 sequence showing a calcified cortical tuber (arrow). (c) T2 sequence showing an RML in the left frontal lobe (arrow). (d) T2 dual echo sequence showing a calcified SEN (arrow). Note the SENs without calcification in the other ventricle (arrowheads). SEN, subependymal nodule; TSC, Tuberous Sclerosis Complex.
More tubers in total (P<0.001) and per lobe (P<0.001 for all lobes) were found in the TSC2 group compared to the TSC1 group. The TSC2 group also had a larger total tuber surface area (P<0.001) but no difference was found when the percentage of tubers in each lobe was compared between these two groups. Individuals in the TSC2 group were more likely to have cystic tubers (P=0.012), and the fraction of cystic tubers was higher in the TSC2 group (P=0.017). Analysis of the TSC1, TSC2p and TSC2x groups showed similar results. The total number of tubers, number of tubers per lobe and tuber surface area were higher in the TSC2p and TSC2x groups compared to the TSC1 group (p≤0.001). The TSC2x group had more temporal lobe tubers than either the TSC2p or TSC1 group (P<0.001). Cystic tubers were found most often in the TSC2x group (P=0.003), and the fraction of cystic tubers was higher in the TSC2x group compared to the TSC1 group (P=0.006).
Radial migration lines
RMLs were present in 62 patients (97%). An example of an RML on MRI is shown in Figure 4c. All individuals in the TSC1 group, 13/16 (81%) from the TSC2p group, and 23/27 (85%) from the TSC2x group had RMLs. Roughly half of all RMLs could be traced to a tuber (Table 1 and Supplementary Figure 3). Cystic RMLs were found in nine patients (14%; range: 1–9; median: 1). Calcified RMLs were present in 12 patients (19%; range: 1–8; median: 2). In two cases RMLs, but no tubers, were found. Both these individuals were from the TSC1 group.
The total number of RMLs was significantly higher in the TSC2 group than in the TSC1 group, although this was no longer significant after correcting for multiple testing (P=0.028, q=0.071). No additional significant differences between the number of RMLs, or their cystic or calcified aspect were identified in the analysis of the TSC1, TSC2p and TSC2x groups.
Subependymal nodules
SENs were identified in 54 patients (84%). In the TSC1 group, 18 individuals (86%) had SENs. In the TSC2p and TSC2x group, 13 (81%) and 23 (85%) individuals respectively had SENs (Table 1 and Supplementary Figure 3). Calcified SENs were present in 22 patients (34%; range: 1–15; median: 3). An MRI of a calcified SEN is shown in Figure 4d. Details on the location of the SENs can be found in Table 1.
The TSC2 group had a higher number of SENs (P=0.009), and these were more often calcified (P=0.015) compared to the TSC1 group. No differences were found in the number and calcification of SENs in the analysis of the TSC1, TSC2p and TSC2x groups.
Subependymal giant cell astrocytoma
An SEGA was identified in seven individuals from the TSC2 group (11%) (Table 1); in 2 (13%) from the TSC2p group and 5 (19%) from the TSC2x group. No significant differences were identified.
Patients with genetic mosaicism
Two individuals from the TSC2 group were mosaic.14 The TSC2 c.2838-122G>A, p.? and TSC2 c.3099C>G, p.(Y1033*) variants were found at a frequency of 11 and 10% respectively in peripheral blood DNA. Both individuals had bilateral tubers, RMLs and SENs, none of which were cystic or calcified.
Discussion
Brain pathology as assessed by MRI was compared between TSC patients with (i) a TSC1 variant that affected function, (ii) a TSC2 variant that affected function but was predicted to encode protein (TSC2p) and (iii) a TSC2 variant that was predicted to prevent TSC2 protein expression (TSC2x). The added value of the results from these analyses was determined compared to analyses between the TSC1 group and the whole TSC2 group. Our results are consistent with previous studies6, 7, 8, 9, 10, 11, 12, 13, 14, 21: the TSC2 group was associated with more and larger tubers, more RMLs, more SEGAs and more SENs. Subdivision of the TSC2 group into TSC2p and TSC2x subgroups did not reveal major differences in TSC-pathology, as detected by MRI, although a higher number and fraction of tubers in the temporal lobe and a higher fraction of cystic tubers in the TSC2x group were observed.
Although the larger numbers of cystic tubers and tubers in the temporal lobe in the TSC2x group might simply be due to chance, it might be clinically relevant. Patients with more temporal tubers have a higher risk of developing autistic features21 and cystic tubers have been associated with a higher incidence of epilepsy22 and autism spectrum disorder.23 The larger number of calcified SENs in the TSC2 group could also be clinically relevant, as calcified SENs are more likely to develop into a SEGA.24
Two patients in our cohort were mosaic. Both had bilateral TSC-related abnormalities. It would be interesting to study genotype-phenotype associations in a larger cohort of mosaic TSC patients, to determine whether these individuals are more likely to have specific types of pathology, as has been suggested previously.25, 26, 27, 28
Our cohort consisted of 40 individuals with a de novo mutation (12 TSC1, 28 TSC2), 12 individuals from 8 different families and 12 individuals (5 TSC1, 7 TSC2) for whom we did not have access to parental DNA. Familial TSC cases are reported to have a milder phenotype than sporadic TSC cases, although ascertainment bias cannot be excluded.13 The presence of the familial and mosaic cases in our cohort might have skewed our results to a less severe phenotype. Population-based cohort studies, such as the Tuberous Sclerosis 2000 study, will hopefully show whether familial cases are indeed milder.7
Overall, more brain abnormalities were found in the TSC2 group. TSC2 encodes the catalytic GAP domain of the TSC complex and is therefore essential for canonical TSC complex function. Individuals with a TSC1 variant that affects function, or a TSC2 variant that affects function but where the GAP domain is expressed, might therefore be expected to have a less severe phenotype due to residual TSC2 GAP activity. Indeed, in our functional assessment, we observed an effect of TSC2 expression on S6K-T389 phosphorylation in the absence of co-expressed TSC1 (Figure 2), but did not observe an effect of TSC1 expression on S6K-T389 phosphorylation in the absence of TSC2 (Figure 3). However, we did not find strong evidence for differences between the TSC2x and TSC2p groups. Our functional study indicated that all the changes predicted to result in expression of altered TSC2 protein led to essentially complete inactivation of the TSC complex-dependent inhibition of mTORC1. Therefore, although the function of the TSC complex when over-expressed in cultured cells might be different from its role in vivo, the similarity between the TSC2x and TSC2p groups is consistent with our in vitro functional assessment. S6K-T389 phosphorylation in the presence of nine TSC2 variants was essentially the same as in the absence of TSC2 (Figure 3). As our cohort consisted of only 64 individuals, we were unable to make more than two subgroups of TSC2 variants. In larger cohorts it may be possible to detect smaller genotype/phenotype correlations; however, small effects are less likely to have prognostic value in the clinic.
The chromosomal location, larger size and more complex structure of TSC2, compared to TSC1, might make the TSC2 locus more susceptible to the second hit mutations that are required for TSC pathology. Indeed, there is considerable phenotypic variation between different individuals with the same TSC1 or TSC2 variant, even within a single family.29 This suggests that it is highly likely that random second hit mutations are the most important cause of variation in brain pathology. This is difficult to show in patients, but may be inferred by excluding other causes for phenotypic variability. Another way to investigate the frequency of these stochastic events is to perform histologic analyses on post mortem brains of TSC patients, to determine the presence of cells that have undergone somatic mutations, as has been done previously.30
A recent study showed that the length of the predicted C-terminal tails of mutant TSC1 and TSC2 proteins might be associated with intelligence.15 We correlated the length of the predicted C-terminal tails with the number of tubers per hemisphere and per lobe, and the number of RMLs and SENs. There were no significant differences. This is in agreement with the study of Wong et al,15 suggesting that IQ is not directly related to brain abnormalities, and implies that the pathogenetic mechanisms underlying brain pathology and cognitive development in TSC are distinct. This was also reported by Goorden et al,31 who showed that Tsc1 mutant mice have cognitive deficits in the absence of overt brain pathology. The functional consequences of a longer or shorter C-terminal tail are unknown. It is not yet clear whether truncated TSC1 or TSC2 are expressed in vivo, or whether NMD prevents their synthesis.
The MRI scans used in our study were acquired during routine diagnostics of patients attending a specialist pediatric clinic at an academic hospital, which may introduce a bias towards more severe brain abnormalities. Not all MRIs were made following a standard protocol, and some abnormalities might have been missed. Nonetheless, the numbers of abnormalities identified in our cohort were mostly similar or higher than those reported in previous studies.9, 10, 24 The number of RMLs in our cohort was lower than that reported in another cohort, possibly because we did not use diffusion tensor imaging or three-directional scans.32
In summary, we compared TSC brain pathology to genotype. TSC2 variants were associated with more tubers, RMLs and SENs than TSC1 variants, and although larger studies might identify clinically relevant subdivisions of TSC1 and TSC2 variants, we found little additional value for the subdivision of TSC2 variants. Our study is consistent with the hypothesis that the frequency of second hit events is the most important driver of the variability in TSC-associated brain lesions, as detected by MRI.
References
Nellist M, Janssen B, Brookcarter PT et al: Identification and characterization of the tuberous sclerosis gene on chromosome-16. Cell 1993; 75: 1305–1315.
van Slegtenhorst M, deHoogt R, Hermans C et al: Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997; 277: 805–808.
Gao XS, Zhang Y, Arrazola P et al: Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat Cell Biol 2002; 4: 699–704.
Bissler JJ, Kingswood JC, Radzikowska E et al: Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2013; 381: 817–824.
Franz DN, Belousova E, Sparagana S et al: Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2013; 381: 125–132.
Au KS, Williams AT, Roach ES et al: Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med 2007; 9: 88–100.
Bolton PF, Clifford M, Tye C et al: Intellectual abilities in tuberous sclerosis complex: risk factors and correlates from the Tuberous Sclerosis 2000 Study. Psychol Med 2015; 45: 2321–2331.
Curatolo PM, Moavero R, Roberto D, Graziola F : Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol 2015; 4: 259–273.
Dabora SL, Jozwiak S, Franz DN et al: Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet 2001; 68: 64–80.
Doherty C, Goh S, Poussaint TY, Erdag N, Thiele EA : Prognostic significance of tuber count and location in tuberous sclerosis complex. J Child Neurol 2005; 20: 837–841.
Jansen FE, Braams O, Vincken KL et al: Overlapping neurologic and cognitive phenotypes in patients with TSC1 or TSC2 mutations. Neurology 2008; 70: 908–915.
Kothare SV, Singh K, Chalifoux JR et al: Severity of manifestations in tuberous sclerosis complex in relation to genotype. Epilepsia 2014; 55: 1025–1029.
Sancak O, Nellist M, Goedbloed M et al: Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype-phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet 2005; 13: 731–741.
van Eeghen AM, Black ME, Pulsifer MB, Kwiatkowski DJ, Thiele EA : Genotype and cognitive phenotype of patients with tuberous sclerosis complex. Eur J Hum Genet 2012; 20: 510–515.
Wong HT, McCartney DL, Lewis JC, Sampson JR, Howe CJ, de Vries PJ : Intellectual ability in tuberous sclerosis complex correlates with predicted effects of mutations on TSC1 and TSC2 proteins. J Med Genet 2015; 52: 815–822.
Hoogeveen-Westerveld M, Wentink M, van den Heuvel D et al: Functional assessment of variants in the TSC1 and TSC2 genes identified in individuals with Tuberous Sclerosis Complex. Hum Mutat 2011; 32: 424–435.
Chantranupong L, Wolfson RL, Orozco JM et al: The sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep 2014; 9: 1–8.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F : Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013; 8: 2281–2308.
Nellist M, Brouwer RWW, Kockx CEM et al: Targeted Next Generation Sequencing reveals previously unidentified TSC1 and TSC2 mutations. BMC Med Genet 2015; 16: e10.
Nagy E, Maquat LE : A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 1998; 23: 198–199.
Bolton PF, Griffiths PD : Association of tuberous sclerosis of temporal lobes with autism and atypical autism. Lancet 1997; 349: 392–395.
Chu-Shore CJ, Major P, Montenegro M, Thiele E : Cyst-like tubers are associated with TSC2 and epilepsy in tuberous sclerosis complex. Neurology 2009; 72: 1165–1169.
Numis AL, Major P, Montenegro MA, Muzykewicz DA, Pulsifer MB, Thiele EA : Identification of risk factors for autism spectrum disorders in tuberous sclerosis complex. Neurology 2011; 76: 981–987.
Michelozzi C, Di Leo G, Galli F et al: Subependymal nodules and giant cell tumours in tuberous sclerosis complex patients: prevalence on MRI in relation to gene mutation. Child Nerv Syst 2013; 29: 249–254.
Boronat S, Caruso P, Thiele EA : Absence of subependymal nodules in patients with tubers suggests possible neuroectodermal mosaicism in tuberous sclerosis complex. Dev Med Child Neurol 2014; 56: 1207–1211.
Kozlowski P, Roberts P, Dabora S et al: Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet 2007; 121: 389–400.
Tyburczy ME, Dies KA, Glass J et al: Mosaic and intronic mutations in TSC1/TSC2 explain the majority of TSC patients with no mutation identified by conventional testing. PLoS Genet 2015; 11: e1005637.
Verhoef S, Bakker L, Tempelaars AM et al: High rate of mosaicism in tuberous sclerosis complex. Am J Hum Genet 1999; 64: 1632–1637.
Lyczkowski DA, Conant KD, Pulsifer MB et al: Intrafamilial phenotypic variability in tuberous sclerosis complex. J Child Neurol 2007; 22: 1348–1355.
Crino PB, Aronica E, Baltuch G, Nathanson KL : Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology 2010; 74: 1716–1723.
Goorden SMI, van Woerden GM, van der Weerd L, Cheadle JP, Elgersma Y : Cognitive deficits in Tsc1(+/−)mice in the absence of cerebral lesions and seizures. Ann Neurol 2007; 62: 648–655.
van Eeghen AM, Teran LO, Johnson J, Pulsifer MB, Thiele EA, Caruso P : The neuroanatomical phenotype of tuberous sclerosis complex: focus on radial migration lines. Neuroradiology 2013; 55: 1007–1014.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
R Swenker reports financial assistance from Novartis. MCY de Wit reports grants from Dutch Epilepsy Foundation, and grants and non-financial support from Novartis outside the submitted work; and the Erasmus MC received honoraria from Novartis for educational lectures presented by the author. The remaining authors declare no conflict of interest.
Additional information
Author contributions
IE Overwater contributed to study design, data collection, data analysis, data interpretation and writing of the report. R Swenker contributed to study design, experimental work, data collection, data analysis, data interpretation and writing of the report. EL van der Ende contributed to data collection, data analysis and writing of the report. KBM Hanemaayer contributed to data collection, data analysis and writing of the report. M Hoogeveen-Westerveld contributed to experimental work, data collection, data analysis and data interpretation, and writing of the report. AM van Eeghen contributed to study design, and writing of the report. MH Lequin contributed to study design, data collection and writing of the report. AMW van den Ouweland contributed to data collection, and writing of the report. HA Moll contributed to study design, data interpretation and writing of the report. M Nellist contributed to study design, experimental work, data collection, data analysis, data interpretation and writing of the report. MCY de Wit contributed to study design, data collection, data analysis, data interpretation and writing of the report.
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Overwater, I., Swenker, R., van der Ende, E. et al. Genotype and brain pathology phenotype in children with tuberous sclerosis complex. Eur J Hum Genet 24, 1688–1695 (2016). https://doi.org/10.1038/ejhg.2016.85
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.85
This article is cited by
-
Tsc2 mutation rather than Tsc1 mutation dominantly causes a social deficit in a mouse model of tuberous sclerosis complex
Human Genomics (2023)
-
An mTORC1-mediated negative feedback loop constrains amino acid-induced FLCN-Rag activation in renal cells with TSC2 loss
Nature Communications (2022)
-
A case report of severe tuberous sclerosis complex detected in utero and linked to a novel duplication in the TSC2 gene
BMC Neurology (2020)
-
An update on the central nervous system manifestations of tuberous sclerosis complex
Acta Neuropathologica (2020)
-
Radiobiological Characterization of Tuberous Sclerosis: a Delay in the Nucleo-Shuttling of ATM May Be Responsible for Radiosensitivity
Molecular Neurobiology (2018)
