
Abstract
The 3M syndrome is a rare autosomal recessive disorder recently ascribed to mutations in the CUL7 gene and characterized by severe pre- and postnatal growth retardation. Studying a series of 33 novel cases of 3M syndrome, we have identified deleterious CUL7 mutations in 23/33 patients, including 19 novel mutations and one paternal isodisomy of chromosome 6 encompassing a CUL7 mutation. Lack of mutations in 10/33 cases and exclusion of the CUL7 locus on chromosome 6p21.1 in six consanguineous families strongly support the genetic heterogeneity of the 3M syndrome.
Similar content being viewed by others


Main
The 3M syndrome (OMIM 273750) is an autosomal recessive condition characterized by pre- and postnatal growth retardation, facial dysmorphism, large head circumference, normal intelligence and skeletal changes including long slender tubular bones and tall vertebral bodies.1, 2, 3, 4, 5 Studying a series of 29 families, we have previously mapped the disease locus to chromosome 6p21.1, and identified disease-causing mutations in the CUL7 gene.6
Here, we report the molecular analysis of the CUL7 gene in a series of 33 additional cases and the identification of mutations in 23/33 cases including one paternal isodisomy of chromosome 6. The absence of CUL7 mutation in the other 10/33 cases and the exclusion of the 6p21.1 locus in consanguineous families support the genetic heterogeneity of the 3M syndrome.
Patients and methods
Patients
All patients included in this study fulfilled the diagnostic criteria for 3M syndrome, namely (1) severe pre- and postnatal growth retardation below −3 SD, (2) large head circumference and (3) facial dysmorphism (Figure 1a) that is, prominent forehead, anteverted nares and full lips. Thirty-three patients (16 boys and 17 girls) from 33 unrelated families were included, ranging in age from birth to 14 years. Among them, 19/33 patients were born to consanguineous parents.

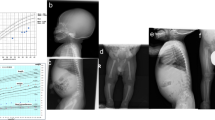
Facial and skeletal features of patient no. 14 at 3 years of age. (a) Note the round face, frontal bossing, short nose and full lips. (b) Note tall vertebral bodies and the hip dislocation.
Skeletal changes were not consistently present at birth but occurred in the course of the disease, namely delayed bone age, slender long bones and tall vertebral bodies (Figures 1b and 2). Two patients also presented with bilateral dislocation of hips (Figure 1) and two others had severe kyphoscoliosis.

Skeletal features of a child at 7 years of age with 3M syndrome and without a CUL7 mutation. Note the long slender tubular bones and tall vertebral bodies.
This series also includes one pregnancy, terminated at 33 weeks of gestation. The foetus presented with severe growth retardation (weight <5th percentile, height<5th percentile), normal head circumference (>50th percentile), characteristic facial features, prominent heels and slender long bones, suggestive of the 3M syndrome (Figure 3a). Analysis of the femoral growth plate of this foetus showed an increased chondrocyte density and size in the resting and proliferative zones but no major abnormalities in the prehypertrophic and hypertrophic zones (Figure 3b). The placenta was reduced in size, with no major abnormality. Apart from reduced liver and lung sizes, no other anomaly was detected at autopsy.

(a) Foetal case of 3M syndrome. The foetus presented with severe growth retardation, normal head circumference, characteristic facial features, prominent heels and long slender tubular bones suggestive of 3M syndrome. (b) Histological study of the femoral growth plate. Note the increased density and size of chondrocytes in the resting and proliferative zones and the defect in matrix production with no major abnormalities in the prehypertrophic or hypertrophic zones (1: resting zone; 2: proliferative zone; 3: hypertrophic zone).
Genomic sequencing
We obtained blood samples with written consent from the affected individuals and their unaffected relatives. Genomic DNA was extracted from peripheral blood by standard procedures. For mutation detection, we used 23 primers to amplify the 25 coding exons of CUL7 (primers available on request). We purified the PCR product with exonuclease I (ExoSAPIT; Amersham Bioscience) according to the manufacturer's instructions. Sequencing reactions were run on an ABI 3130 sequencer using a dye terminator cycle sequencing kit (Applied Biosystems) and analysed by sequencing analysis (Applied Biosystems).
Microsatellite analyses
Genotyping was performed using seven repeat-containing microsatellite markers from the CUL7 region on chromosome 6p21.1 (markers available on request). Microsatellite analysis was performed in consanguineous families, in the affected foetus and his parents using markers of the ABI PRISM linkage mapping set (Applied Biosystems). HEX or FAM fluorescently labelled PCR products were run on an ABI 3130 sequencer and analysed using GeneMapper (Applied Biosystems).
FISH analyses
Lung cells from the affected foetus were submitted to FISH analyses. Bacterial artificial chromosome (BAC) clones were selected from the UCSC genome browser (www.genome.ucsc.edu) database and obtained from the Sanger Institute (www.sanger.ac.uk). FISH experiments were performed on interphase nuclei preparations using a bac RP11-653G5 clone corresponding to the CUL7 gene on 6p21.1 and a control BAC RP11-39C2 clone on 6p12.3. BAC clone DNA was amplified by rolling circle amplification using TempliPhiTM Large Construct kit (GE Healthcare) following the manufacturer's instructions. BAC probes, RP11-653G5 and RP11-39C2, were labelled in red (tetramethyl rhodamine) and green (fluorescein isothiocyanate), respectively, by nick translation.
Results
Direct sequencing of CUL7 identified deleterious mutations in 23/33 patients. Twelve out of 33 patients were homozygotes and 11/33 patients were compound heterozygotes. The mutations were located throughout the gene and included 8 missense, 19 nonsense and 6 splice-site mutations (Table 1, families 30–52). Among them, 19 were novel mutations (Table 1).
Only one mutation inherited from the father was detected in the affected foetus (I19: c.3645+1G>A). These findings were suggestive of either a deletion or a uniparental disomy. Microsatellite analyses of the foetus at the 3M locus was consistent with either homozygosity or hemizygosity, but FISH analysis using the probe RP11-653G5 (6p21.1) excluded a large-scale deletion (data not shown). Extensive microsatellite analysis of chromosome 6 revealed that the foetus had complete isodisomy of chromosome 6, of paternal origin (data not shown). Finally, among the 10 cases without any CUL7 mutations, 7/10 patients were born to related parents. Microsatellite analysis at the 3M locus revealed that 6/7 inbred children were heterozygotes at the 6p21.1 region, ruling out CUL7 as the disease gene in these families (data not shown). No clinical or radiological differences were observed between patients with and without CUL7 mutation (Figures 1 and 2, respectively).
Discussion
Studying a large series of 33 additional cases of 3M syndrome, we identified CUL7 mutations in 23/33 families. The combination of these data with our previous results supports the view that CUL7 is the major disease gene in the 3M syndrome, accounting for 84% of our cases (52/62).
Failure to identify CUL7 mutations in 10 patients and exclusion of the 6p21.1 region in six consanguineous families argue in favour of genetic heterogeneity. This observation is in contrast with clinical homogeneity of the 3M syndrome. Indeed, no distinctive feature was observed in patients, whether or not CUL7 mutations were observed. These data suggest that a second disease-causing gene is closely related to the CUL7 pathway.
CUL7 belongs to the cullin family and plays a scaffold role in the E3 ubiquitin ligase complex, in which CUL7 interacts with both a heterodimer (composed of Skp1 bound to a member of the F-box protein family named Fbx29) and the ROC1 RING-finger protein.7, 8, 9, 10, 11 The exact function and specific substrates of CUL7 are unknown. At high expression levels, CUL7 may interfere with the function of other cullins by sequestering ROC1 in the cytoplasm.12 On the other hand, in normal cells, the growth-promoting activity of CUL7 is attributed to its ability to bind p53,12 and the increased p53 activity could partly explain the delayed growth of Cul7−/− mouse embryo fibroblasts.17 Finally, in cells expressing T antigen, CUL7 acts as a tumour suppressor,7, 13 and T antigen may inhibit CUL7 activity by inhibiting the ubiquitination of a substrate or its binding to CUL7.14
We have shown earlier that nonsense and missense CUL7 mutations (R1445X and H1464P) impaired the ability of CUL7 to recruit ROC1, suggesting that impaired ubiquitination may play a role in the pathogenesis of prenatal growth retardation in humans.6 In addition, the association of pre- and postnatal growth retardation with skeletal changes in 3M syndrome suggests that CUL7 may play a specific role in the endochondral ossification process. In keeping with this, analysis of the femoral growth plate of the 3M foetus revealed an increased in chondrocyte density and a defect in matrix production in the resting and proliferative zones. These preliminary data suggest that CUL7 is involved in chondrocyte growth and proliferation. However, one cannot exclude the responsibility of the paternal isodisomy of chromosome 6 in the phenotype observed in the foetus. Apart from low birth weight, none of the other features described in the paternal isodisomy of chromosome 6 was present in the foetus (ie, macroglossia, heart defect and, obviously, neonatal diabetes).15, 16 The prenatal growth retardation and growth plate anomalies might be due to the combination of the paternal isodisomy of chromosome 6 and 3M syndrome.
Additional studies are ongoing and will hopefully lead to the understanding of CUL7 function as well as to the identification of its partners.
References
Spranger J, Opitz JM, Nourmand A : A new familial intrauterine growth retardation syndrome the ‘3M syndrome’. Eur J Pediatr 1976; 123: 115–124.
Winter RM, Baraitser M, Grant DB, Preece MA, Hall CM : The 3M syndrome. J Med Genet 1984; 21: 124–128.
Hennekam RC, Biljlsma JB, Spranger J : Further delineation of the 3M syndrome with review of the literature. Am J Med Genet 1987; 28: 195–209.
Mueller RF, Buckler J, Arthur R et al: The 3-M syndrome: risk of intracerebral aneurysm ? J Med Genet 1992; 29: 425–427.
Van der Wal G, Otten BJ, Brunner HG, Van der Burgt I : 3M syndrome: description of 6 new patients with review of the literature. Clin Dysmorphol 2001; 10: 241–252.
Huber C, Dias-Santagata D, Glaser A et al: Identification of mutations in CUL7 in 3-M syndrome. Nat Genet 2005; 37: 1119–1124.
Kohrman DC, Imperiale MJ : Simian virus 40 large T antigen stably complexes with a 185-kilodalton host protein. J Virol 1992; 66: 1752–1760.
Daud AI, Lanson Jr NA, Claycomb WC, Field LJ : Identification of SV40 large T-antigen-associated proteins in cardiomyocytes from transgenic mice. Am J Physiol 1993; 264: 1693–1700.
Dias DC, Dolios G, Wang R, Pan ZQ : CUL7: a DOC domain-containing cullin selectively binds Skp1.Fbx29 to form an SCF-like complex. Proc Natl Acad Sci USA 2002; 99: 16601–16606.
Zheng N, Schulman BA, Song L et al: Structure of the Cul1-Rbx1-Skp1-FboxSkp2 SCF ubiquitin ligase complex. Nature 2002; 416: 703–709.
Petroski MD, Deshaies RJ : Function and regulation of Cullin-Ring ubiquitin ligases. Nat Rev Mol Cell Biol 2005; 6: 9–20.
Andrews P, He YJ, Xiong Y : Cytoplasmic localized ubiquitin ligase cullin 7 binds to p53 and promotes cell growth by antagonizing p53 function. Oncogene 2006; 25: 4534–4548.
Kasper JS, Kuwabara H, Arai T, Ali SH, DeCaprio JA : Simian virus 40 large T antigen's association with the CUL7 SCF complex contributes to cellular transformation. J Virol 2005; 79: 11685–11692.
Tsai SC, Pasumarthi KB, Pajak L et al: Simian virus 40 large T antigen binds a novel Bcl-2 homology domain 3-containing proapoptosis protein in the cytoplasm. J Biol Chem 2000; 275: 3239–3246.
Hermann R, Laine A-P, Johansson C et al: Transient but not permanent neonatal diabetes mellitus is associated with paternal uniparental isodisomy of chromosome 6. Pediatrics 2000; 105: 49–52.
Diatloff-Zito C, Nicole A, Marcelin G et al: Genetic and epigenetic defects of the 6q24 imprinted locus in a cohort of 13 patients with transient neonatal diabetes: new hypothesis raised by the finding of a unique case with hemizygotic deletion in the critical region. J Med Genet 2007; 44: 31–37.
Arai T, Kasper JS, Skaar JR, Ali SH, Takahashi C, DeCaprio JA : Targeted disruption of p185/Cul7 gene results in abnormal vascular morphogenesis. Proc Natl Acad Sci USA 2003; 100: 9855–9860.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Huber, C., Delezoide, AL., Guimiot, F. et al. A large-scale mutation search reveals genetic heterogeneity in 3M syndrome. Eur J Hum Genet 17, 395–400 (2009). https://doi.org/10.1038/ejhg.2008.200
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.200
Keywords
This article is cited by
-
Utility of next-generation sequencing in genetic testing and counseling of disorders involving the musculoskeletal system—trends observed from a single genetic unit
Journal of Orthopaedic Surgery and Research (2022)
-
Ubiquitin ligases: guardians of mammalian development
Nature Reviews Molecular Cell Biology (2022)
-
The functional analysis of Cullin 7 E3 ubiquitin ligases in cancer
Oncogenesis (2020)
-
Murine obscurin and Obsl1 have functionally redundant roles in sarcolemmal integrity, sarcoplasmic reticulum organization, and muscle metabolism
Communications Biology (2019)
-
Clinical Utility Gene Card for: 3-M syndrome - Update 2013
European Journal of Human Genetics (2014)
