
Abstract
The receptor activator of NF-κB (RANK) and immunoreceptor tyrosine-based activation motif (ITAM)-containing adaptors are essential factors involved in regulating osteoclast formation and bone remodeling. Here, we identify early estrogen-induced gene 1 (EEIG1) as a novel RANK ligand (RANKL)-inducible protein that physically interacts with RANK and further associates with Gab2, PLCγ2 and Tec/Btk kinases upon RANKL stimulation. EEIG1 positively regulates RANKL-induced osteoclast formation, likely due to its ability to facilitate RANKL-stimulated PLCγ2 phosphorylation and NFATc1 induction. In addition, an inhibitory peptide designed to block RANK-EEIG1 interaction inhibited RANKL-induced bone destruction by reducing osteoclast formation. Together, our results identify EEIG1 as a novel RANK signaling component controlling RANK-mediated osteoclast formation, and suggest that targeting EEIG1 might represent a new therapeutic strategy for the treatment of pathological bone resorption.
Similar content being viewed by others


Introduction
Bone homeostasis is maintained by balancing the activities of osteoclasts (bone resorbing) and osteoblasts (bone forming). Imbalances in bone remodeling cause a variety of bone diseases1,2. Two cytokines are essential for osteoclast differentiation: receptor activator of nuclear factor κB (NF-κB) ligand (RANKL), which belongs to the tumor necrosis factor (TNF) family; and macrophage colony-stimulating factor (M-CSF)3,4. RANKL initiates its signal transduction through binding to receptor activator of NF-κB (RANK) that is expressed in osteoclast precursors. RANK then recruits the adaptor molecule TNF receptor-associated factor 6 (TRAF6), leading to the activation of NF-κB and mitogen-activated protein kinases (MAPKs), including c-Jun N-terminal kinase (JNK) and p385,6. RANKL-induced activation of NF-κB and c-Fos is required for the induction of nuclear factor of activated T-cells cytoplasmic 1 (NFATc1), a key transcription factor for osteoclast formation7,8,9. Recent findings indicate that the activation of JNK and NF-κB pathways requires the phosphorylation of growth factor receptor-bound protein 2 (Grb2)-binding adaptor protein (Gab2), and its subsequent recruitment to RANK. Moreover, phospholipase Cγ2 (PLCγ2) forms a complex with Gab2, is required for Gab2 phosphorylation and modulates Gab2 recruitment to RANK10,11.
NFATc1 is also induced by calcium signaling that is triggered by the phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM)-containing adaptors, such as DNAX-activating protein 12 (DAP12) and the γ chain of Fc receptor (FcRγ). These adaptors associate with osteoclast-associated receptor (OSCAR), an immunoglobulin-like receptor12,13,14. Phosphorylation of ITAM-containing adaptors results in the recruitment of spleen tyrosine kinase (Syk), leading to the activation of PLCγ and subsequent calcium mobilization10,12,13,15. Therefore, osteoclastogenesis is regulated by both RANK and ITAM signalings. It has been recently demonstrated that Btk/Tec kinases act downstream of RANK, and form a signaling complex with the adaptor molecule B-cell linker protein (BLNK), which is activated by Syk downstream of ITAM signaling; activated BLNK acts as a scaffold to bridge Btk/Tec kinases and PLCγ to form an osteoclastogenic signaling complex, thus providing a mechanism of integrating RANK and ITAM signaling in osteoclast differentiation16. However, the mediator molecule(s) between RANK and the downstream molecules, such as Gab2, PLCγ2 and Tec/Btk have not been fully characterized.
Here, we identify early estrogen-induced gene 1 (EEIG1) as a novel RANK signaling component that is required for osteoclastogenesis. EEIG1 knockdown results in an obvious reduction of RANKL-induced osteoclast formation, which is caused by impaired RANKL-stimulated PLCγ2 activation and NFATc1 induction. In response to RANKL stimulation, RANK forms a signaling complex containing EEIG1, Gab2, PLCγ2 and Tec/Btk. We also show that intervention of the RANK-EEIG1 interaction by an inhibitory peptide could serve as a new therapeutic strategy for treating bone diseases.
Results
RANKL induces EEIG1 expression during osteoclastogenesis
To further explore RANKL-induced molecules that are potentially involved in osteoclast formation, we performed a genome-wide microarray screening to examine the gene expression profiles during the differentiation from bone marrow-derived macrophages (BMMs) to osteoclasts17. We found that the mRNA expression level of EEIG1, also known as Fam102A, was increased by 4.8-fold 24 h after RANKL stimulation (Figure 1A). We further analyzed the time course of EEIG1 mRNA expression in RANKL-treated BMMs by RT-PCR analyses, and found that the expression of EEIG1 continued to increase until 72 h after RANKL stimulation. The mRNA level of Fam102B, an isoform of EEIG1, was barely changed during osteoclastogenesis (Figure 1B). We also confirmed the induction of EEIG1 expression in calvarial bones of RANKL-injected mice (Supplementary information, Figure S1). The time course of RANKL-induced EEIG1 expression was also confirmed by immunoblotting (Figure 1C). Moreover, 3 days after RANKL stimulation, cells with multiple nuclei (an indicator for osteoclasts) became strongly positive for EEIG1 protein expression (Figure 1D). The expression of EEIG1 protein occurs predominantly in the cytoplasm (Supplementary information, Figure S2). These data show that EEIG1 expression is induced by RANKL during the differentiation from BMMs to osteoclasts.

Identification of EEIG1 as a novel RANKL-inducible gene. (A) Genechip analysis of EEIG1 mRNA upon RANKL stimulation. (B) RT-PCR analyses of the mRNA levels for EEIG1, Fam102B, NFATc1, TRAP and GAPDH in RANKL-stimulated BMMs. (C) Expression of EEIG1 and NFATc1 proteins in RANKL-stimulated BMMs. GAPDH served as the loading control. (D) Immunofluorescence staining of EEIG1 (green) in osteoclasts. The nuclei (blue) were stained with DAPI. Scale bar, 50 μm. (E) Quantitative real-time PCR analyses to assess the mRNA level of EEIG1 relative to that of β-actin upon stimulation with LPS, estradiol (E2) or RANKL in BMMs. (F) BMMs were stimulated with RANKL, LPS or E2 for the indicated periods. The stimulated cells were lysed and subjected to western blot analyses with antibodies against EEIG1, NFATc1 and tubulin. Tubulin served as the loading control. (G) Effect of NFATc1 on the expression of EEIG1. BMMs infected with retroviruses expressing control vector or constitutively active NFATc1 (NFATc1-CA) were cultured with M-CSF in the absence or presence of RANKL for 2 days, and then western blot analyses were performed with antibodies against EEIG1, NFATc1 and GAPDH. (H) BMMs were incubated with DMSO (Mock) or CsA (5 μM) in the absence or presence of RANKL for 2 days. Total RNAs were extracted and EEIG1 mRNA levels were examined by quantitative real-time PCR. In all the experiments, BMMs cultured in the presence of M-CSF were stimulated with RANKL, LPS or E2 as indicated.
As EEIG1 was originally identified as an early estrogen-inducible gene in breast cancer cell lines18, we compared the EEIG1 expression in BMMs stimulated with either RANKL, lipopolysaccharides (LPS), or estradiol (E2). EEIG1 mRNA was marginally increased upon estradiol stimulation, and EEIG1 expression was not detected in BMMs stimulated with LPS. However, the expression of EEIG1 was obviously induced 24 h after RANKL stimulation and continued to increase through 72 h (Figure 1E). Consistently, the protein expression of EEIG1 was induced by RANKL stimulation, but not by estradiol or LPS (Figure 1F).
We further investigated whether RANKL-induced EEIG1 expression involves NFATc1, an important downstream target of RANK signaling. We found that EEIG1 expression was upregulated by overexpression of a constitutively active form of NFATc1 (NFATc1-CA), even without RANKL stimulation (Figure 1G, lane 3). RANKL-induced EEIG1 expression was markedly decreased by Cyclosporin A (CsA), an inhibitor of NFATc1 (Figure 1H). A luciferase reporter gene containing a 1.2-kb EEIG1 promoter fragment was also activated by NFATc1 overexpression. Consistently, a putative NFAT-binding DNA element19 is present in the promoter region of EEIG1 (Supplementary information, Figure S3). Together, these data suggest that RANKL induces EEIG1 expression via NFATc1 during osteoclastogenesis.
EEIG1 positively regulates RANKL-induced osteoclast differentiation
To investigate the role of EEIG1 in RANKL-induced osteoclast differentiation, primary BMMs were infected with retroviruses expressing EEIG1. Transduced BMMs were cultured in the presence of RANKL and M-CSF, and were stained for tartrate-resistant acid phosphatase (TRAP). EEIG1 overexpression increased the number of TRAP+ MNCs (> 5 nuclei) by 2-fold upon RANKL stimulation (Figure 2A). We then examined the expression level of NFATc1, an important transcription factor of osteoclast formation. As compared with the control, overexpression of EEIG1 increased the NFATc1 expression in the presence or absence of RANKL stimulation (Figure 2B). Next, we examined the effects of EEIG1 overexpression on bone resorption. The area of the resorbed pit was significantly increased by EEIG1 overexpression, suggesting that EEIG1 enhances the bone-resorbing activity of mature osteoclasts (Figure 2C).

Role of EEIG1 in RANKL-induced osteoclastogenesis and bone resorption. (A-C) BMMs were transduced with pMX-puro (control) or FLAG-tagged EEIG1 retrovirus and cultured for 4 days (A), 2 days (B) or 7 days (C) in the presence of M-CSF with or without RANKL. (A) Cells were fixed and stained for TRAP (left panel). Numbers of TRAP+ MNCs (> 5 nuclei) were counted (right panel). (B) Cells were harvested and the lysates were analyzed by western blot analyses using antibodies against FLAG, EEIG1, NFATc1 and GAPDH. (C) After culturing on dentin slices for 7 days, BMMs were stained with hematoxylin for visualization of pit formation (left panel). The area of resorption pits was calculated with Image-Pro Plus 4.5 (right panel). (D, E) BMMs were infected with retroviruses expressing control shRNA (control) or EEIG1-specific shRNA (shEEIG1), and cultured for 4 days (D) or 2 days (E). The numbers of TRAP+ MNCs were counted (D). Western blot analyses were performed to detect the expression of EEIG1, NFATc1 and GAPDH (E). (F) BMMs expressing control shRNA or EEIG1-specific shRNA were cocultured with calvarial osteoblasts for 7 days in the presence of 1α, 25-dyhydroxy vitamin D3 (VitD3) and prostaglandin E2 (PGE2). The numbers of TRAP+ MNCs were counted. Data are presented as mean ± SD (A, C, D, F) and are representative of at least 3 experiments. Scale bar, 200 μm (A, D, F), 100 μm (C). *P < 0.05, **P < 0.01.
We further analyzed the effect of shRNA-mediated EEIG1 knockdown on RNAKL-induced osteoclast formation. Knockdown of EEIG1 in BMMs suppressed the formation of TRAP+ MNCs (Figure 2D and Supplementary information, Figure S4) and attenuated the expression of NFATc1 induced by RANKL (Figure 2E). Consistently, in a time-course experiment, depletion of EEIG1 by shRNA significantly inhibited osteoclast formation (Supplementary information, Figure S5). Moreover, EEIG1 knockdown also led to a severe defect in osteoclast formation when BMMs were co-cultured with osteoblasts (Figure 2F). These results suggest that EEIG1 plays a critical role in RANKL-induced osteoclast differentiation, and its role may be closely related to the induction of NFATc1.
EEIG1 directly interacts with RANK upon RANKL stimulation
To investigate the underlying mechanism by which EEIG1 regulates RANKL-induced osteoclastogenesis, we tested whether EEIG1 interacts with RANK upon RANKL stimulation. The results of coimmunoprecipitation experiments revealed that endogenous EEIG1 interacts with RANK following RANKL stimulation (Figure 3A). We further used yeast two-hybrid assay to determine whether EEIG1 directly interacts with RANK. The RANK-TRAF6 interaction served as a positive control. Similar to the positive control, we found that EEIG1 indeed interacts with RANK (Figure 3B). These data suggest that EEIG1 likely regulates osteoclast differentiation through a physical interaction with RANK.

Direct interaction of EEIG1 with RANK. (A) BMMs were stimulated with RANKL for 2 days. Cells were lysed and immunoprecipitated with antibody against EEIG1. The resulting immunoprecipitated samples were subjected to western blot analyses with antibody against RANK. An aliquot of the lysates was reserved for the blotting of the total EEIG1 and RANK. (B) Direct interaction of EEIG1 with RANK was determined by yeast two-hybrid assay. The yeast EGY48 transformed with p80p-LacZ cells were co-transfected with pB42AD-EEIG1 (amino acids 1-384) and pLexA-RANK (amino acids 235-625). Following the selection, β-gal activity was tested in the induction medium. Representative streaks of strains expressing the two-hybrid fusions are shown. The negative control is the yeast cell line co-transfected with pLexA and pB42AD vectors; the positive control is the yeast cell line EGY48/p80p-LacZ co-transfected with pLexA-RANK and pB42AD-TRAF6.
Mapping the interaction domains between RANK and EEIG1
RANK is a transmembrane protein containing a very long cytosolic domain (mouse, 391 amino acids; human, 383 amino acids). To identify EEIG1-binding sites in the RANK cytoplasmic domain that interacts with EEIG1, several GST-tagged RANK deletion constructs were utilized, as shown in Figure 4A. Similar to the full-length RANK cytoplasmic domain, the deletion construct containing the C-terminal region (RANK 532-625) interacted with EEIG1 (Figure 4B). Further deletion within the C-terminal region revealed that the region spanning the amino acids 532-542 of RANK is necessary for EEIG1 interaction (Figure 4C, 4D and Supplementary information, Figure S6A and S6B). To further specify EEIG1-binding site in the RANK cytoplasmic domain, we generated several RANK mutants, in which certain amino acids were mutated to alanines, and tested their ability to bind EEIG1. Only the mutations of RANK 532-537 (GDIIVV) abolished RANK-EEIG1 interaction (Figure 4E and 4F). The functional relevance of the interaction between the RANK 532-537 region and EEIG1 was investigated by using RAW 264.7 cell lines stably expressing FLAG-tagged wild-type RANK (RANK WT) or FLAG-tagged RANK 532-537 mutant (RANK 532-537 Mt) (Figure 4G). As the FLAG motif was fused to the amino termini of RANK, anti-FLAG antibody was used to induce the aggregation of RANK to activate downstream signaling17. After stimulation, cells stably expressing RANK WT formed TRAP+ MNCs (Figure 4G). However, cells stably expressing the RANK 532-537 Mt failed to undergo osteoclast differentiation. NFATc1 induction was also decreased in cells expressing RANK 532-537 Mt compared with that of the RANK WT cells (Figure 4H), whereas RANKL-induced NF-κB activation was not affected by the overexpression of RANK 532-537 Mt (Supplementary information, Figure S7), which suggests the possibility that the RANK-EEIG1 interaction may specifically contribute to the induction of NFATc1. Together, these data indicate that the 532-537 region within the RANK cytoplasmic domain is crucial for RANK-EEIG1 interaction, and this interaction likely plays an important role in the regulation of osteoclast differentiation.

The C-terminal region of RANK spanning amino acids 532-537 is critical for its interaction with EEIG1. (A, C, E) Schematic diagrams of wild-type and mutant constructs of GST-RANK. Numbers shown above or beneath each protein diagram denote amino acid numbers in RANK. Constructs that can interact with EEIG1 are indicated as +. (B, D, F) 293T cells were transfected with FLAG-tagged full-length EEIG1 together with the indicated GST-tagged RANK or its mutant constructs. The GST-RANK proteins were precipitated with Glutathione-Sepharose beads, and the associated EEIG1 was detected by anti-FLAG antibody. An aliquot of the lysates was reserved to blot the total EEIG1. The GST-RANK expression was detected by Coomassie Brilliant Blue staining (CBB) in B or western blot analyses with anti-GST in D and F. (G, H) RAW 264.7 cells that stably express RANK WT or RANK 532-537 Mt were stimulated with anti-FLAG antibody to induce osteoclast formation. (G) Cells were stained for TRAP activity 4 days after stimulation (left panel). Numbers of TRAP+ MNCs (> 5 nuclei) were counted (right panel). Data are presented as mean ± SD and are representative of at least 3 experiments. Scale bar, 200 μm. *P < 0.001. (H) RAW 264.7 cells were cultured with or without an anti-FLAG antibody for 2 days as indicated. Cells were then lysed and subjected to western blot analyses with antibodies against EEIG1, NFATc1, RANK or GAPDH. GAPDH served as the loading control.
To identify the specific region of EEIG1 required for its interaction with RANK, we generated several deletion mutants of EEIG1 (Figure 5A). These mutants were transfected into 293T cells together with GST-tagged full-length RANK. RANK interacted with wild-type EEIG1 and two deletion mutants (EEIG1 ΔSer and EEIG1 ΔC), but not the N-terminal deleted EEIG1 (EEIG1 ΔN) (Figure 5B), suggesting that the N-terminal region spanning amino acids 1-157 of EEIG1 is necessary for RANK binding. We further narrowed down the RANK-binding region within EEIG1 by analyzing cells co-transfected with GST-RANK and several serial-deletion constructs of FLAG-tagged EEIG1 (ΔN1-32, ΔN1-64, ΔN1-96, ΔN1-128, ΔN1-138 and ΔN1-148). Only ΔN1-138 and ΔN1-148 failed to interact with RANK. These results demonstrate that the region spanning amino acids 129-138 of EEIG1 is necessary for RANK-EEIG1 interaction (Figure 5C and 5D).

The N-terminal region of EEIG1 is necessary for RANK interaction. (A, C) Schematic diagrams of FLAG-tagged EEIG1 and its mutant constructs. Numbers shown above each protein diagram denote the amino acid numbers in EEIG1. Constructs that can interact with RANK are indicated as +. (B, D) Interaction of GST-RANK with various FLAG-EEIG1 constructs was analyzed by a GST pull-down assay as described in Figure 4B. An aliquot of the lysates was reserved to blot the total EEIG1. The GST-RANK expression was detected by CBB in B or western blot analysis with anti-GST in D.
RANKL induces the formation of a complex containing RANK, EEIG1, Gab2, PLCγ2 and Tec/Btk
It has been reported that RANK associates with Gab2 and PLCγ2 upon RANKL stimulation10,11,20. As our data indicate that EEIG1 physically interacts with RANK, we asked whether endogenous EEIG1, Gab2 and PLCγ2 form complexes with RANK. BMMs were incubated with RANKL and M-CSF for 2 days to generate pre-osteoclasts (pre-OCs). Endogenous EEIG1 was immunoprecipitated with anti-EEIG1, and endogenous RANK, Gab2 or PLCγ2 was observed in the EEIG1-immunoprecipitates, but not in the IgG-immunoprecipitates (Figure 6A). We further showed that endogenous RANK immunoprecipitated with EEIG1, PLCγ2 and Gab2 following RANKL stimulation (Figure 6B). These results suggest that RANKL induces the formation of a complex composed of RANK, EEIG1, Gab2 and PLCγ2. We further confirmed the association of RANK with EEIG1, Gab2, and PLCγ2 by co-transfection studies in human 293T cells. As shown in Figure 6C and 6D, EEIG1 interacts with PLCγ2 and Gab2. Moreover, we found that PLCγ2 associates with both EEIG1 and RANK (Figure 6E), and we also observed the association between PLCγ2 and EEIG as well as Gab2 (Figure 6F).

RANK forms a signaling complex containing EEIG1, Gab2, PLCγ2 and Tec/Btk. (A) BMMs were incubated with M-CSF and RANKL for 2 days. Cells were lysed and subjected to immunoprecipitation by antibody against EEIG1 or normal rabbit IgG. Western blot analyses were performed with antibodies against PLCγ2, Gab2, RANK and EEIG1. An aliquot of the lysates was reserved to blot the total PLCγ2, Gab2, RANK and EEIG1. (B) BMMs were incubated with M-CSF and RANKL for 2 days to generate preosteoclasts. After serum-starvation for 6 h, cells were lysed and subjected to immunoprecipitation by antibody against RANK. Western blot analyses were performed to detect the indicated proteins. (C-F) 293T cells were co-transfected with FLAG-EEIG1 and various expression plasmids as indicated. Cell lysates were precipitated with antibody against PLCγ2 (C, E, F) or Gab2 (D). The associated proteins were detected by the indicated antibodies. (G-I) 293T cells were co-transfected with Flag-tagged or GST-tagged proteins as indicated. Cell lysates were harvested and GSTpull-down assays were performed. Westen blot analyses using anti-GST and anti-Flag were performed to detect the presence of the indicated proteins in the immunoprecipitates. (J) BMMs were cultured with M-CSF in the absence or presence of RANKL for 2 days. Cells were lysed and fractionated by a discontinuous sucrose density gradient ultracentrifugation. Lipid raft fractions and non-raft fractions were concentrated and analyzed by western blotting with antibodies specific for EEIG1, RANK, PLCγ2, Gab2 and Btk. Flotillin 2 served as the marker for lipid rafts. The data shown in A, B and J are representatives of 3 independent experiments.
Tec/Btk kinases are the molecules that link RANK and ITAM pathways to activate calcium signaling16. However, how RANK is connected to Tec/Btk is not yet understood. We hypothesized that EEIG1 may bridge RANK and Tec/Btk. Indeed, results from GST pull-down assays demonstrated that EEIG1 could bind ectopically-expressed Tec or Btk (Figure 6G and 6H). However, EEIG1 failed to interact with Syk (Figure 6I). To further study the RANK signaling complex, we isolated membrane lipid rafts after RANKL stimulation and observed increased levels of EEIG1, RANK, PLCγ2, Gab2 and Btk recruited to lipid rafts (Figure 6J). Together, these data indicate that RANKL stimulation induces the formation of a signaling complex containing RANK, EEIG1, Gab2, PLCγ2 and Tec/Btk.
EEIG1 facilitates RANKL-induced PLCγ2 activation
Given that PLCγ2 is required for the activation of osteoclastogenic signaling pathways downstream of RANK10,16, we investigated whether EEIG1 is potentially involved in regulating the tyrosine phosphorylation of PLCγ2 induced upon RANKL stimulation. We found that overexpression of EEIG1 increased PLCγ2 phosphorylation and NFATc1 expression in the presence of RANKL stimulation (Figure 7A). Conversely, RANKL-induced PLCγ2 phosphorylation and NFATc1 expression were markedly inhibited by shRNA-mediated knockdown of EEIG1 (Figure 7B). Collectively, these data point to the critical importance of EEIG1 in RANK signaling.

Requirement of EEIG1 for the activation of PLCγ2. (A) BMMs were transduced with pMX-puro (control) or FLAG-EEIG1 retrovirus and cultured for 2 days with M-CSF in the absence or presence of RANKL. (B) BMMs were infected with retroviruses expressing control shRNA (control) or EEIG1-specific shRNA (shEEIG1) and were cultured in the same condition as in A. Cells were lysed and analyzed by western blotting with antibodies against phospho-PLCγ2, NFATc1, EEIG1 and PLCγ2. GAPDH served as the loading control.
Inhibitory effect of EEIG1 inhibitor peptide (EIP) on osteoclast formation and RANKL-induced bone loss
We hypothesized that blocking the RANK-EEIG1 interaction may serve as a new strategy for treating resorption diseases. Therefore, we designed a cell-permeable EIP, in which the RANK-binding region of EEIG1 (amino acids 129-138) was fused to a cell-permeable sequence derived from the human transcription factor Hph-117 (Figure 8A). We tested whether EIP could inhibit osteoclastogenesis, and found that RANKL/M-CSF-induced osteoclast formation was inhibited by EIP in a dose-dependent manner, whereas no inhibition was observed for the control peptide (Figure 8B). Importantly, EIP significantly inhibited RANKL-induced EEIG1 and NFATc1 expression. Consistently, RANKL-induced PLCγ2 phosphorylation was severely decreased by EIP (Figure 8C).

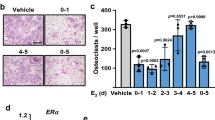
EIP inhibits osteoclast formation and RANKL-induced bone destruction. (A) Structure of EIP. (B) In vitro differentiation of osteoclasts from BMMs treated with increasing concentrations of EIP. TRAP+ MNCs (>5 nuclei) were counted 4 days after RANKL stimulation. Left panel, TRAP staining; right panel, numbers of TRAP+ MNCs. Data represent mean ± SD. Scale bar, 200 μm. (C) BMMs were stimulated with RANKL in the absence or presence of EIP (10 μM). Cells were lysed and analyzed by western blotting with antibodies against EEIG1, NFATc1, phospho-PLCγ2 and PLCγ2. GAPDH served as the loading control. (D-G) EIP suppresses RANKL-induced (2 mg RANKL/kg body weight) osteoclast formation and bone destruction. TRAP staining was carried out on the sections of mouse calvaria bone (D). Bone morphometric analyses indicate that RANKL-induced increases in the number of osteoclasts (E), the eroded surface (F), and the marrow cavity area (G) were significantly suppressed by the local administration of EIP. Data represent mean ± SD (n = 5). *P <0.01, **P < 0.001.
We next investigated the effects of EIP on the pathological formation of osteoclasts by injecting the subcutaneous tissue over the periosteum of mouse calvaria with vehicle alone (PBS), RANKL plus control peptide, or RANKL plus EIP. EIP or control peptide was injected for 5 days at 1-day intervals. RANKL was administered for 3 days at 2-day intervals. Whole calvaria was fixed in 4% paraformaldehyde and used for TRAP staining (Figure 8D). The formation of TRAP+ MNCs was greatly suppressed in EIP-treated mice (Figure 8D and 8E), and the extent of bone erosion was significantly reduced by EIP treatment (Figure 8F and 8G), suggesting that EIP can inhibit RANKL-induced bone destruction in vivo. Together, these results suggest that using cell-permeable peptides targeting the EEIG1-RANK interaction may represent a promising new strategy for anti-resorptive medicine development.
Discussion
Since the discovery of the RANKL/RANK system, intensive studies have focused on elucidating RANK signaling mechanisms involved in osteoclast differentiation, function and survival21,22,23. It has become clear that RANK transduces key signals by directly interacting with TRAF624,25. In addition, it has been reported that PLCγ2 binds to Gab2 in response to RANKL, mediates Gab2 recruitment to RANK, and is required for osteoclast formation10,20. Moreover, PLCγ2 and Tec/Btk kinases are activated by RANK and ITAM signals, thereby activating calcium signaling required for the induction of NFATc1, the key transcription factor for osteoclast differentiation11,16,26. It remains to be determined whether Gab2 and PLCγ2 can directly associate with RANK, and whether such binding is mediated by additional intermediates such as TRAFs. Also, how RANK links to PLCγ2 and Tec/Btk kinases is still unclear. In this regard, we have identified EEIG1 as a novel RANK pathway component that has the potential to mediate RANK signaling. The RANK-EEIG1 signaling complex is necessary for PLCγ2 phosphorylation that is required for the induction of NFATc1.
EEIG1 was originally identified as an early estrogen-inducible gene in breast cancer cells containing the estrogen receptor18. In our study, neither estrogen nor LPS induced the expression of EEIG1 gene in osteoclast precursors (BMMs), whereas RANKL treatment markedly increased mRNA and protein levels of EEIG1. Moreover, EEIG1 expression was upregulated by the transduction of a constitutively active form of NFATc1, and was markedly decreased by an NFATc1 inhibitor (Figure 1G and 1H). These observations suggest that EEIG1 gene expression is induced by RANKL stimulation in osteoclast precursors through NFATc1. In previous reports, RANKL-induced activation of NF-κB and c-Fos induction was deemed important for the initial induction of NFATc17,25. However, NFATc1 is more strongly activated by calcium signaling and binds to its own promoter, thereby initiating its own auto-amplification6,25,27. We thus hypothesize that the initial induction of NFATc1 is a prerequisite for RANKL-induced EEIG1 expression. The induced EEIG1 enhances RANK signaling pathway(s), further triggering the upregulation of NFATc1. In support of this hypothesis, we found that overexpression of EEIG1 enhances NFATc1 expression, while depletion of EEIG1 abrogates RANKL-induced NFATc1 expression. Therefore, it might be possible that a positive feedback circuit involving RANK, NFATc1, and EEIG1 promotes the efficient differentiation of osteoclasts.
The question remains as to how EEIG1 regulates the differentiation of osteoclasts. We found that EEIG1 physically interacts with the highly conserved domain (HCD) of RANK, which is distinct from TRAF6-binding sites in RANK28. A previous report suggested that the HCD of RANK recruits Gab2 that further associates with PLCγ210,11. In this context, our data suggest that EEIG1 mediates RANK signaling to activate PLCγ2. It should be noted that Gab2 deficiency results in reduced activation of NF-κB and JNK, and a partial reduction in NFATc1 expression20, while targeted deletion of PLCγ2 leads to decreased RANKL-induced signaling to AP1 and NF-κB, and a defective upregulation of NFATc110. Notably, in contrast to the loss of Gab2 or PLCγ2, depletion of EEIG1 abrogated RANKL-induced NFATc1 expression (Figure 2E), but did not inhibit the activation of NF-κB and MAPKs (Supplementary information, Figure S8), indicating that EEIG1 links RANK to a specific signaling cascade. In agreement with this hypothesis, a downregulation of EEIG1 function either by RNAi or by an inhibitory peptide could inhibit RANKL-induced PLCγ2 activation (Figures 7B and 8C). Moreover, EEIG1 could associate with Tec/Btk but not Syk. Further, we observed that after RANKL stimulation, EEIG1, Gab2, Btk, as well as RANK, were recruited to membrane lipid rafts (Figure 6J), which are crucial signaling domains for RANK signal transduction29,30. Thus, it is likely that the RANK signaling complex generated upon RANKL stimulation contributes to the RANK-mediated osteoclastogenic signaling. Although the RANK-EEIG1 interaction is necessary for certain downstream signaling events such as PLCγ2 phosphorylation and NFATc1 activation, EEIG1 knockdown does not induce a dissociation of the RANK signaling complex, suggesting that the interaction is not essential for the formation or maintenance of the RANK signaling complex (Supplementary information Figure S9). Further studies are required to elucidate the mechanisms by which EEIG1 regulates downstream molecules or signaling events in the context of the RANK signaling complex.
In summary, we show that EEIG1 is a novel RANK signaling component that is required for osteoclastogenesis. Moreover, a peptide that targets the EEIG1-RANK interaction has an inhibitory effect on osteoclast formation and RANKL-induced bone destruction. Thus, our study defines an important role of EEIG1 in osteoclast differentiation, and the knowledge gained could contribute to the development of new therapeutics with high efficacy for the treatment of pathological bone resorption.
Materials and Methods
Reagents and plasmids
Recombinant human M-CSF was purchased from R&D systems (Minneapolis, MN, USA). RANKL was purchased from Peprotech EC (London, UK). Antibodies used in the study include: monoclonal anti-FLAG (M2) (Sigma-Aldrich), anti-RANK (Abchem Corporation, Cambridge, UK), anti-phospho-PLCγ2, anti-Flotillin 2 (Cell Signaling Technology, Beverly, MA, USA), anti-GST, anti-PLCγ2, anti-NFATc1, anti-Btk, anti-Tec (Santa Cruz Biotechnology Inc), anti-Gab2 (Millipore), anti-GAPDH (Yeongin Frontier, Seoul, Korea) and rabbit polyclonal anti-EEIG1 (Young-in Frontier, Seoul, Korea and Sigma-Aldrich). DNA fragments encoding the mouse RANK and various truncated mutants were prepared by PCR and cloned into the pEBG expression vector. Site-directed mutagenesis of mouse RANK was carried out using PCR. Mouse EEIG1 was cloned into pFLAG-CMV2, pEBG and pMX-puro expression vectors. The mouse EEIG1 and various truncated mutants were generated by PCR and were cloned into the pFLAG-CMV2 expression vector. Scrambled-shRNA and EEIG1-shRNA (shEEIG1) in the lentiviral pLKO.1-puro vector (Sigma-Aldrich) were subcloned into pMX-puro retroviral expression vector using ClaI/BamHI restriction sites. The target sequence of shEEIG1 is as follow: 5′-GCTTTTGCAAACACGGATG-3′. A retroviral vector, pMX-puro was provided by Dr T Kitamura (University of Tokyo, Tokyo, Japan). The retroviral vector containing a constitutively active form of NFATc1 (NFATc1-CA) has been previously described17.
Cell culture and resorption assay
Murine BMMs were prepared as described previously31. In brief, bone marrow cells were cultured in α-MEM (Hyclone) containing 10% FBS (Hyclone) with M-CSF (30 ng/ml) for 3 days. The culture medium containing M-CSF (30 ng/ml) was replaced every 2 days. The non-adherent cells were removed and adherent cells (BMMs) were harvested. For osteoclst formation, isolated BMMs were treated with or without 200 ng/ml of RANKL in the presence of M-CSF (30 ng/ml) for 3-4 days. The cells were fixed and stained using TRAP staining kit (Sigma). Pink-colored TRAP-positive multinucleated (> 5 nuclei) cells (MNCs) were counted as osteoclast-like cells. Data are presented as the averages of 2 separate experiments done in triplicate ± SD. Plat-E cells (a gift from Dr T Kitamura, University of Tokyo, Tokyo, Japan), 293T cells and RANK 532-537 Mt-expressing RAW 264.7 cells were cultured in DMEM (Hyclone) containing 10% FBS with antibiotics. In the coculture experiments, BMMs (1 × 105 cells/well in 48-well plates) were cultured with calvarial osteoblast cells (1 × 104 cells/well) in the presence of 20 nM 1α, 25-dyhydroxy vitamin D3 and 1 μM prostaglandin E2 to induce the expression of osteoclastogenic M-CSF and RANKL, and to permit the osteoclast differentiation for 7 days. To analyze the bone pit formation, BMMs (1 × 104 cells/well in 96-well plates) were seeded onto dentine slices and allowed to differentiate into osteoclasts in the presence of M-CSF and RANKL for 7 days, with a change of medium after 2 days. Cells on the dentin slices were removed by washing with PBS, and stained with hematoxylin. Pit areas were photographed under a light microscope and analyzed using Image-Pro Plus version 4.5 software (Media-Cybernetics). Dentin slices were kindly provided by M Takami (Showa University, Tokyo, Japan).
GeneChip analysis
For GeneChip analysis (Illumina), total RNA (550 ng) was used for cDNA synthesis by reverse transcription followed by the synthesis of biotinylated cRNA using the Ambion Illumina RNA Amplification Kit (Ambion). The labeled cRNA samples were hybridized to the mouse-6 expression bead array according to the manufacturer's instructions (Illumina).
Western blot analysis and immunoprecipitation
BMMs were incubated with M-CSF (30 ng/ml) in the presence or absence of RANKL (200 ng/ml), LPS (100 ng/ml) or estradiol (E2) (10 nM) for the indicated periods. Then the cells were washed with ice-cold PBS, and lysed in cell extraction buffer (20 mM HEPES pH 7.9, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 5 mM EDTA, 1 mM Na3VO4, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 mM PMSF). The cell lysates harvested at the indicated time points were subjected to western blot analyses using specific antibodies. Immunoprecipitations were performed on whole-cell extracts that were incubated with indicated antibodies overnight, followed by the incubation with protein G-Sepharose (GE Healthcare Life Science) for 2 h. The immunoprecipitates were then subjected to SDS-PAGE and western blotting. 293T cells were processed for analyses 48 h after transfection. Cells were harvested and lysed in cell extraction buffer. The whole cell lysates were subjected to western blot analyses or immunoprecipitation as described above.
Reverse transcription-polymerase chain reaction (RT-PCR)
RT-PCR was performed as previously described32. PCR primers are as follows: EEIG1 sense, 5′-CTCAGTATGGCTGTAGAGGG-3′; antisense, 5′-ATCTTCTCCACAATGTCGTC-3′; Fam102B sense 5′-CTCAAAGTACATCTTGGAAT-3′; antisense, 5′- CGGAAGCTTCCTCTTTA-3′; NFATc1 sense, 5′-CGATTCTCGGACTCTCCAGAGTCG-3′; antisense, 5′-ACCGTGCTCGCAAATTCATCGTT-3′; TRAP sense, 5′-CACGATGCCAGCGACAAGAG-3′; antisense, 5′-TGACCCCGTATGTGGCTAAC-3′; GAPDH sense, 5′-GTGGAGATTGTTGCCATCAACG-3′; antisense, 5′-CAGTGGATGCAGGGATGATGTTCTG -3′
Immunostaining
Cells were placed on poly-L-lysine-coated slides, fixed in 4% paraformaldehyde for 10 min and treated with 0.1% Triton X-100 for 5 min. After blocking with 1% rabbit serum in cold PBS containing 0.2% Triton X-100, cells were sequentially incubated in 5% BSA/PBS for 30 min, 5 μg/ml rabbit anti-EEIG1 peptide antibody or control IgG for 60 min, and then FITC-conjugated anti-rabbit IgG. Cells were mounted using Vectashield medium with DAPI to visualize the nuclei. Images were obtained using a Zeiss Axiovert 200 microscope (Carl Zeiss, Oberkochen, Germany) equipped with a Plan-Neofluor 20×/0.50 objective lens (Carl Zeiss) and Axioversion 3.1 software (Carl Zeiss).
Retroviral-mediated gene transduction
Plat-E cells were transfected with pMX-puro, pMX-puro-EEIG1, pMX-puro-EEIG1shRNA or pRC-NFATc1-CA retroviral vectors by VivaMagic transfection reagent (Vivagen, Seoul, Korea), and the supernatants collected from 24 h to 48 h after transfection were used as the viral stocks. For retroviral infection, BMMs were incubated with the retrovirus stock and polybrene (10 μg/ml) for 6 h. 2 days after exposure to virus, the infected cells were analyzed to determine the infection efficiency, and the rest of the cells were stimulated for western blot analyses or in vitro assays for osteoclast formation.
Real-time quantitative PCR
BMMs were cultured with M-CSF (30 ng/ml) with or without RANKL (200 ng/ml), LPS (100 ng/ml) or estradiol (E2) (10 nM) for the indicated periods. Total RNA was extracted from cultured cells using TRIzol (Invitrogen). 5 μl of cDNA template (1/20 dilution) was used for real-time PCR. Real-time PCR was performed using the KAPA SYBR® FAST universal qPCR Kit (green) (Kapa Biosystems, Boston, MA, USA) and an ABI Prism 7000 sequence detection system (Applied Biosystems). The endogenous EEIG1 mRNA was amplified with specific primers. PCR primers are as follows: EEIG1 sense, 5′-GCACCAATTCCCTAACTGGA-3′; antisense, 5′-AAGTCAGACAGGCTGGAGGA-3′; GAPDH sense, 5′- GTGGAGATTGTTGCCATCAACG-3′; antisense, 5′-CAGTGGATGCAGGGATGATGTTCTG-3′.
GST pull-down assay
293T cells were transfected with various expression vectors as indicated by VivaMagic transfection reagent. After 48 h of transfection, cells were lysed in GST lysis buffer (20mM HEPES pH 7.4, 150 mM NaCl, 150 nM KCl, 10 mM EDTA, 10% glycerol, 1% Nonidet P-40, 1 mM PMSF, 1 μg/ml leupeptin, and 1 μg/ml aprotinin). The whole-cell extracts were pull downed by Glutathione-Sepharose 4B (GE Healthcare Life Science) for 4 h at 4 °C, and analyzed by western blotting.
RAW 264.7 cells with stable expression of the RANK 532-537 Mt
RAW 264.7 cells were transduced with pMX-puro-FLAG-RANK WT, pMX-puro-FLAG-RANK 532-537 Mt vectors using microporation (Invitrogene). Cells were selected in DMEM containing 10% FBS and 4 μg/ml of puromycin. Puromycin-resistant clones were examined for osteoclast formation and were subjected to western blot analyses.
Peptide synthesis and in vitro peptide assay
Control peptides or EIPs were synthesized by Peptron (Daejeon, Korea). Cell-permeable peptides were purified by reverse-phase HPLC with the purity higher than 95% determined by mass spectrometry analysis. The concentrated stock solution of peptides was 10 mM. For osteoclast formation, BMMs were incubated with M-CSF (30 ng/ml) and RANKL (200 ng/ml) in the presence of control peptide or EIP for 4 days and subjected to in vitro assay for osteoclast formation. For western blot analyses, cells were incubated with M-CSF (30 ng/ml) in the presence or absence of RANKL (200 ng/ml), together with control peptide or EIP (10 μM) for 2 days and the whole-cell lysates were subjected to western blot analyses.
RANKL-induced bone destruction
All animal study protocols were approved by the Animal Care Committee of Ewha Laboratory Animal Genomics Center. In these models, more than 5 mice were examined in each group. Seven-week-old C57BL/6 female mice were administered with a local calvarial injection of RANKL (2 mg/kg body weight) 3 times at 2-day intervals. EIP or control peptide (20 mg/kg body weight) was injected five times at 1-day intervals. 1 day after the final injection, all the mice were sacrificed and analyzed as previously described33. We measured the osteoclast number per millimeter of calvarial bone surface, ES/BS (%), and marrow cavity.
Yeast two-hybrid mating assay
A yeast two-hybrid screen was performed using the Matchmaker LexA two-hybrid system (Clontech, CA, USA). For detecting RANK-EEIG1 interactions in yeast, the coding regions containing the test sequence were amplified by PCR, using primers with an EcoRI, or a XhoI site. The PCR product of RANK was subcloned into pLexA for expressing a LexA fusion gene. The EEIG1 and TRAF6 were amplified by PCR using primers with an EcoRI and XhoI site, and subcloned into pB42AD. The LexA-RANK with pB42AD-EEIG1 or LexA-RANK with pB42AD-TRAF6 were cointroduced into yeast strain EGY48 (p8op-lacZ), and the cotransformants were selected in SD/-His/-Trp/-Ura plates. These colonies were subjected to β-galactosidase assays on an SD plate containing 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal). Interaction was demonstrated by growth of the cotransformants as blue colonies.
Isolation of lipid rafts
Lipid rafts were isolated by a discontinuous sucrose density gradient ultracentrifugation. Cells were washed with ice-cold PBS and lysed in 1 ml of ice-cold TNE buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 μg/ml aprotinin, 1 μg/ml leupeptin and 1 mM PMSF) containing 0.5% Brij 98. The lysate was incubated on ice for 30 min and mixed with an equal volume of 85% (w/v) sucrose in TNE. The mixture was overlaid with 6 ml of 35% sucrose, which in turn was topped with 3 ml of 5% sucrose. The gradient was subjected to ultracentrifugation at 40 000 rpm in an SW 41 Ti rotor (Beckman Instruments) for 18 h at 4 °C. After centrifugation, 11 equal fractions of 1 ml each were collected from the top of the gradient and lipid raft fractions (fraction 3-5) and non-raft fractions (fraction 9-12). The fractions were concentrated by trichloroacetic acid (TCA) precipitation. An equal volume of 100% TCA was added to each fraction and incubated at −20 °C for 20 min. Samples were then centrifuged at 12 000 rpm for 15 min at 4 °C. Pellets were washed with 1 ml of ice-cold acetone. Those pellets were resuspended in cell extraction buffer (20 mM HEPES pH 7.9, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 5 mM EDTA, 1 μg/ml aprotinin, 1 μg/ml leupeptin and 1 mM PMSF), and western blot analysis was performed. Fractions were analyzed for the lipid raft marker protein flotillin 2.
Statistics
The results are presented as mean ± SD from at least 3 independent experiments. The statistical significances of differences were determined using Student's t-test. P < 0.05 was considered statistically significant.
References
Harada S, Rodan GA . Control of osteoblast function and regulation of bone mass. Nature 2003; 423:349–355.
Teitelbaum SL . Bone resorption by osteoclasts. Science 2000; 289:1504–1508.
Boyle WJ, Simonet WS, Lacey DL . Osteoclast differentiation and activation. Nature 2003; 423:337–342.
Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ . Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev 1999; 20:345–357.
Wong BR, Josien R, Lee SY, Vologodskaia M, Steinman RM, Choi Y . The TRAF family of signal transducers mediates NF-κB activation by the TRANCE receptor. J Biol Chem 1998; 273:28355–28359.
Asagiri M, Takayanagi H . The molecular understanding of osteoclast differentiation. Bone 2007; 40:251–264.
Asagiri M, Sato K, Usami T, et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med 2005; 202:1261–1269.
Wagner EF, Eferl R . Fos/AP-1 proteins in bone and the immune system. Immunol Rev 2005; 208:126–140.
Jang HD, Shin JH, Park DR, et al. Inactivation of glycogen synthase kinase-3β is required for osteoclast differentiation. J Biol Chem 2011; 286:39043–39050.
Mao D, Epple H, Uthgenannt B, Novack DV, Faccio R . PLCγ2 regulates osteoclastogenesis via its interaction with ITAM proteins and GAB2. J Clin Invest 2006; 116:2869–2879.
Taguchi Y, Gohda J, Koga T, Takayanagi H, Inoue J . A unique domain in RANK is required for Gab2 and PLCγ2 binding to establish osteoclastogenic signals. Genes Cells 2009; 14:1331–1345.
Koga T, Inui M, Inoue K, et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature 2004; 428:758–763.
Mocsai A, Humphrey MB, Van Ziffle JA, et al. The immunomodulatory adaptor proteins DAP12 and Fc receptor γ-chain (FcRγ) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci USA 2004; 101:6158–6163.
Kaifu T, Nakahara J, Inui M, et al. Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J Clin Invest 2003; 111:323–332.
Faccio R, Takeshita S, Zallone A, Ross FP, Teitelbaum SL . c-Fms and the αvβ3 integrin collaborate during osteoclast differentiation. J Clin Invest 2003; 111:749–758.
Shinohara M, Koga T, Okamoto K, et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell 2008; 132:794–806.
Kim H, Choi HK, Shin JH, et al. Selective inhibition of RANK blocks osteoclast maturation and function and prevents bone loss in mice. J Clin Invest 2009; 119:813–825.
Wang DY, Fulthorpe R, Liss SN, Edwards EA . Identification of estrogen-responsive genes by complementary deoxyribonucleic acid microarray and characterization of a novel early estrogen-induced gene: EEIG1. Mol Endocrinol 2004; 18:402–411.
Kim K, Kim JH, Lee J, et al. Nuclear factor of activated T cells c1 induces osteoclast-associated receptor gene expression during tumor necrosis factor-related activation-induced cytokine-mediated osteoclastogenesis. J Biol Chem 2005; 280:35209–35216.
Wada T, Nakashima T, Oliveira-dos-Santos AJ, et al. The molecular scaffold Gab2 is a crucial component of RANK signaling and osteoclastogenesis. Nat Med 2005; 11:394–399.
Takayanagi H, Kim S, Koga T, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 2002; 3:889–901.
Takayanagi H, Kim S, Matsuo K, et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-β. Nature 2002; 416:744–749.
Wada T, Nakashima T, Hiroshi N, Penninger JM . RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med 2006; 12:17–25.
Kobayashi N, Kadono Y, Naito A, et al. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J 2001; 20:1271–1280.
Gohda J, Akiyama T, Koga T, Takayanagi H, Tanaka S, Inoue J . RANK-mediated amplification of TRAF6 signaling leads to NFATc1 induction during osteoclastogenesis. EMBO J 2005; 24:790–799.
Lee SH, Kim T, Jeong D, Kim N, Choi Y . The tec family tyrosine kinase Btk regulates RANKL-induced osteoclast maturation. J Biol Chem 2008; 283:11526–11534.
Nakashima T, Takayanagi H . The dynamic interplay between osteoclasts and the immune system. Arch Biochem Biophys 2008; 473:166–171.
Darnay BG, Ni J, Moore PA, Aggarwal BB . Activation of NF-κB by RANK requires TNF receptor-associated factor (TRAF) 6 and NF-kappaB-inducing kinase. Identification of a novel TRAF6 interaction motif. J Biol Chem 1999; 274:7724–7731.
Ha H, Kwak HB, Le SW, Kim HH, Lee ZH . Lipid rafts are important for the association of RANK and TRAF6. Exp Mol Med 2003; 35:279–284.
Ha H, Kwak HB, Lee SK, et al. Membrane rafts play a crucial role in receptor activator of nuclear factor κB signaling and osteoclast function. J Biol Chem 2003; 278:18573–18580.
Choi HK, Kim TH, Jhon GJ, Lee SY . Reactive oxygen species regulate M-CSF-induced monocyte/macrophage proliferation through SHP1 oxidation. Cell Signal 2011; 23:1633–1639.
Han SY, Lee NK, Kim KH, et al. Transcriptional induction of cyclooxygenase-2 in osteoclast precursors is involved in RANKL-induced osteoclastogenesis. Blood 2005; 106:1240–1245.
Takayanagi H, Ogasawara K, Hida S, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature 2000; 408:600–605.
Acknowledgements
We thank T Kitamura (University of Tokyo, Japan) for PLAT-E cells and pMX vectors. This work was supported by grants from the National Research Foundation of Korea (NRF) funded by the Korean government (MEST, R0A-2008-000-20001-0, R31-2008-000-10010-0, No 2012R1A5A1048236, No 2012M3A9C5048708). SYL is a Yonam Foundation Scholar. HK, HK, EJ and JL were supported by the second stage of the Brain Korea 21 Project.
Author information
Authors and Affiliations
Corresponding author
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Expression of EEIG1 in calvarial bones injected with RANKL. (PDF 31 kb)
Supplementary information, Figure S2
EEIG1 localizes in the cytoplasm. (PDF 41 kb)
Supplementary information, Figure S3
Transactivation of EEIG1 promoter construct by NFATc1. (PDF 51 kb)
Supplementary information, Figure S4
Effect of siRNA specific for EEIG1 on osteoclast formation. (PDF 71 kb)
Supplementary information, Figure S5
Effect of EEIG1 knock-down on osteoclast formation at multiple time points. (PDF 421 kb)
Supplementary information, Figure S6
The C-terminal region of RANK spanning amino acids 532-592 interacts with EEIG1. (PDF 60 kb)
Supplementary information, Figure S7
Effect of RANK 532-537 Mt expression on NF-κB activation. (PDF 42 kb)
Supplementary information, Figure S8
Effect of EEIG1 silencing on NF-κB and MAP kinase activation. (PDF 186 kb)
Supplementary information, Figure S9
EEIG1 silencing does not induce dissociation of RANK signaling complex. (PDF 45 kb)
Supplementary information
Materials and Methods (PDF 161 kb)
Rights and permissions
About this article
Cite this article
Choi, H., Kang, H., Jung, E. et al. Early estrogen-induced gene 1, a novel RANK signaling component, is essential for osteoclastogenesis. Cell Res 23, 524–536 (2013). https://doi.org/10.1038/cr.2013.33
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2013.33
