
Abstract
Toll-like receptor (TLR)-mediated inflammatory response could negatively affect bone metabolism. In this study, we determined how osteogenesis is regulated during inflammatory responses that are downstream of TLR signaling. Human primary osteoblasts were cultured in collagen gels. Pam3CSK4 (P3C) and Escherichia coli lipopolysaccharide (EcLPS) were used as TLR2 and TLR4 ligand respectively. Porphyromonas gingivalis LPS having TLR2 activity with either TLR4 agonism (Pg1690) or TLR4 antagonism (Pg1449) and mutant E. coli LPS (LPxE/LPxF/WSK) were used. IL-1β, SH2-containing inositol phosphatase-1 (SHIP1) that has regulatory roles in osteogenesis, alkaline phosphatase and mineralization were analyzed. 3α-Aminocholestane (3AC) was used to inhibit SHIP1. Our results suggest that osteoblasts stimulated by P3C, poorly induced IL-1β but strongly upregulated SHIP1 and enhanced osteogenic mediators. On the contrary, EcLPS significantly induced IL-1β and osteogenic mediators were not induced. While Pg1690 downmodulated osteogenic mediators, Pg1449 enhanced osteogenic responses, suggesting that TLR4 signaling annuls osteogenesis even with TLR2 activity. Interestingly, mutant E. coli LPS that induces weak inflammation upregulated osteogenesis, but SHIP1 was not induced. Moreover, inhibiting SHIP1 significantly upregulated TLR2-mediated inflammatory response and downmodulated osteogenesis. In conclusion, these results suggest that induction of weak inflammatory response through TLR2 (with SHIP1 activity) and mutant TLR4 ligands could enhance osteogenesis.
Similar content being viewed by others


Introduction
Osteoblasts play a central role in bone remodeling by directly regulating osteogenesis and also mediate bone resorption by coordinating with bone resorbing osteoclasts. During physiologic conditions, this dynamic bone remodeling is controlled by precise orchestration of osteoblasts and osteoclasts.1 An imbalance in this homeostasis is linked to metabolic bone diseases, such as osteoporosis and inflammatory bone diseases.2 During chronic inflammatory conditions, such as arthritis and periodontitis, bone homeostasis is skewed towards osteoclast-induced bone resorption.1
Toll-like receptors (TLRs) are principal pattern recognition receptors that recognize a broad category of microbial structures. TLR2 recognizes structures from Gram-positive bacteria and TLR4 receptor complex recognizes lipopolysaccharide (LPS) from Gram-negative bacteria.3 TLR signaling can indirectly or directly affect bone homeostasis. As an indirect mechanism, inflammatory cytokines that are downstream of TLR signaling mediate bone loss. For instance, osteoblasts also express TLRs4 and secrete inflammatory cytokines, which in turn induce osteoclast activation. Moreover, osteogenic differentiation can be inhibited during inflammation which also contributes to inflammatory bone loss.5
Osteoblasts, similar to macrophages,6 downmodulate cytokine secretion during chronic TLR challenge.7 Monocytes or macrophages, for instance, upregulate anti-inflammatory SH2-containing inositol phosphatase-1 (SHIP1)8 and regulate inflammatory response. SHIP1 also regulates osteoblast proliferation, differentiation and survival via the PI3K/Akt signaling.9 Currently, little is known as to how direct TLR activation in osteoblasts regulates TLR downstream events and osteogenic functions. The objectives of this study were to determine how inflammatory responses downstream of TLR2 and TLR4 signaling and SHIP1 activity regulate osteogenesis.
Materials and methods
Human primary osteoblast and monocyte culture
Human primary osteoblasts were obtained from ScienCell Research Laboratories (Carlsbad, CA, USA). Osteoblasts (from three different donors) were grown in α-MEM supplemented with penicillin, streptomycin and 10% heat inactivated fetal bovine serum and all reagents were purchased from Sigma-Aldrich (St Louis, MO, USA). Osteoblasts were employed between passages of three - five and culture media was further supplemented with 100 µg·mL−1 of ascorbic acid and β-glycerophosphate (Sigma-Aldrich) to induce differentiation. Three-dimensional collagen gels with osteoblasts were cultured in cell culture wells of 12 mm diameter containing a 0.4 mm pore sized polyester membrane trans-wells (Corning Incorporated, Corning, NY, USA) as previously described.10 Collagen gels were prepared by mixing 3 mg·mL−1 of type I rat tail collagen (Invitrogen, Carlsbad, CA, USA), α-MEM with 10% heat inactivated fetal bovine serum and osteoblast suspension (final concentration of 1.33×105 cells per mL). Three milliliters of gel solution was added to each transwell resulting in 4×105 cells per transwell. The gels were allowed to undergo initial contraction by incubating at 37 °C for 2 h. Two milliliters of culture medium was subsequently added and the cultures were incubated at 37 °C in a humidified atmosphere with 5% CO2 for 7 days. Half of the media was changed and replenished every 2 days during the culture. Peripheral blood monocytes were isolated from mononuclear fraction by means of adherence and cultured in RPMI1640 as described previously.6,11 The percentage of viable osteoblasts or monocytes (typically >90%) was monitored by trypan blue (Invitrogen) exclusion.
TLR ligands, bacterial strains and LPS purification
Pam3CSK4 (P3C) and Escherichia coli LPS (EcLPS) were employed as pure TLR2 and TLR4 ligands respectively and were obtained from Invitrogen. LPS preparations from E. coli mutant strains and from P. gingivalis were previously described12,13 and are summarized as follows: the E. coli msbB strain contains an inactivating mutation in the genetic locus that encodes the lipid A secondary myristoyl transferase, msbB. As a consequence, these bacteria produce penta-acylated, bis-phosphorylated lipid A lacking the secondary myristate at the 3′ position of the glucosamine disaccharide backbone. Therefore, this lipid A resembles the structure of the penta-acylated lipid A. The coding sequences for Francisella novicida LpxE and LpxF lipid A phosphatases were previously cloned into the expression vector pWSK29, yielding the plpxE and plpxF constructs, respectively. To create the recombinant E. coli msbB, the vectors pWSK29, plpxE and plpxF were transformed into the E. coli msbB strain by electroporation. The resulting recombinant strains were designated as WSK, LPxE and LPxF respectively. LPS was isolated from these E. coli mutant strains and from Porphyromonas gingivalis ATCC 33277 strain using cold MgCl2–C2H5OH procedure. LPS was purified by using TRI Reagent approach and crude LPS was subjected to modified Folch extraction to remove phospholipids and further treated to remove trace amounts of proteins detected by enhanced colloidal gold staining. Total fatty acid content of LPS was analyzed by gas chromatography and the purified LPS preparations were subsequently analyzed by negative ion MALDI–TOF–MS for the structural determination. Two detected ion peaks that were clustered around mass of 1 690 and 1 435/1 449 were designated as penta-acylated P. gingivalis LPS (Pg1690) and tetra-acylated P. gingivalis (Pg1449), respectively. All LPS preparations were employed at 1 μg·mL−1 (unless otherwise specified).
Cytokines, antibodies and microplate assays
Recombinant TNF-α was purchased from R and D Systems (Minneapolis, MN, USA) and 1–100 ng·mL−1 of TNF-α was used in cell cultures. Anti-TLR2 (clone TL2.1) and anti-TLR4 (clone HTA125) directly conjugated (for flow cytometry) or purified (for functional blocking) antibodies were purchased from eBiosciences (San Diego, CA, USA) and were employed at a concentration of 10 µg·mL−1. 3 α-aminocholestane (3AC), an inhibitor of SHIP1 was purchased from Echelon (Salt Lake City, UT, USA). Malachite Green assay, a fluorescent polarization assay that detects the 5′-inositol phosphatase activity of SHIP1, was purchased from Echelon (Salt Lake City, UT, USA) and employed to measure SHIP1 activity.
Real-time PCR analysis
The cultures were washed twice with PBS and were incubated in 300 µL PBS containing 1 mg·mL−1 type II collagenase as previously described.6,11 Total RNA was extracted using QIAGEN (Germantown, MD, USA) RNeasy mini kits according to the manufacturer’s instructions. Avian reverse transcriptase (RT)-PCR first-strand kits (Sigma, St Louis, MO, USA) were used to synthesize cDNA from total RNA. The concentration of total RNA was determined at 260 nm (OD260) and the purity of the cDNA was determined at OD260/OD280. Nucleotide sequences were obtained from PubMed (National Center for Biomedical Information) and the primers were custom designed using primer3 software. The following primers were employed: TLR2, 5′-GGA GGC TGC ATA TTC CAA GG-3′ and 5′-GCC AGG CAT CCT CAC AGG-3′; TLR4, 5′-CTT GAC CTT CCT GGA CCT CTC-3′ and 5′-ACT TGG AAA ATG CTG TAG TTC C-3′; IL-1beta, 5′-GGC AGA AAG GGA ACA GAA AGG-3′ and 5′-AGT GAG TAG GAG AGG TGA GAG AGG-3′; SHIP1, 5′-CTG AGG CTG GAA GAA AAA CG-3′ and 5′-GCA CTT AGC ACA GCA CTT GG-3′; beta-actin, 5′-ACT CTT CCA GCC TTC CTT CC-3′ and 5′-GTT GGC GTA CAG GTC TTT GC-3′. Real-time RT-PCR analysis was performed using the MyCycler (Bio-Rad, Hercules, CA, USA) with SYBR Green kits (Bio-Rad) by standard curve method as previously described. For each transcript analyzed, a standard curve with predetermined concentrations and serial diluted respective PCR amplification products from 0.1 ng to 0.000 01 ng was constructed. This approach allows the standards to be amplified in the same way as the template cDNA in the unknown samples since the product sequence and size are identical. Levels of β-actin mRNA served as an internal control to normalize samples for variations in sample volume loading, presence of inhibitors and nucleic acid recovery during RNA extraction and cDNA synthesis procedures. All analyses were performed in triplicate.
Flow cytometry analysis
Flow cytometry analysis was performed as previously described.14,15 Briefly, cultured osteoblasts or monocytes were incubated with directly conjugated TLR2 and TLR4 monoclonal antibodies or their respective isotype matched controls for 30 min at 4 °C, washed and the cells were fixed in 1% paraformaldehyde and protected from direct light until analyzed by flow cytometry. Analysis was performed with LSR-II (Becton Dickinson, San Jose, CA, USA) and the expression of TLR2 and TLR4 was determined as the percentage of positive cells in the relevant population defined by forward scatter and side scatter characteristics. Expression levels were also evaluated by assessing mean fluorescence intensity indices calculated by relating mean fluorescence intensity noted with the relevant monoclonal antibody to that of isotype control monoclonal antibodies for samples labeled in parallel and acquired using identical settings.
Determining alkaline phosphatase levels in the cell culture
Alkaline phosphatase activity was analyzed by both (i) histochemical and (ii) biochemical/colorimetric methods (all reagents purchased from Sigma-Aldrich) as previously described.10 (i) Histochemical staining procedure was performed by employing nitro blue tetrazolium (NBT) and 5-bromo-4-chloro-3-indolyl phosphate (BCIP) and a substrate buffer (0.1 mol·L−1 Tris, 100 mmol·L−1 sodium chloride, 5 mmol·L−1 MgCl2, pH 9.5). The cultures were washed with hanks balanced salt solution and incubated with BCIP/NBT substrate solution for 10 min. The color development was visually monitored and the cultures were washed with distilled water and analyzed by capturing images with Nikon Eclipse 400 microscope (Tokyo, Japan) at ×1 magnification. (ii) For the biochemical assay, the cultures were washed twice with PBS and were incubated in 300 µL PBS containing 1 mg·L−1 type II collagenase. The cell culture digest was transferred to a 96-well flat bottom plate and incubated with 5 mmol·L−1 p-nitrophenyl phosphate in 50 mmol·L−1 glycine and 1 mmol·L−1 MgCl2, pH 10.5 at 37 °C for 2 h. The alkaline phosphatase activity was detected by measuring the absorbance of the p-nitrophenol product formed at OD405 on a microplate reader. The alkaline phosphatase activity was quantitated from known concentrations of alkaline phosphatase and generating a standard curve. All analyses were performed in triplicate.
In vitro mineralized nodule formation assay and quantification of calcium accumulation
In vitro mineralized nodule formation and calcium accumulation was analyzed by both (i) histochemical and (ii) biochemical/colorimetric methods (all reagents were purchased from Sigma-Aldrich) as previously described.10 For histochemical analyses, cultures were washed three times in distilled water and stained with 2% alizarin red solution (pH 4.1–4.3) for 3 min. The excess dye was washed and the amount of orange–red calcium deposits were analyzed by capturing images with Nikon Eclipse 400 microscope at ×1 magnification. For biochemical analyses of calcium deposition, cell cultures were washed thoroughly in deionized water and incubated overnight in 0.5 mL of 2 mol·L−1 HCl. The following day, the acidic solution was neutralized with 0.5 mL of 2 mol·L−1 NaOH and 5 μL of the neutralized solution from each group was placed in 96-well plate. A standard curve was generated by using serial dilutions of CaCO3. One hundred microliters of color reagent (0.1% o-cresolphthalein complexone and 1% 8-hydroxyquinoline) and 100 μL of buffer reagent (500 mmol·L−1 2-amino-2-methyl-1-propanol, pH 10.7) was added and incubated for 15 min at room temperature. Calcium deposition was measured by determining the absorbance at OD570 using a microplate reader. All analyses were performed in triplicate.
Data analysis
All the experimental data were prepared and statistical analysis performed by employing Sigma Plot 11.0 software (San Jose, CA, USA). Student’s t-test and one way ANOVA (all pair-wise multiple comparison procedures) were performed to determine statistical significance.
Results
Osteoblasts resist upregulation of TLR2 and TLR4 in response to TLR ligands
Osteoblasts express TLRs and play a role in inflammatory response by secreting cytokines. However, little is known about the constitutive expression of TLRs and how TLR expression is regulated in osteoblasts. In this study, the expression of TLR2 and TLR4 in osteoblasts was compared with that of peripheral blood monocytes. Synthetic P3C and EcLPS were employed as TLR2 and TLR4 ligands respectively. Unstimulated cells or osteoblasts/monocytes stimulated with either P3C or EcLPS were analyzed for the expression of TLR2 and TLR4 through real-time RT-PCR and flow cytometry. Relative to blood monocytes (Figure 1i–1k and 1n), osteoblasts (Figure 1a–1c and 1f) have low constitutive mRNA and protein levels for TLR2 and TLR4. P3C was a poor inducer of either TLR2 or TLR4 in osteoblasts (Figure 1a–1b, 1d and 1g). P3C upregulated TLR2 relative to TLR4 in monocytes (Figure 1i, 1j, 1l and 1o). Monocytes upregulated TLR2 and TLR4 in response to EcLPS (Figure 1i, 1j, 1m and 1p). However, relative to monocytes, osteoblasts did not upregulate TLR2 and TLR4 when stimulated with EcLPS (Figure 1a, 1b, 1e and 1h).

Osteoblasts resist upregulation of TLR2 and TLR4 in response to TLR ligands. Human primary osteoblasts between passages of 3–5 and blood monocytes were cultured as described in the section on ‘Materials and methods’. Osteoblasts or monocytes were either unstimulated or stimulated with either P3C (1 µg·mL−1) or EcLPS (1 µg·mL−1) for 1 week. Total RNA was extracted and quantitated and its purity confirmed as described in the section on ‘Materials and methods’. cDNA was synthesized from identical quantities of toral RNA from all the samples. The purity of cDNA was determined by analysis of the OD260/OD280 ratio. mRNA quantitation was performed by real-time RT-PCR analysis. Levels of β-actin mRNA served as an internal control to normalize samples. All analyses were performed in triplicate. RT-PCR was performed as described in the section on ‘Materials and methods’ to determine the expression of TLR2 (a, i) and TLR4 (b, j). Surface expressions of TLR2 (c–e; k–m) and TLR4 (f–h; n–p) were analyzed through flow cytometry as described in materials and methods. Isotype-matched antibody controls (Iso) are shown for TLR2 (c, k) and TLR4 (f, n) in osteoblasts and monocytes. Shown is representative data from two individual experiments. *P=0.017 and **P=0.004; t-test.
TLR2- but not TLR4-activated osteoblasts enhances osteogenic functions
Expression of alkaline phosphatase (ALP) and in vitro mineralization are suggestive of osteogenic functions and these could be negatively regulated when osteoblasts are challenged with microbial stimuli.16 Osteoblasts were either unstimulated or stimulated with 1 μg·mL−1 of either P3C or EcLPS and were cultured for 1 week in three-dimensional collagen gels as previously reported10 and summarized in materials and methods. The expression of ALP and in vitro mineralization were analyzed through histochemical (Figure 2a and 2c) and biochemical approaches (Figure 2b and 2d). The data suggests that P3C significantly upregulated ALP (Figure 2a and 2b) and mineralization (Figure 2c and 2d) and on the contrary, EcLPS did not alter the expression of ALP (Figure 2a and 2b) and mineral deposition relative to that of unstimulated control (Figure 2c and 2d). It is known that TLR2, relative to that of TLR4 signaling, is a weak inducer of inflammation which suggests that regulatory mechanisms during inflammatory response could modulate osteogenesis. We have previously reported that SHIP1 is a negative regulator of inflammatory responses during inflammatory conditions6 and SHIP1 has been reported to play a role in osteogenesis.9 We therefore determined the kinetics of inflammatory cytokines such as IL-1β and SHIP1 when osteoblasts were stimulated with P3C and EcLPS (Figure 2e–2h). The data suggests that, EcLPS significantly upregulated IL-1β (Figure 2e), but SHIP1 was not induced (Figure 2f). On the contrary, P3C poorly induced the expression of IL-1β (Figure 2e), but significantly upregulated SHIP1 (Figure 2f). Expression of SHIP1 and IL-1β bear an inverse relationship trend (although not statistically significant) when osteoblasts were stimulated with P3C (Figure 2g) but not with EcLPS (Figure 2h).

TLR2 but not TLR4 activated osteoblasts enhances osteogenic functions. Human primary osteoblasts were cultured in three-dimensional collagen gels and were either unstimulated or stimulated for one week with either P3C or EcLPS at a concentration of 1 μg·mL−1. Regulation of ALP was determined by (a) employing NBT and BCIP and a Substrate buffer (0.1 mol·L−1 Tris, 100 mmol·L−1 sodium chloride, 5 mmol·L−1 MgCl2, pH 9.5) and (b) biochemical method by measuring the absorbance of the p-nitrophenol product formed at OD405 nm and by histochemical method. In vitro mineralization and calcium deposition was analyzed by histochemical method by using alizarin red staining (c) and biochemical method (d) by using color reagent (0.1% o-cresolphthalein complexone and 1% 8-hydroxyquinoline) and buffer reagent (500 mmol·L−1 2-amino-2-methyl-1-propanol, pH 10.7) and determining the absorbance at OD570 (nm). All analyses were performed in triplicate and images were captured at ×1 magnification. Kinetics of expressions of IL-1β (e) and SHIP1 (f) were quantitated through real-time RT-PCR by generating a standard curve and through normalization with β-actin levels. Correlation of the expression of IL-1β and SHIP1 when stimulated with P3C (g) and EcLPS (h). The data is representative of four separate experiments. *P≤0.005; t-test.
Weak TLR4 agonist and antagonist LPS structures enhance osteogenic functions
Increased levels of hemin produce mutant P. gingivalis lipid A structures.17 Of these, Pg1690 is a TLR4 agonist and Pg1449 is a TLR4 antagonist.13 However, both these LPS preparations have residual lipopeptides with equivalent TLR2 activity.13 Due to structural monophosphorylation and the number and length of fatty acid chains, Pg1690 is a weak TLR4 agonist relative to the canonical EcLPS and Pg1449 is weaker LPS relative to Pg1690 and thereby, exerts TLR4 antagonism. These LPS preparations are especially relevant as they enable us to know how TLR2 responses are regulated during TLR4 activation or TLR4 antagonism. We employed Pg1690 and Pg1449 LPS preparations to determine how weak or antagonistic TLR responses regulate osteogenesis. Relative to EcLPS (Figure 2a–2d), Pg1690 moderately induced ALP (Figure 3a and 3b) and mineral deposition (Figure 3c and 3d). Interestingly, Pg1449, being a TLR4 antagonist (and a TLR2 agonist) was a potent inducer of ALP (Figure 3a and 3b) and mineralization (Figure 3c and 3d). Also, increasing concentration of Pg1449 relative to Pg1690, enhanced ALP (Figure 3a and 3b) and mineralization (Figure 3c and 3d) in a dose-dependent manner. An interesting finding is that, Pg1690, being both TLR2 and TLR4 agonist, was a poor inducer of SHIP1 (Figure 3f), but significantly upregulated IL-1β (Figure 3e). On the contrary, Pg1449 being a TLR2 agonist but a TLR4 antagonist, significantly upregulated SHIP1 (Figure 3f) with poor IL-1β expression (Figure 3g). These results suggest that TLR4 signaling could annul TLR2-induced SHIP1 and upregulate inflammatory response. TLR2 (T2), TLR4 (T4) and isotype-matched (Iso) functional blocking antibodies were employed (Figure 3g and 3h) and the data suggest that SHIP1 expression is induced by TLR2 but not by TLR4. It should be noted that the blocking antibodies might also have some TLR activity, as stimulation with Pg1449 along with T4 did not induce SHIP1 expression (Figure 3h). The studies with EcLPS (Figure 2) and with LPS structures from P. gingivalis (Figure 3) suggest that the potency of TLR4 activation could be a key regulator in negatively interfering with osteogenesis.

Weak TLR4 agonist and antagonist LPS structures enhance osteogenic functions. Osteoblasts cultured in three-dimensional collagen gels were either unstimulated or stimulated for one week with either Pg1690 or Pg1449 LPS at a concentration of 1 μg·mL−1. Also, Pg1690 and Pg1449 were combined and employed at 2:1 and 1:2 ratios with the final total concentration of 1 μg·mL−1. Regulation of ALP was determined by histochemical (a) and through biochemical (b) methods. In vitro mineralization and calcium deposition was analyzed by histochemical (c) and biochemical (d) methods. All analyses were performed in triplicate and images were captured at ×1 magnification. Expressions of IL-1β (e) and SHIP1 (f) were quantitated through real-time RT-PCR by generating a standard curve and through normalization with β-actin levels. TLR2 (T2), TLR4 (T4) or isotype-matched (Iso) functional blocking antibodies were employed at a concentration of 20 μg·mL−1 to determine how IL-1β (g) and SHIP1 (h) are regulated with TLR activation. **All pair-wise multiple comparison procedures (Holm–Sidak method) with overall significance of 0.05. The differences in the mean values among all the groups were statistically significant (P<0.001) except that of unstimulated control and Pg1690; representative data from two separate experiments is presented. *All pair-wise multiple comparison procedures (Dunn’s Method), P≤0.006 relative to unstimulated control; •P=0.004, t-test.
Altering the potency of TLR4 responses enhances osteogenic functions
In this study, we aimed to determine whether low doses of EcLPS have any positive influence on osteogenic functions. The data suggests that EcLPS employed at 1 ng·mL−1 moderately induced ALP (Figure 4a and 4b) and mineralization (Figure 4c and 4d) and this was downmodulated in a dose-dependent manner. We subsequently determined how mutant LPS structures that are weak TLR4 agonists (and therefore, weak inducers of inflammatory response) regulate osteogenic mediators. Mutant EcLPS structures namely, LPxF, LPxE and WSK were used along with P3C (positive control) and canonical EcLPS (negative control). When compared with EcLPS, osteoblasts stimulated with LPxF, LPxE or WSK significantly induced the expression of ALP (Figure 4e and 4f) and upregulated in vitro mineralization (Figure 5g and 5h). However, the induction of ALP and mineral deposition was more potent when osteoblasts were stimulated with P3C (Figure 5e and 5h). Moreover, SHIP1 was not significantly upregulated when osteoblasts were stimulated with any of the mutant LPS (i.e., LPxF, LPxE or WSK) relative to stimulating with P3C (Figure 5j). Despite lower induction of SHIP1, IL-1β was also not strongly induced by these mutant LPS structures relative to stimulation with the canonical EcLPS (Figure 5i). These results suggest that, irrespective of the potency of LPS structures that target TLR4, SHIP1 expression is not under the control of TLR4 signaling. Therefore, upregulation of osteogenic mediators in response to LPxF, LPxE or WSK, could be facilitated due to a weak inflammatory response mediated by these mutant LPS structures.

Altering the potency of TLR4 responses enhances osteogenic functions. Osteoblasts cultured in three-dimensional collagen gels were either unstimulated or stimulated for one week with either 1 000 ng·mL−1 P3C or 1–1 000 ng·mL−1 of EcLPS (a–j). Additionally, mutant LPS structures namely LPxF, LPxE and WSK were purified as described in materials and methods and were employed to stimulate osteoblasts at a concentration of 1 μg·mL−1 (e–j). Regulation of ALP was determined through histochemical (a, e) and biochemical (b, f) methods and regulation of in vitro mineralization/calcium deposition was analyzed by histochemical (c, g) and biochemical (d, h) methods. All analyses were performed in triplicate and images were captured at ×1 magnification. Expressions of IL-1β (i) and SHIP1 (j) were quantitated through real-time RT-PCR by generating a standard curve and through normalization with β-actin levels. Expression of ALP (a, b) and mineralization (c, d) were downmodulated with EcLPS in a dose-dependent manner. Relative to EcLPS, mutant LPS structures (LPxF, LPxE and WSK) were potent inducers of ALP (e, f) and in vitro mineralization (g, h). Real-time PCR data suggests that mutant LPS structures (LPxF, LPxE and WSK) were weak inducers of both IL-1β (i) and SHIP1 (j) relative to EcLPS-induced IL-1β (i) and P3C-induced SHIP1 (j). The experiments were repeated twice and representative data is shown. *P≤0.001, t-test; **P≤0.05, t-test.

Potency of the inflammatory response negatively regulates and overrules TLR2-induced osteogenic functions. Three-dimensional osteoblasts cultured in collagen gel cultures were stimulated for one week with either 1 μg·mL−1 of P3C along with 0–100 ng·mL−1 of TNF-α (a–d). Regulation of ALP was determined through histochemical (a) and biochemical (b) methods. In vitro mineralization and calcium deposition was analyzed by histochemical (c) and biochemical (d) methods. All analyses were performed in triplicate and images were captured at ×1 magnification. When stimulated with TNF-α, a dose-dependent downmodulation of ALP expression and Ca deposition that is induced by P3C is shown. *P≤0.001, t-test.
Potency of the inflammatory response negatively regulates and overrules TLR2-mediated osteogenic mediators
During inflammatory conditions, cytokines such as TNF-α negatively affect osteogenic functions.18 In order to determine how osteogenesis mediated by TLR2 is affected by the potency of the inflammatory response, TNF-α was simultaneously added in doses from 0–100 ng·mL−1 to osteoblasts that were unstimulated or stimulated with either P3C or EcLPS (Figure 5a and 5d). Relative to unstimulated controls, TNF-α at a dose of 100 ng·mL−1 downmodulated ALP expression (Figure 5a and 5b) and in vitro mineralization (Figure 5c and 5d). ALP (Figure 5a and 5b) and mineralization (Figure 5c and 5d) upregulated by P3C were inhibited by TNF-α in a dose-dependent manner. These findings suggest that osteogenic mediators induced by TLR2 signaling could be negatively affected by the potency of the inflammatory response mediated by strong TLR4 responses (Figures 2–4) and cytokines such as TNF-α (Figure 5).
Inhibiting SHIP1 enhances TLR2-induced inflammatory response
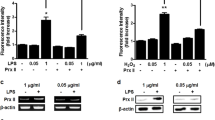
Data in Figures 2 and 3 suggest that TLR2 enhanced osteogenic mediators and levels of SHIP1. We therefore aimed to determine the role of SHIP1 in regulating osteogenesis. 3AC, an inhibitor of SHIP1,19 was employed to determine how SHIP1 regulated IL-1β (Figure 6) and osteogenic mediators (discussed in Figure 7). On the bases of dose-dependent cytotoxicity in osteoblasts (Figure 6a), 1–10 µmol·mL−1 3AC was employed and expression of IL-1β that was weakly induced by TLR2 was significantly upregulated when SHIP1 was inhibited by 3AC in a dose-dependent manner (data not shown). P3C and Pg1449 (TLR2 agonist but TLR4 antagonist) were employed as these ligands significantly induced SHIP1 and EcLPS was used as a negative control to determine how inhibiting SHIP1 regulates the inflammatory response. Since P3C and Pg1449 were potent inducers of SHIP1 (by activating TLR2), a significant upregulation of IL-1β was noted when 3AC was employed to inhibit SHIP1 (Figure 6b). Since SHIP1 was not potently upregulated through TLR4 when stimulated with EcLPS, IL-1β was only slightly upregulated when SHIP1 was inhibited (Figure 6b). Malachite Green assay, a fluorescent polarization assay that detects the 5′-inositol phosphatase activity of SHIP1, was employed to measure SHIP1 levels. Inhibition of SHIP1 activity with 3AC was profound when osteoblasts were stimulated with P3C or Pg1449 (Figure 6c).

SHIP1 inhibition enhances inflammatory response in osteoblasts. Human primary osteoblasts between passages of 3–5 were cultured and incubated with 3AC at doses of 0–100 µmol·mL−1. 3AC dose-dependent cytotoxicity in osteoblasts was determined through tryphan blue exclusion method (a). Subsequently, osteoblasts were stimulated with 1 µg·mL−1 of P3C and were incubated with varying doses of 3AC (1–10 µmol·mL−1) and the levels of IL-1β gene expression and SHIP1 protein expression were determined and 3AC induced dose-dependent downmodulation of SHIP1 and upregulation of IL-1β (data not shown). Unstimulated osteoblasts or osteoblasts stimulated with P3C/Pg1449 alone or stimulated P3C/Pg1449 along with 10 µmol·mL−1 of 3AC were employed to determine the gene expression of IL-1β (b) and protein expression of SHIP1 (c). P3C- and Pg1449-induced SHIP1 expression was downregulated when osteoblasts were stimulated with AC3 (c). Osteoblasts stimulated with P3C/Pg1449 in the presence of AC3 to inhibit SHIP1 further upregulated the levels of IL-1β expression (b). All experiments were performed in triplicate and shown are the means along with standard error.

Inhibiting SHIP1 disrupts osteoblast homeostasis. Three-dimensional osteoblasts cultured in collagen gel cultures were stimulated for 1 week with 1 μg·mL−1 of P3C, Pg1449 or EcLPS (a, c, d, f). Additionally, 10 µmol·mL−1 of AC3 was employed, while osteoblasts were stimulated with these TLR ligands (b, c, e, f). The expression of ALP was determined through histochemical (a, b) and biochemical (c) methods and regulation of in vitro mineralization/calcium deposition was analyzed by histochemical (d, e) and biochemical (f) methods. P3C- and Pg1449-induced ALP (a, c) and mineralization (d, f) were downregulated when osteoblasts were concomitantly stimulated with AC3 (b, c, e, f). EcLPS was a poor inducer of ALP and in vitro mineralization and stimulation with AC3 did not further affect the levels of ALP or mineralization. The data is representative of three individual experiments.
SHIP1 inhibition disrupts osteoblast homeostasis
Inactivating SHIP1 enhanced TLR2-induced IL-1β in a manner similar to that of TLR4 responses (Figure 6). We subsequently aimed to determine how inhibiting SHIP1 regulates osteogenic mediators. Osteoblasts were stimulated with P3C, Pg1449 or EcLPS either in the presence or absence of 3AC to inhibit SHIP1. As shown in Figures 2–5, stimulating osteoblasts with P3C or with Pg1449 significantly upregulated the expression of ALP (Figure 7a and 7c) and in vitro mineralization (Figure 7d and 7f). EcLPS did not alter (relative to unstimulated osteoblasts) the expression of ALP (Figure 7a and 7c) and in vitro mineralization (Figure 7d and 7f). Relative to stimulation with P3C or Pg1449 alone (Figure 7a, 7c, 7d and 7f), inhibiting SHIP1 with AC3, significantly downregulated ALP expression and in vitro mineralization (Figure 7b, 7c, 7e and 7f). These data suggest that TLR2 signaling requires concurrent induction of SHIP1 for enhancing osteogenic mediators and inactivating SHIP1 made TLR2 responses similar to that of TLR4 activation.
Discussion
We have presented novel findings that induction of SHIP1 in osteoblasts was under the control of TLR2 but not TLR4 signaling. Subsequently, we showed that TLR2 signaling upregulated osteogenic mediators (induction of ALP and in vitro mineralization) and these were downmodulated when SHIP1 was inhibited. We further showed that potent TLR4 activation downregulated osteogenic mediators that were induced by TLR2. These results reinforce the notion that the skeletal and immune systems share cellular receptors, cytokine networks and transcription factors. For instance, TLR signaling and inflammatory response in the innate immune system can negatively regulate bone homeostasis. However, osteoblasts also express TLRs and can directly respond to microbial stimuli20 and secrete inflammatory cytokines such as IL-1 and TNF-α.21 In an effort to curtail inflammatory response, studies have shown that prolonged exposure of osteoprogenitors to microbial infections resulted in downregulation of TLR expression.22 Our data suggests that osteoblasts have low constitutive expression of TLR2 and TLR4 and are resistant to upregulation (relative to monocytes) when stimulated with TLR ligands. Low constitutive expression of TLRs has been demonstrated in mucosal11,23 systems. This is implicated as an adaptive measure to prevent excessive inflammatory host tissue damage. On the contrary, increased expression of TLRs has been implicated in inflammatory bowel disease.24
The role of TLR4 in disrupting osteogenesis is supported by a study that demonstrated LPS stimulated osteoblasts resulted in reduced osteogenesis in wild-type (WT) mice, whereas osteoblasts from myd88−/− mice (MyD88 being an essential TLR downstream adaptor) sustained osteogenic functions.4 In contrast, another study that employed LPS from P. gingivalis concluded that TLR2 activation resulted in poor differentiation of osteoblasts.25 However, this study lacked evidence that the P. gingivalis LPS specifically targeted TLR2 as P. gingivalis produces heterogeneous LPS that can target both TLR226 and also TLR4.27 In this study, we employed Pg1690 and Pg1449 and these LPS preparations have equivalent TLR2 activity, but Pg1690 is a TLR4 agonist and Pg1449 is a TLR4 antagonist. Osteoblasts when stimulated with Pg1690 moderately upregulated osteogenic mediators, but was not a potent inducer of IL-1β relative to EcLPS. Moreover, Pg1449 (being a weaker LPS relative to Pg1690 and also a TLR4 antagonist) significantly upregulated ALP and in vitro mineralization and IL-1β was poorly induced. These novel findings suggests that the potency of the inflammatory response mediated by TLR ligands indirectly correlated with the regulation of osteogenic functions. By employing inflammatory cytokines such as TNF-α, we demonstrated that potent inflammatory stimuli downregulate osteogenic functions that were upregulated by TLR2 responses. In mice microglial cells which are the innate immune cells of the central nervous system, TNF-α has been shown to upregulate the expression of TLR2.28 Our data suggest that unlike monocytes, osteoblasts have low constitutive expression of TLR2 and TLR4 and these receptors are resistant to upregulation when stimulated with their ligands (Figure 1). Further, EcLPS when employed at a 1 000-fold lower concentration moderately induced ALP and mineralization and moreover, mutant EcLPS structures induced a poor inflammatory response (relative to EcLPS) but significantly upregulated osteogenic functions. It should be noted that the composition of fatty acid chains and the number and position(s) of the phosphate groups in lipid A of LPS structure determines the TLR4 immunostimulatory potency, presumably due to their ability to engage MD2, the coreceptor of TLR4. The canonical EcLPS is hexa-acylated, 1,4-bis-phosphorylated glucosamine disaccharide and the employed mutant LPS, namely, LPxE (lipid A 1′-phosphate) and LPxF (lipid A 4′-phosphate) are penta-acylated mono-phosphorylated.12
TLR4 has a broader signaling mechanism relative to that of TLR2. TLR4 along with the Toll/interleukin receptor domain associates with a family of five adaptor proteins.29 These adapter molecules couple with downstream protein kinases and activate transcription factors such as nuclear factor-κB and members of the interferon-regulatory factor. The adaptors can be broadly divided into MyD88-dependent (which also uses MAL or TIRAP) and MyD88-independent pathways which employ adaptors, namely, TRIF and TRAM. SRIF is the fifth adaptor which is a negative regulator of TRIF/TRAM signaling.30 Unlike TLR4, TLR2 signaling is limited and uses only the MyD88-dependent pathways. LPS stimulated osteoblasts that were derived from myd88−/− mice retained matrix mineralization and ALP activity that was comparable to unstimulated controls.4 Also, murine osteoblasts do not have TRIF signaling31 and these studies suggest that TLR signaling in osteoblasts only use MyD88-dependent pathway. This evidence is relevant to our current studies as SHIP1 specifically targets the MyD88 pathway.
We have shown that SHIP1 is upregulated in osteoblasts in response to TLR2 but not TLR4 ligands. Moreover, inhibiting SHIP1 enhanced inflammatory response mediated by TLR2 ligands and these downmodulated osteogenic functions in a manner similar to TLR4 ligands. SHIP1−/− mice demonstrate increased osteoclastogenesis and bone resorption during inflammatory conditions such as arthritis.32 Animal models that lack SHIP1 have large hyper-active osteoclasts and demonstrate severe osteoporosis.33 Also, studies suggest that SHIP1 is essential for osteoblast development and SHIP1−/− mice demonstrate reduced bone growth and density.9,34 Induction of SHIP1 is also downstream of TGF-β signaling8 and TGF-β has diverse cellular and anti-inflammatory functions.35 Studies have suggested that TGF-β downmodulates IL-1R/TLR signaling and thereby suppress inflammatory response.36 Also, evidence suggests that TGF-β-mediated anti-inflammatory effects are limited to MyD88-dependent TLR signaling37. This evidence makes us speculate that TLR2 responses which employ only the MyD88 pathway could be effectively antagonized by TGF-β–SHIP1 induction. On the contrary, TLR4 might require MyD88-independent pathways for inducing TGF-β and SHIP1 and at least murine osteoblast lack MyD88-independent pathways. This speculation is supported by studies that show LPS–TLR4 signaling-induced IL-1β but not TGF-β38 and LPS hyper-responsiveness has been shown in transgenic mice that lack TGF-β signaling.39 TLR knockout studies suggest that TLR2 but not TLR4 is associated with bone loss. For instance, peritoneal macrophages cultured with P. gingivalis differentiated into osteoclasts in response to TLR2 signaling. Moreover, culture supernatants obtained from the TLR2−/− macrophages induced differentiation of much fewer osteoclasts relative to supernatants obtained from WT or TLR4−/− macrophages.40 These findings are supported by another study where P. gingivalis utilizes TLR2 and MyD88 in inducing osteoclast differentiation.41 TLR2−/− mice resist alveolar bone loss relative to WT mice following oral challenge with P. gingivalis.42 Another study presented data suggesting that WT and TLR4−/− but not TLR2−/− mice were susceptible to periodontal bone loss.43 These studies concluded the role of TLR2 in inducing bone loss through osteoclast activation. However, these studies lack comprehensive analysis of bone loss that could be induced by disrupted osteogenesis. Figure 8 presents a schematic summary of our research findings in this study.

Schematic diagram summarizing this study. When osteoblasts are stimulated with TLR2 ligand (P3C) or with mutant LPS structures (LPxF, LPxE and WSK) or with TLR4 antagonizing LPS (Pg1449), the inflammatory response as a measure of IL-1β is moderate, but osteogenesis (levels of ALP and mineralization) is upregulated (a). SHIP1 is upregulated with TLR2 but not with TLR4 signaling (a, b). When osteoblasts are stimulated with potent TLR4 ligand (EcLPS) or when SHIP1 is inhibited (using 3AC) or when osteoblasts are additionally stimulated with TNF-α, IL-1β is upregulated and osteogenic mediators are downmodulated even in the presence of TLR2 activity (b).
Osteoimmunology is an interdisciplinary field that explores the links associated with immune and skeletal systems. Newer approaches attempting to modulate immune responses44 and bone metabolism9,34 by targeting SHIP1 are currently considered. Preventing osteoclast bone resorption45 or enhancing osteogenesis through anabolic agents46 are being explored. Therapeutic approaches that aim to enhance osteogenesis by manipulating the potency of inflammatory response would be novel. For instance, monophosphorylated LPS structures are employed as vaccine adjuncts as these altered LPS structures induce a weak inflammatory response with little host tissue damage.47 Our studies with mutant LPS structures provide novel evidence that these molecules could enhance osteogenesis and thereby have a potential to restore bone homeostasis during inflammatory bone diseases.
References
Caetano-Lopes J, Canhao H, Fonseca JE . Osteoimmunology—the hidden immune regulation of bone. Autoimmun Rev 2009; 8: 250–255.
Harada S, Rodan GA . Control of osteoblast function and regulation of bone mass. Nature 2003; 423: 349–355.
Beutler B, Hoebe K, Du X, Ulevitch RJ . How we detect microbes and respond to them: the Toll-like receptors and their transducers. J Leukoc Biol 2003; 74: 479–485.
Bandow K, Maeda A, Kakimoto K et al. Molecular mechanisms of the inhibitory effect of lipopolysaccharide (LPS) on osteoblast differentiation. Biochem Biophys Res Commun 2010; 402: 755–761.
Kadono H, Kido J, Kataoka M, Yamauchi N, Nagata T . Inhibition of osteoblastic cell differentiation by lipopolysaccharide extract from Porphyromonas gingivalis. Infect Immun 1999; 67: 2841–2846.
Muthukuru M, Cutler CW . Upregulation of immunoregulatory Src homology 2 molecule containing inositol phosphatase and mononuclear cell hyporesponsiveness in oral mucosa during chronic periodontitis. Infect Immun 2006; 74: 1431–1435.
Maruyama K, Sano G, Matsuo K . Murine osteoblasts respond to LPS and IFN-gamma similarly to macrophages. J Bone Miner Metab 2006; 24: 454–460.
Sly LM, Rauh MJ, Kalesnikoff J, Song CH, Krystal G . LPS-induced upregulation of SHIP is essential for endotoxin tolerance. Immunity 2004; 21: 227–239.
Iyer S, Margulies BS, Kerr WG . Role of SHIP1 in bone biology. Ann NY Acad Sci 2013; 1280: 11–14.
Muthukuru M, Sun J . Doxycycline counteracts bone morphogenic protein 2-induced osteogenic mediators. J Periodontol 2013; 84: 656–665.
Muthukuru M, Jotwani R, Cutler CW . Oral mucosal endotoxin tolerance induction in chronic periodontitis. Infect Immun 2005; 73: 687–694.
Coats SR, Berezow AB, To TT et al. The lipid A phosphate position determines differential host Toll-like receptor 4 responses to phylogenetically related symbiotic and pathogenic bacteria. Infect Immun 2011; 79: 203–210.
Jain S, Coats SR, Chang AM, Darveau RP . A novel class of lipoprotein lipase-sensitive molecules mediates Toll-like receptor 2 activation by Porphyromonas gingivalis. Infect Immun 2013; 81: 1277–1286.
Muthukuru M . Technical advance: decreased helper T cells and increased natural killer cells in chronic periodontitis analyzed by a novel method for isolating resident lymphocytes. J Leukoc Biol 2012; 92: 683–692.
Muthukuru M, Cutler CW . Antigen capture of Porphyromonas gingivalis by human macrophages is enhanced but killing and antigen presentation are reduced by endotoxin tolerance. Infect Immun 2008; 76: 477–485.
Xing Q, Ye Q, Fan M, Zhou Y, Xu Q, Sandham A . Porphyromonas gingivalis lipopolysaccharide inhibits the osteoblastic differentiation of preosteoblasts by activating Notch1 signaling. J Cell Physiol 2010; 225: 106–114.
Al-Qutub MN, Braham PH, Karimi-Naser LM, Liu X, Genco CA, Darveau RP . Hemin-dependent modulation of the lipid A structure of Porphyromonas gingivalis lipopolysaccharide. Infect Immun 2006; 74: 4474–4485.
Gilbert L, He X, Farmer P et al. Inhibition of osteoblast differentiation by tumor necrosis factor-alpha. Endocrinology 2000; 141: 3956–3964.
Brooks R, Fuhler GM, Iyer S et al. SHIP1 inhibition increases immunoregulatory capacity and triggers apoptosis of hematopoietic cancer cells. J Immunol 2010; 184: 3582–3589.
Bar-Shavit Z . Taking a toll on the bones: regulation of bone metabolism by innate immune regulators. Autoimmunity 2008; 41: 195–203.
Marriott I . Osteoblast responses to bacterial pathogens: a previously unappreciated role for bone-forming cells in host defense and disease progression. Immunol Res 2004; 30: 291–308.
Mo IF, Yip KH, Chan WK, Law HK, Lau YL, Chan GC . Prolonged exposure to bacterial toxins downregulated expression of toll-like receptors in mesenchymal stromal cell-derived osteoprogenitors. BMC Cell Biol 2008; 9: 9–52.
Melmed G, Thomas LS, Lee N et al. Human intestinal epithelial cells are broadly unresponsive to Toll-like receptor 2-dependent bacterial ligands: implications for host-microbial interactions in the gut. J Immunol 2003; 170: 1406–1415.
Sartor RB . Pathogenesis and immune mechanisms of chronic inflammatory bowel diseases. Am J Gastroenterol 1997; 92 (12 Suppl): 5S–11S.
Wang YH, Jiang J, Zhu Q et al. Porphyromonas gingivalis lipids inhibit osteoblastic differentiation and function. Infect Immun 2010; 78: 3726–3735.
Hajishengallis G, Martin M, Schifferle RE, Genco RJ . Counteracting interactions between lipopolysaccharide molecules with differential activation of Toll-like receptors. Infect Immun 2002; 70: 6658–6664.
Darveau RP, Arbabi S, Garcia I, Bainbridge B, Maier RV . Porphyromonas gingivalis lipopolysaccharide is both agonist and antagonist for p38 mitogen-activated protein kinase activation. Infect Immun 2002; 70: 1867–1873.
Syed MM, Phulwani NK, Kielian T . Tumor necrosis factor-alpha (TNF-alpha) regulates Toll-like receptor 2 (TLR2) expression in microglia. J Neurochem 2007; 103: 1461–1471.
O’Neill LA, Bowie AG . The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 2007; 7: 353–364.
Banerjee A, Gerondakis S . Coordinating TLR-activated signaling pathways in cells of the immune system. Immunol Cell Biol 2007; 85: 420–424.
Sato N, Takahashi N, Suda K et al. MyD88 but not TRIF is essential for osteoclastogenesis induced by lipopolysaccharide, diacyl lipopeptide, and IL-1alpha. J Exp Med 2004; 200: 601–611.
Lisignoli G, Piacentini A, Toneguzzi S et al. Osteoblasts and stromal cells isolated from femora in rheumatoid arthritis (RA) and osteoarthritis (OA) patients express IL-11, leukaemia inhibitory factor and oncostatin M. Clin Exp Immunol 2000; 119: 346–353.
Ross FP . M-CSF, c-Fms, and signaling in osteoclasts and their precursors. Ann NY Acad Sci 2006; 1068: 110–116.
Iyer S, Viernes DR, Chisholm JD, Margulies BS, Kerr WG . SHIP1 regulates MSC numbers and their osteolineage commitment by limiting induction of the PI3K/Akt/β-catenin/Id2 axis. Stem Cells Dev 2014; 23: 2336–2351
Roberts AB, Sporn MB . Physiological actions and clinical applications of transforming growth factor-beta (TGF-beta). Growth Factors 1993; 8: 1–9.
Lee YS, Kim JH, Kim ST et al. Smad7 and Smad6 bind to discrete regions of Pellino-1 via their MH2 domains to mediate TGF-beta1-induced negative regulation of IL-1R/TLR signaling. Biochem Biophys Res Commun 2010; 393: 836–843.
Choi KC, Lee YS, Lim S et al. Smad6 negatively regulates interleukin 1-receptor-Toll-like receptor signaling through direct interaction with the adaptor Pellino-1. Nat Immunol 2006; 7: 1057–1065.
Merry K, Gowen M . The transcriptional control of TGF-beta in human osteoblast-like cells is distinct from that of IL-1 beta. Cytokine 1992; 4: 171–179.
McCartney-Francis N, Jin W, Wahl SM . Aberrant Toll receptor expression and endotoxin hypersensitivity in mice lacking a functional TGF-beta 1 signaling pathway. J Immunol 2004; 172: 3814–3821.
Ukai T, Yumoto H, Gibson FC 3rd, Genco CA . Macrophage-elicited osteoclastogenesis in response to bacterial stimulation requires Toll-like receptor 2-dependent tumor necrosis factor-alpha production. Infect Immun 2008; 76: 812–819.
Zhang P, Liu J, Xu Q et al. TLR2-dependent modulation of osteoclastogenesis by Porphyromonas gingivalis through differential induction of NFATc1 and NF-kappaB. J Biol Chem 2011; 286: 24159–24169.
Burns E, Bachrach G, Shapira L, Nussbaum G . Cutting Edge: TLR2 is required for the innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol 2006; 177: 8296–8300.
Gibson FC 3rd, Genco CA . Porphyromonas gingivalis mediated periodontal disease and atherosclerosis: disparate diseases with commonalities in pathogenesis through TLRs. Curr Pharm Des 2007; 13: 3665–3675.
Keck S, Freudenberg M, Huber M . Activation of murine macrophages via TLR2 and TLR4 is negatively regulated by a Lyn/PI3K module and promoted by SHIP1. J Immunol 2010; 184: 5809–5818.
Jones B . Bone. Regulating osteoclast differentiation to prevent bone loss. Nat Rev Rheumatol 2014; 10: 66.
Rosen CJ, Rackoff PJ . Emerging anabolic treatments for osteoporosis. Rheum Dis Clin North Am 2001; 27: 215–233 viii
Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC . The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 2007; 316: 1628–1632.
Acknowledgements
These studies were supported by Elam M. and Georgina E. Hack Memorial Research Funds, Department of Periodontics, University of Washington, Seattle, WA, USA. The publication costs are supported by WVCTSI funds, West Virginia University, Morgantown, WV, USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Muthukuru, M., Darveau, R. TLR signaling that induces weak inflammatory response and SHIP1 enhances osteogenic functions. Bone Res 2, 14031 (2014). https://doi.org/10.1038/boneres.2014.31
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/boneres.2014.31
This article is cited by
-
LY450139 Inhibited Ti-Particle-Induced Bone Dissolution via Suppressing Notch and NF-κB Signaling Pathways
Calcified Tissue International (2022)
-
Effect of Different Titanium Surfaces on Maturation of Murine Bone Marrow-Derived Dendritic Cells
Scientific Reports (2017)
-
Compressive mechanical stress may activate IKK-NF-κB through proinflammatory cytokines in MC3T3-E1 cells
Biotechnology Letters (2015)
