
Abstract
Gastric cancer (GC) is a life-threatening disease worldwide. Despite remarkable advances in treatments for GC, it is still fatal to many patients due to cancer progression, recurrence and metastasis. Regarding the development of novel therapeutic techniques, many studies have focused on the biological mechanisms that initiate tumours and cause treatment resistance. Tumours have traditionally been considered to result from somatic mutations, either via clonal evolution or through a stochastic model. However, emerging evidence has characterised tumours using a hierarchical organisational structure, with cancer stem cells (CSCs) at the apex. Both stochastic and hierarchical models are reasonable systems that have been hypothesised to describe tumour heterogeneity. Although each model alone inadequately explains tumour diversity, the two models can be integrated to provide a more comprehensive explanation. In this review, we discuss existing evidence supporting a unified model of gastric CSCs, including the regulatory mechanisms of this unified model in addition to the current status of stemness-related targeted therapy in GC patients.
Similar content being viewed by others

Main
Gastric cancer (GC) is the third leading cause of death and the fifth most frequently diagnosed cancer in adults worldwide, with an estimated 0.72 million deaths and 0.95 million new cases in 2012 (Torre et al, 2015). Apparent tumour heterogeneity exists not only between sexes but also regarding the spatial-temporal distribution of the tumour. Overall, the incidence and mortality of GC are nearly twice as high in males as in females, and the disease usually occurs in individuals who are 45 years or older and predominantly from developing countries and regions, such as East Asia (Magee et al, 2012). Despite considerable advances in GC treatment and decreasing trends in incidence and mortality rates, many patients still die due to cancer progression, recurrence and metastasis. Due to a lack of effective screening methods, more than 50% of patients are in advanced stages at initial diagnosis, at which time most of these patients do not have the opportunity for radical surgery and are reluctant to undergo treatment with adjuvant therapy. The response rate (RR) of patients with advanced GC (AGC) to first-line chemotherapy is only 50%, and the median overall survival (OS) is less than 12 months (Cunningham et al, 2008; Kang et al, 2009). Furthermore, the OS of patients after second-line therapy is only ∼6 months (Janowitz et al, 2016). In view of the failure of conventional therapy, substantial attention has been focused on the genetic and molecular mechanisms that initiate tumours and cause drug resistance. More evidence is emerging to indicate that tumours are characterised by a hierarchical organisational structure, with cancer stem cells (CSCs) at the apex, and additional evidence suggests that successful eradication of CSCs may be a promising therapeutic approach for treating cancer.
Through improvements in cell sorting, gene labelling and lineage tracing, substantial evidence has emerged in support of the hierarchical model hypothesis. For example, CD34+CD38− leukemia cells, which were the first CSCs identified using flow cytometry, demonstrated self-renewal ability and the capacity to differentiate into mature leukemic cells and initiate human acute myeloid leukemia after serial transplantation into immunocompromised mice (Bonnet and Dick, 1997). The lineage tracing of cells containing stemness markers has provided more direct, functional evidence for the presence of CSCs (Schepers et al, 2012; Hayakawa et al, 2015). Hence, it has been gradually accepted that the central features of CSCs are their unlimited self-renewal capacity and pluripotency and that this small number of cancer cells is responsible for tumour initiation, progression, recurrence and metastasis (Clarke et al, 2006). However, this model has been challenged based on results obtained using advances in high-throughput sequencing of multiple tumour genomes for detecting large variations in gene mutations within or between tumours (Gerlinger et al, 2012; Nik-Zainal et al, 2012) and using functional assays combined with genetic analyses. The diversity of driver mutations implies the presence of subclones and their role in tumour heterogeneity. In addition, the use of single-nucleus sequencing to investigate the genetic structure of single cancer cells by accurately quantifying the genomic copy number revealed the existence of distinct subclones among individual cells (Navin et al, 2011). Likewise, the single-cell DNA/RNA sequencing of circulating tumour cells (CTCs) has allowed the detection of heterogeneity in genome mutations and signalling pathways that may contribute to tumour progression and treatment failure (Yu et al, 2012; Heitzer et al, 2013; Miyamoto et al, 2015). Generally, the identification of different genetic subclones suggests that gene mutations and clonal evolution also have key roles in tumour initiation, progression and metastasis.
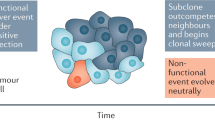
As discussed above, both the stochastic and hierarchical models are reasonable hypotheses that have been proposed to describe tumour heterogeneity, but alone, each model is insufficient to explain the diversity within tumours. In light of growing controversy, Kreso and Dick (2014) proposed that the two models could be integrated into a more comprehensive explanation. By combining genetic analyses with functional assays of tumour-initiating cells (T-ICs), it was found that T-ICs also contain genetic subclones, which influence their properties (Notta et al, 2011; Chow et al, 2014); that is, T-ICs can also evolve. Hence, it is reasonable to imagine that an early-stage tumour containing rare CSCs and other cells becomes more heterogeneous and invasive as advantageous mutations accumulate and that as the CSCs develop into several subclones, they achieve a higher capacity for self-renewal and thus form a greater proportion of the total cells (Figure 1). In addition, some studies have shown that sorted non-T-IC subpopulations can transform into T-ICs under specific conditions (Chaffer et al, 2013) via two possible mechanisms: by dedifferentiation of non-T-ICs (Schwitalla et al, 2013) or by occasional mutations in non-T-ICs that endow them with stemness. These studies highlight the plasticity of cancer cells and the dynamic process of tumour evolution, during which T-ICs and non-T-ICs can interconvert. Collectively, a unified model that is dynamically regulated by other determinants such as epigenetics (Baer et al, 2013; Klutstein et al, 2016), gene expression stochasticity (Elowitz et al, 2002; Kaern et al, 2005; Spencer et al, 2009), the CSC niche (Plaks et al, 2015) and the tumour microenvironment (TME; Medema and Vermeulen, 2011) is proposed, and this model may provide an overall explanation for the biological properties of the tumour (Kreso and Dick, 2014).

The unified model of cancer.(A) The most primitive CSCs continuously divide, self-renew, differentiate into non-CSCs and eventually form a tumour mass with functional diversity, during the course of which the cancer cells occasionally develop genetic mutations that can endow non-CSCs with stemness. (B) The tumour becomes more heterogeneous and invasive as advantageous mutations in the CSCs accumulate; in addition, several subclones develop, and the tumour cells confer a higher capacity for self-renewal and begin to make up a greater proportion of the total cells. (C) Most of the CSCs are drug-resistant and survive after chemotherapy or radiotherapy. (D) The residual CSCs cause tumour relapse, and more subclones are generated.
In this review, we discuss the evidence for a unified model in describing gastric CSCs (GCSCs) and the regulatory mechanisms of this model in addition to the current status of stemness-related targeted therapy in GC patients.
Origin of GCSCs
On the basis of the Lauren classification, GC is primarily divided into two distinct categories: intestinal type and diffuse-type. The pathological process of intestinal type GC has been well studied; the gastric epithelium develops through sequential stages of chronic gastritis, atrophic gastritis, metaplasia, dysplasia and eventually cancer. However, it is unclear which determinants are involved and how those determinants result in the emergence of these morphologic changes, especially regarding the mechanism by which GCSCs originate. According to the unified model of carcinogenesis, tumour cells are composed of several subpopulations of CSCs and highly homogeneous non-CSCs, and these subclones could be produced by the mutation of a single CSC. Therefore, the following questions have been raised: what is the precursor of CSCs, and how does a normal cell obtain the ability to infinitely self-renew? Current evidence suggests that GCSCs likely originate from multiple cell types, including gastric stem cells (GSCs), glandular cells and bone marrow-derived cells (Figure 2A).

The combination of the hierarchical and stochastic theories of GC.(A) GC stem cells originate from gastric stem cells, dedifferentiated epithelial cells or bone marrow-derived cells. (B) The accumulation of cancer-associated mutations and chromosomal aberrations in the initiating cells, which are induced by multiple factors, such as diet, alcohol, H. pylori infection and smoking, promotes the pathological process of gastric adenocarcinoma, in which the gastric epithelium develops through sequential stages of chronic gastritis, atrophic gastritis, metaplasia (intestinal or spasmolytic polypeptide-expressing), dysplasia and eventually cancer. (C) The formation, invasion and metastasis of GC occur according to the unified model, which is dynamically regulated by other determinants such as epigenetic alternation, gene expression stochasticity, immune escape, niche, signalling pathways and networks of soluble factors. CAF = cancer-associated fibroblast.
GCSCs originate from GSCs
In the human stomach, the epithelium of gastric units or glands is primarily composed of four types of functional cells: chief cells, parietal cells, mucous cells and enteroendocrine cells. The majority of these differentiated cells are short-lived and are rapidly replaced by new cells, and each gland is considered to be formed by polyclonal expansion of multiple stem cells (McDonald et al, 2008). Hence, it is widely accepted that the integrity of the fast-growing gastric epithelium is sustained by quiescent stem cells. Early evidence revealed by radiolabelling with 3H-thymidine and electron microscopy identified certain immature, granule-free cells with the highest labelling index in a region near the isthmus of units that were presumed to be GSCs (Karam and Leblond, 1993). The first specific identification of stem cells in situ by the Villinβ-gal/+ marker demonstrated that this rare subpopulation is primarily located at or below the isthmus of pyloric glands (Qiao et al, 2007). Moreover, based on lineage tracing, cells in the isthmus that express trefoil factor family 2 (TFF2) messenger RNA have also been proposed as progenitor cells (Quante et al, 2010). Multiple specific markers have also been used to distinguish stem cells from mature epithelial cells in different sites within the stomach. Lgr5 is a well-established stem cell marker in the intestine and colon (Barker et al, 2007) that has been shown to be expressed strictly in the bottom of the adult human pyloric glands. Via stimulation of gastric units in vitro and lineage tracing in vivo, Lgr5+ cells have been shown to possess self-renewal capacity; therefore, Lgr5 can be considered an exclusive marker for GSCs (Barker et al, 2010). Above the bottom of the pyloric and fundic glands, Sox2+ stem cells without a marked overlap in Lgr5 expression were identified by using double-labelling methods and marker tracing (Arnold et al, 2011). Recently, some studies have found that a specific subpopulation of chief cells had the potential to function as reserve stem cells. A single Troy+ mature chief cell located at the base of the gastric corpus glands could generate an entire gland, based on lineage tracing, and could form long-lived organoids in a three-dimensional culture system (Stange et al, 2013). Likewise, Mist1-expressing cells in the isthmus but not in the lower third of the gastric corpus units have also been observed to have self-renewal ability and to differentiate into multiple lineages of mature cells, such as mucous cells and chief cells. In addition, the use of transgenic mouse models also showed that Mist1+ isthmus cells could expand and evolve independently of Lgr5+ cells and are the origin of mature epithelial cells in the corpus units (Hayakawa et al, 2015). These results show the heterogeneity of stem cells in different regions of the stomach and even in different zones of an identical unit, which implies that GCSCs are heterogeneous.
The mechanism of carcinogenesis is diverse in distinct phenotypes of GSCs, but the model of field cancerisation has been well documented. This model holds that a mutated GSC expands to the whole gland and forms a clonal patch by unit fission, which has been demonstrated previously by the detection of mitochondrial DNA mutations in normal and intestinal metaplastic mucosa adjacent to the tumour tissue of GC patients (McDonald et al, 2008). With the identification of GSC-specific markers, numerous driver mutations in the malignant progression of GSCs were verified by functional analysis in transgenic mouse models. A recent study showed that the inactivation of the tumour suppressor gene Krüppel-like factor 4 (Klf4) in Villin+ gastric stem-like cells accelerated the malignant transformation of gastric mucosa in the carcinogen-induced Villin-Cre+;Klf4fl/fl mice (Li et al, 2012). The effect of aberrant Wnt pathway activity, such as the deletion of the adenomatous polyposis coli (APC) gene, on the carcinogenesis of Lgr5+ stem cells was evaluated in the Lgr5-CreERT2;APCfl/fl mouse model. The results showed that β-cateninhi adenomas only in the pylorus were rapidly formed by the expansion of Lgr5+ transformed stem cells after the mice were treated with tamoxifen to induce the loss of APC (Barker et al, 2010). A study regarding the effect of Helicobacter pylori (H. pylori) infection on Lgr5+ stem cells also revealed that these bacteria could directly act on stem cells, promote proliferation and increase the expression of tumour-associated genes, such as the Wnt target gene Axin2 and the anti-apoptosis gene Olfactomedin4 (Sigal et al, 2015). Similarly, it has been demonstrated that mutations in distinct pathway components in isthmus Mist1+ stem cells of the corpus gland were responsible for the differentiation of intestinal type and diffuse-type GC (IGC and DGC). Such effects have been observed for mutations in the Kras gene, facilitating the metaplastic/dysplastic transformation and expansion of Mist1+ isthmus cells; aberrant Notch activation, resulting in the development of IGC; and mutations in E-cadherin and transformation-related protein 53 (TRP53), inducing the formation and invasion of DGC containing numerous lineage-traced signet-ring cells in the context of chronic inflammation (Hayakawa et al, 2015). Collectively, it is presumed that GCSCs are likely derived from aberrant GSCs and that they possess various genetic and functional properties in disparate primary sites. This hypothesis would be more convincing if more in-depth genomic analyses and additional advanced bioinformatics techniques were applied to these stem cells.
GCSCs originate from dedifferentiated epithelial cells
Dedifferentiation is a cellular process by which mature, differentiated cells transform back to a primitive or fetal stage and lose certain characteristics, for example, their specific function or phenotype. Some researchers have suggested that the dedifferentiation of gastric epithelial cells (GECs) may be an aberrant change in the developmental cycle that gives rise to metaplasia, dysplasia and ultimately GC. A study showed that malignant changes in the gastric epithelium of Mongolian gerbils were associated with dedifferentiated mature cells defined by the expression of cytokeratin-7, which is mainly present in fetal epithelial cells (Kirchner et al, 2001). Recently, utilising lineage tracing to analyse the origin of the spasmolytic polypeptide-expressing metaplasia (SPEM) cells induced by L-635 treatment in the Mist1-CreERT2 mouse model, a study showed that the loss of acid-secreting cells under an induced inflammatory environment resulted in the rapid production and spread of SPEM that developed from transdifferentiated mature chief cells, which demonstrated the capacity of mature GECs to regain ‘stemness’ and to initiate a tumour in the presence of inflammation (Nam et al, 2010). To explore the mechanism by which gastric mucosa converts to intestinal type, research revealed that the homeobox protein CDX1 could directly bind to and activate the expression of genes encoding reprogramming proteins, such as SALL4 and KLF5, which endow GECs with intestinal stem-like features. The long-term expression of CDX1 induced the generation of intestinal stem cell markers and the subsequent expression of intestinal mature cell markers, which indicated that mature GECs could dedifferentiate into stem-like cells and then transdifferentiate into intestinal type cells via the aberrant expression of stemness-related genes (Fujii et al, 2012). In addition, another four transcription factors, Klf4, Oct 3/4, c-MYC and Sox2, have also been demonstrated to induce the reprogramming of differentiated GECs into pluripotent stem cells (Aoi et al, 2008). Furthermore, the involvement of H. pylori in the generation of GCSCs via inducing the epithelial–mesenchymal transition (EMT) of GECs has been reported previously (Bessede et al, 2014; Choi et al, 2015). In particular, the CagA protein of H. pylori was responsible for the transformation of GECs into a subset of cells with mesenchymal phenotypes and CSC features, including the activation of mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) signalling pathways (Bessede et al, 2014).
Although the above studies support the idea that mature GECs are likely to reacquire stemness properties and act as sources of GCSCs, the experimental techniques used in these studies were did not provide sufficient direct evidence of the capacity of mature GECs to dedifferentiate and initiate tumorigenesis. For example, the identification of cells by using specific markers could not definitively distinguish a bona fide stem cell from other cells expressing the identical marker; the observation of stemness characteristics may simply be due to the activation of stem cells among the studied cells. Hence, more rigorous and stronger evidence is needed to validate these data.
GCSCs originate from bone marrow-derived cells
Bone marrow-derived cells (BMDCs) are well known for their striking plasticity regarding their phenotype and function; they constantly migrate to peripheral tissues via chemotaxis towards chronic inflammation and injury, and they display the ability to differentiate into multiple lineages. To date, many biological effects on the gastric mucosa and stroma exerted by BMDCs have been demonstrated, including (1) contributing to the formation of a mesenchymal or perivascular stem cell niche (Quante et al, 2011; Hayakawa et al, 2015); (2) involvement in the repair of damage caused by chronic inflammation and the regeneration of glandular epithelium (Okamoto et al, 2002; Ferrand et al, 2011b; Bessede et al, 2015); (3) promotion of the proliferation and metastasis of GC cells (Quante et al, 2011; Hayakawa et al, 2015; Zhu et al, 2016a); and (4) acting as a potential source of epithelial cancer (Bessede et al, 2015). Despite massive investigative efforts, the mechanism underlying malignant changes in BMDCs is still not well-established. Early research using a mouse model revealed that chronic infection with H. felis, rather than acute injury or inflammation, resulted in the recruitment and repopulation of BMDCs into the gastric mucosa, and the progeny of the BMDCs developed subsequently through metaplasia, dysplasia and finally into epithelial cancer (Houghton et al, 2004). Recently, using a female mouse model treated with male mouse BMDCs expressing green fluorescent protein (GFP) and infected with different mouse-adapted H. pylori strains, metaplastic and dysplastic glands with GFP+ cells were detected in most of the mice after 1 year of infection with H. pylori, and nearly a quarter of the foci contained these BMDCs (Varon et al, 2012). In addition, fluorescence in situ hybridisation (FISH) for the Y chromosome was also performed on the stomach tissue sections of Helicobacter-infected mice in the above-mentioned two studies to further confirm that donor-derived engrafted BMDCs were the origin of the gastric mucosal cells expressing GFP and epithelial markers (Houghton et al, 2004; Varon et al, 2012). Thus, chronic inflammation and injury of the gastric mucosa may be a primary cause of BMDC homing. Moreover, the mechanism of BMDC migration in the epithelium is likely associated with the involvement of the nuclear factor-kappa B (NF-κB) pathway via the secretion of chemokines (tumour necrosis factor-α, TNF-α; Ferrand et al, 2011a). However, some results conflicting with the above evidence have also been presented. A study showed that syngeneic BMDCs labelled with GFP were only marginally scattered in stroma and not in glands of gastric neoplastic lesions or tumours induced by N-nitroso-N-methylurea (MNU) and H. felis infection in recipient mice (Yang et al, 2013). Likewise, opposite-sex donor BMDCs were only identified by FISH for sex chromosomes in perineoplastic stroma of secondary GC in an aplastic anaemia patient treated with allogeneic hematopoietic stem cell transplantation (Worthley et al, 2009). The opposite conclusion is also possible because in the human case, chronic inflammation likely existed before transplantation, and in animal experiments, it is difficult to fully model the true conditions. However, the evidence for the properties of stem cells, which are presumed to originate from BMDCs, would be more reasonable if single-cell studies, lineage tracing and optimised animal models were performed.
Identification and functional verification of GCSCs
Although many experimental investigations have been performed on the identification and functional verification of GCSCs, the hierarchical model for stomach remains indefinite because of a lack of direct evidence. To date, there are several major methods that can be used to isolate GCSCs, including (1) identifying GCSCs by specific markers (Qiao and Gumucio, 2011); (2) separating GCSCs from side population (SP) cells (Fukuda et al, 2009; Nishii et al, 2009; Ehata et al, 2011; She et al, 2012; Hasegawa et al, 2014); (3) screening for GCSCs through drugs or radiation treatment (Xue et al, 2012; Xu et al, 2015b); and (4) capturing tumour cells from circulating blood, metastatic sites or peritoneal milky spots (Cao et al, 2009; Chen et al, 2012). For these selected CSC-rich subpopulations, two general approaches are used to validate the properties of stemness. One approach is the spheroid body formation assay in vitro; the other is the tumour-formation assay in vivo.
Identification by specific markers
The antigen CD44 was the first putative marker used to identify GCSCs. The first study showed that CD44+ cells separated from three GC cell lines (GCCLs) through fluorescence-activated cell sorting (FACS) exhibited self-renewal capacity as indicated by spheroid formation in vitro and tumour formation in mice, while both the CD44− cells and short-hairpin RNA (shRNA) CD44-knockdown cells exhibited a remarkable decline in tumorigenicity (Takaishi et al, 2009). Subsequently, CD44v8-10 was identified as the predominant CD44 variant and served as a GCSC marker through a series of experiments in primary tumour samples including the isolation and culture of primary GC cells (GCCs), limiting dilution assays and xenografts in NOD/SCID/IL2Rγ− (NSG) mice (Lau et al, 2014). Strikingly, CD44+CD54+ CTCs captured in the peripheral blood of GC patients have also exhibited the ability to self-renew. Chen et al (2012), using an antibody combined with magnetic beads, separated up to 103 CD44+ cells from the blood samples of seven chemotherapy patients, and cells from six of those patients successfully formed tumour spheres when passaged in vitro. Furthermore, after transplantation into SCID mice, CD44+CD54+ cells from these primary cultured cells quickly developed into tumours that were similar to the original lesions, and the remainder of the cells were negative for these markers. To improve specificity, the co-recognition of multiple markers with CD44 has been widely studied, and the relevant data are presented in Table 1. Recently, studies have focused on the mechanism of CD44+ cells functioning as GCSCs. Hedgehog (HH) signalling has been proven to have an important role in the maintenance of stemness of CD44+ CSCs, as HH signalling proteins are upregulated in CD44+ GCCs, and the inhibition of HH signalling by Smoothened (Smo) shRNA or vismodegib results in a decrease in tumorigenesis and drug resistance of CD44+ cells and the full suppression of cancer cell proliferation, migration and invasion (Yoon et al, 2014). Another study also demonstrated the same functions of the SHH-GLI1 pathway and the effector protein ABCG2 in CD44+Musashi-1+ GCSCs (Xu et al, 2015a).
CD90, another universal marker of multi-tissue stem cells and CSCs, has also been extensively investigated in GC. Jiang et al (2012) first observed that the CD90+ subpopulation in GC could be characterised as CSCs. In that study, the tumour hierarchy was successfully reconstructed using single cells that were selected from high-tumorigenicity mouse models established by using a different source of 103 primary GCCs. The stemness properties of these single cells were also shown through the formation of in vitro spheroids and in vivo tumour masses. The comparison of RNA expression between spheroids and primary tumour cells revealed a marked upregulation of CD90. Then, after single CD90+ cells isolated via FACS were implanted into nude mice, nearly 90% developed into tumours. Recently, although an increasing number of specific GCSC markers have been found using the similar experimental techniques mentioned above (Table 1), studies utilising direct methods such as lineage tracing and single-cell analysis have not yet been reported. Furthermore, the expression of CSC markers is not always stable and reliable in different cells and at different times, perhaps due to the variability in mutations, origin of cells, frequency of tumorigenic cells, regulation of the TME or experimental techniques, resulting in the inability to purify true CSCs (Meacham and Morrison, 2013; Kreso and Dick, 2014). Hence, there are still many unknown variables in the hierarchical model of GC, and additional investigations in the heterogeneity and plasticity of CSC phenotypes are needed.
Separation from side population cells
Side population cells, the subpopulation of cells separated from the main population (MP) of cells by flow cytometric markers, have exhibited CSC-like features in many studies (Patrawala et al, 2005; Wang et al, 2007). With respect to GC, one study showed higher in vivo tumorigenicity of SP cells from GCCLs and from GC samples (Fukuda et al, 2009). Another study also supports this possibility through the use of spheroid- and tumour-formation assays with SP cells (She et al, 2012). However, our group (Zhang et al, 2013) previously found that only some of the SP cells possess stemness properties. Thus, the mechanism for distinguishing between SP cells and MP cells requires additional investigation.
Screening through the use of drugs or radiation treatment
Drug resistance, an inherent characteristic of quiescent cells, such as CSCs, may be a feasible approach for acquiring cancer stem-like cells. Confirming the CSC properties of drug-preconditioned GCCLs, recent research found that vincristine-treated cancer cells exhibited mesenchymal features, multi-drug resistance, high tumorigenicity and the abilities to self-renew and differentiate in vitro (Xue et al, 2012). Likewise, the stemness characteristics of residual cells from 5-fluorouracil (5-FU)-preconditioned GCCLs were also demonstrated by single-cell clonogenic assays and tumorigenicity assays (Xu et al, 2015b). Nevertheless, this method may be difficult to implement in primary tumour tissues due to a mix of various drug-resistant cells, such as normal tissue stem cells; therefore, it would be helpful to combine this technique with other identification methods.
Capturing from circulating blood, metastasis sites or peritoneal milky spots
Metastatic cells from original tumours are well known for their capacity to initiate tumours. Milky spots, namely lymphoid tissues in the abdominal cavity, have been indicated as early infiltration sites of the peritoneal dissemination of GCCs (Tsujimoto et al, 1996). A milky spot has been assumed to be a ‘natural filter’ for capturing GCSCs, as a location in which abundant macrophages are cytotoxic against non-CSCs and contribute to a niche for CSC survival (Cao et al, 2009). A recent study reported many macrophages and metastatic lesions observed by electron microscopy and DiI-labelled MFCs (a murine GCCL) in omental milky spots (OMSs) in a mouse model, and the researchers verified that OMS-derived cancer cells exhibited remarkable CSC properties, such as higher tumorigenicity through a limiting dilution assay in vivo and higher expression of CSC markers (Cao et al, 2011). Furthermore, milky spots were also considered to provide a hypoxic microenvironment for GCSCs through the overexpression of hypoxia inducible factor-1α (HIF-1α) and to enhance the survival and self-renewal capacity of the CSCs in them (Miao et al, 2014). These reports offer an alternative route for investigating the mechanism of cancer cell metastasis and the heterogeneity of CSCs between primary and metastatic sites.
Evidence of the clonal evolution model of gastric cancer
The heterogeneity, the phenotypic characteristics and the biological function of stomach tumours have been described in many studies over the past several decades; however, genetic variation was not investigated based on a comprehensive molecular evaluation until the emergence of next-generation high-throughput sequencing technology. The first study using whole-exome sequencing found massive somatic mutations including 20 candidate driver genes in 22 matched pairs of GC samples and healthy controls. They reported that mutations of a top putative driver gene, ARID1A, were more frequent in the microsatellite instability (MSI) and Epstein-Barr virus (EBV)-infected types of GC and showed a link with a trend of better prognosis (Wang et al, 2011). Similarly, another exome sequencing study showed recurrent gene mutations including in the proto-oncogene PIK3CA and the tumour suppressor genes TP53 and ARID1A in 15 GC samples, as well as frequent genetic abnormalities including in the E-cadherin family gene FAT4 and the chromatin remodelling genes (ARID1A, MLL3 and MLL), in a prevalence screening among 110 GC samples. The functional assays suggested that inactivation of the FAT4 and ARID1A genes was correlated with malignant behaviour of GC such as cellular proliferation, invasion and migration (Zang et al, 2012). The first whole-genome analysis using massively parallel signature sequencing and DNA paired-end tag sequencing in 2 gastric adenocarcinomas (GACs) provided the first detailed reconstruction of a mutant genome and revealed the integrative characteristics of mutational processes in GC compared with exome sequencing (Nagarajan et al, 2012). Subsequently, many mutations and epigenetic alterations were identified using similar techniques in different subtypes of GC, and the corresponding functional verification implied their core roles in initiation and progression of GC (Jaiswal et al, 2013; Kakiuchi et al, 2014; Liang et al, 2014; Shimizu et al, 2014; Wang et al, 2014; Wong et al, 2014; Zhou et al, 2014; Hu et al, 2016; Li et al, 2016a, 2016b; Lim et al, 2016b). Recently, based on the molecular sequencing and analysis platforms, a robust and comprehensive classification was presented, dividing GC into four molecular subtypes and characterising somatic genomic mutations, alterations in gene expression and signalling pathways by analysing 295 human GACs (Cancer Genome Atlas Research Network, 2014). The following subtypes were defined: (1) EBV-infected tumours, which are mainly characterised by recurrent PIK3CA mutations, amplification of the genes for the non-receptor tyrosine kinase JAK2 and the immune suppressive proteins PD-L1/2, and extensive DNA hypermethylation; (2) MSI tumours, which exhibit increased mutation frequency and hypermethylation, including the epigenetic silencing of the mismatch repair gene hMLH1; (3) genomically stable tumours, which are rich in the diffuse histological subtype, E-cadherin and RHOA (a member of Ras superfamily) gene mutations and structural rearrangements, especially in the fusions between the CLDN18 (a member of the claudin family) gene and the ARHGAP26/6 (Rho GTPase-activating proteins) gene; and (4) chromosomal instability (CIN) tumours, which are distinguished by frequent aneuploidy, the overexpression of p53 (TP53 mutation) and recurrent genomic amplifications of receptor tyrosine kinases (RTKs), such as EGFR and VEGFR, and downstream effector RAS proteins. Later, the Asian Cancer Research Group proposed a different type of molecular classification (Cristescu et al, 2015) to establish an association of the diverse patterns of molecular alterations with tumour progression and prognosis. This proposal defined four subtypes: (1) the mesenchymal-like type, with the worst prognosis and highest recurrence rates; (2) the MSI type, with the best OS and the lowest recurrence rates; (3) the TP53-active; and (4) TP53-inactive types with intermediate clinical outcomes. Furthermore, distinct somatic mutations in peritoneal metastatic tumours vs primary tumours were identified (Lim et al, 2016a; Liu et al, 2016). In general, numerous indisputable pieces of evidence have proven the genetic heterogeneity among different subtypes and even within the same subtype of GC, and functional tests have supported that various driver mutations contributed to the clonal expansion of cancer cells endowed with advantageous functions and characteristics toward a favourable direction for survival. Consequently, recurrent genetic alterations constantly change the phenotypic and functional properties of GC for adapting to hostile environments, implying an important role of clonal evolution in GC heterogeneity and development.
The combination of the hierarchical and stochastic theories of gastric cancer
In light of the studies on the origination and identification of GCSCs, the existence of a functional hierarchy in GCCs may be confirmed. However, the GCSCs identified based on specific markers also possess distinct functions and phenotypes. Despite the lack of an exclusive marker, the deficiency of cell separation and purification technologies and the disparity among cell sources, the combination of the hierarchical and stochastic theories is likely to provide a reasonable explanation for how primitive GCSCs continually optimise their own genome, improve the pattern of gene expression and generate various subclones to adapt to the ever-changing TME (Figure 2B). According to the evidence on the origin of GCSCs, the accumulation of diverse cancer-related mutations induced by carcinogenic factors in stem cells that are derived from the epithelium, mesenchyme or dedifferentiated mature cells results in the formation of the most primitive CSCs, which show distinct genotypes in a variety of histopathological and anatomic sites, even within a single gastric unit, due to the presence of different types of stem cells and genetic alterations. Primitive CSCs acquire the properties of unlimited self-renewal and multi-lineage differentiation potential and gradually create a hierarchical structure in the tumour lesion. Concurrently, these CSCs evolve into different subclones to survive and to evade the immune system, and the dominant clones generate their respective hierarchical organisations, while the remaining cells are eliminated. Although this hypothesis has been supported by single-cell analyses of other cancers (Navin et al, 2011; Lawson et al, 2015), genetic and functional studies on GCSCs at the single-cell level are lacking. Hence, using identification, isolation, dynamic observation and deep sequencing techniques with single GCSCs is expected to contribute to existing knowledge and to allow for the study of different subclones of GCSCs via the combination of genomic analyses with functional assays.
Regulatory mechanisms of the unified model of GC
CSC niche
It is generally accepted that dormant stem cells in a given tissue reside in a specific anatomic location, namely the stem cell niche, where a specialised microenvironment is provided to regulate stem cell fate (Voog and Jones, 2010). Likewise, according to previous studies, there is also a niche around CSCs in tumours (Plaks et al, 2015). The CSC niche, containing distinct stromal cells such as fibroblasts, endothelial cells, extracellular matrix and networks of soluble factors and blood vessels (Figure 2C), has an important role in the maintenance of CSC properties and plasticity (Borovski et al, 2011). However, the components and the principally regulated pathways of niches in GC are largely unknown. Some studies have suggested that bone marrow-derived myofibroblasts (BMFs), which are involved in the formation of the niche, promote the development and progression of GC (Worthley et al, 2009; Quante et al, 2011; Shibata et al, 2013; Zhu et al, 2016a). Using an inflammation-induced GC model, Quante et al (2011) found the expansion of MFs in the BM niche at the earliest stage of GC development and subsequent migration into injury sites. The BMFs created a niche that promoted tumorigenesis in TGF-β- and Cxcr4/Cxcl12-dependent pathways. Similarly, another study revealed that the activation of the IL-6/HGF/STAT3 pathway in BMFs-stimulated cancer cells to upregulate TGF-β1, and vice versa. This interaction between BMFs and GCCs is responsible for the regulation of stemness and tumorigenesis (Zhu et al, 2016a). Recent research confirmed that BM-derived Cxcr4+ intraepithelial gastric innate lymphoid cells (ILCs) adjacent to Mist1+ stem cells in the isthmus area functioned as niche cells to maintain the properties of GSCs (Hayakawa et al, 2015). The other essential aspects of the CSC niche, such as hypoxia (Lu et al, 2015; Zhang et al, 2016a), tumour-associated macrophages (Wan et al, 2014; Zhou et al, 2015; Jia et al, 2016; Raggi et al, 2016), blood vessels (Borovski et al, 2011) and vasculogenic mimicry (Sun et al, 2016), have also been studied for their effects on the stemness of CSCs in various types of tumours. Although the relevant research on GC is insufficient, it can nevertheless be speculated that the niche is important for the survival and evolution of GCSCs.
Epigenetic alterations
Epigenetics refers to heritable alterations in gene expression without the involvement of changes in nucleotide sequences. Similar to genetic mutations, epigenetic alterations also have an irreplaceably important function in the malignant transformation of somatic cells and in the development and progression of cancer. In GAC, many mechanisms of variation have been studied, including DNA hypermethylation, histone modification and epigenetic silencing of noncoding RNAs. Using combined transcriptome and epigenome sequence analyses in EBV-infected GCCLs, it was revealed that the transcription of 216 genes was downregulated due to EBV-related hypermethylation and that tumour-associated proteins, such as Indian hedgehog and TraB domain-containing, were overexpressed with a remarkable increase in gene methylation (Liang et al, 2014). In addition, the epigenetic silencing of mismatch repair genes and tumour suppressor genes, including MLH1, CDH1 and CDKN2A, was also identified in distinct subtypes of GC (Ushijima and Hattori, 2012; Cancer Genome Atlas Research Network, 2014). Furthermore, many studies (Peterson et al, 2010; Tomita et al, 2011; Haam et al, 2014; Chen et al, 2015; Guo et al, 2015; Park et al, 2015; Kim et al, 2016; Yang et al, 2016; Yoshida et al, 2017) have demonstrated the effects of aberrant epigenetic activation and of the inactivation of diverse genes, including Tff1, Tff2, BACH2, Sema3E, OLFM4, TET1, TLR4, GDF1, FAT4 and so on, on GC carcinogenesis and progression through in vivo and in vitro functional experiments. For instance, Tomita et al (2011) observed an extensive suppression of Tff1, a tumour suppressor gene, which is associated with DNA methylation and histone modification at the Tff1 promoter in epithelial cells of MNU-treated wild-type mice, and subsequent GC initiation. Additionally, there was an enhanced tumorigenic capacity in gastrin- and Tff1-deficient mice and, conversely, a repression in hypergastrinemic mice. Hence, it was suggested that the progressive epigenetic silencing of Tff1 by promoter methylation, which could be reversed by gastrin, contributed to GC initiation (Tomita et al, 2011).
The ability of long noncoding RNA (lncRNA; Endo et al, 2013; Hu et al, 2014; Okugawa et al, 2014; Fan et al, 2016; Guo et al, 2016; Lu et al, 2016) and microRNA (miRNA) to regulate GC development and progression is also being elucidated (Table 2). In CD44+ GC stem-like cells, it was found that the marked upregulation of miR-106b and the related downregulation of its target protein Smad7 resulted in activation of the TGF-β/Smad pathway, thereby enhancing stemness characteristics such as self-renewal, EMT and invasion (Yu et al, 2014). Likewise, another study showed that the overexpression of miR-19b/20a/92a promoted sustained stem-like properties through activating the Wnt-β-catenin pathway in EpCAM+CD44+ GCCs (Wu et al, 2013). Furthermore, our group and other researchers revealed an important role of lncRNA and miRNA in regulating CD44 expression of GCCs (Hashimoto et al, 2010; Hu et al, 2014; Wei et al, 2016). Recently, Bintu et al (2016) investigated the regulatory mechanism of epigenetics at the single-cell level and proposed a three-state dynamic model of epigenetic silencing, in which the state of gene expression was stochastically converted among activation, reversible silencing and irreversible silencing in an all-or-none manner, namely, by regulating the fraction of repressed cells but not the level of gene expression. Hence, epigenetic aberrations contributed to the distinct functional types and phenotypes of individual GCCs, particularly the initiation, persistence and progression of CSCs; however, additional functional assays are necessary to validate the emergence of epigenetic alterations in identifying potential therapeutic targets.
Other regulatory mechanisms
Almost all of the genetic and epigenetic functions of cells are performed and regulated in the context of various types of signalling pathways, and the dysregulation of such pathways contributes to tumorigenesis. For example, the Wnt pathway significantly influences the development of various tumours (Takahashi-Yanaga and Kahn, 2010). Many studies have shown that the canonical Wnt/β-catenin pathway is involved in the acquisition and maintenance of the stem-like features of GCCs (Wang et al, 2013; Mao et al, 2014; Yong et al, 2016; Zhou et al, 2016), and non-canonical Wnt pathways, especially those activated by Wnt5a, are important in regulating EMT and initiating GCSCs (Kanzawa et al, 2013; Hayakawa et al, 2015). The HH family, as another key regulator during embryonic development, has also been implicated in the formation of diverse tumours (McMillan and Matsui, 2012). In human gastric glands, the SHH pathway has important roles in self-renewal (Song et al, 2011), chemotherapy resistance of CSCs (Yoon et al, 2014; Xu et al, 2015a) and tumour angiogenesis (Chen et al, 2011). In addition, multiple signalling pathways, including those associated with Notch-1 (Yeh et al, 2009; Hong et al, 2014)/Notch2 (Tseng et al, 2012; Huang et al, 2015), STAT3 (Jenkins et al, 2005; Ernst et al, 2008; Tye et al, 2012), TGF-β/Smad (Yoo et al, 2008; Yu et al, 2014; Yang et al, 2016), TNF-α/TNFR1 (Oshima et al, 2014), PI3K/AKT/mTOR (Yun et al, 2014), c-Met (Yashiro et al, 2013) and Ras/Raf/MEK/ERK (Bessede et al, 2014), are likely to be associated with the malignant behaviour of GCSCs. In total, all of these pathways form a complex signalling network inside and outside cancer cells via crosstalk between different axes and via mutual regulation by their products, such as cytokines, chemokines, transcription factors and other regulatory proteins. In addition, there are many unknown or unclear mechanisms, such as the stochasticity of gene expression and the plasticity of cancer cells that promote CSC development and evolution. Therefore, it is impossible for a single theory to elucidate this complicated biological system, whereas a unified model is better able to present a comprehensive and reasonable explanation.
Clinical targeted therapy
The primary methods of targeted therapy in GC involve small molecules, tumour vaccines, monoclonal antibodies (mAbs) and adaptive cellular immunotherapies, including the chimeric antigen receptor T-cell (CAR-T) strategy. Regarding GCSCs, the major drug treatment targets are specific antigens and stemness-related signalling pathways.
Targeting specific antigens
CD44 is a surface marker that is expressed in GC stem-like cells. A study found that CD44v promoted glutathione synthesis and inhibited the activity of reactive oxygen species, thereby resulting in tumour growth via interaction with xCT (a glutamate-cystine transporter; Ishimoto et al, 2011). Accordingly, a dose-escalation clinical study for targeting CD44v+ CSCs by the oral administration of sulfasalazine (SSZ, an inhibitor of xCT) in patients with AGC was conducted. The results showed that SSZ was safe and efficacious in patients, some of whom exhibited decreased levels of CD44v+ cells and glutathione in their biopsy samples (Shitara et al, 2016). Moreover, several types of mAbs, such as bivatuzumab mertansine (BIMI1) and RO5429083, have been designed to fight CD44+ cells. However, multiple phase I clinical trials in different tumours showed that a few patients treated with CD44v6-targeting BIMI1 had fatal drug-associated adverse events, and the overall assessment of the benefits and risks was negative (Tijink et al, 2006; Rupp et al, 2007; Sauter et al, 2007; Riechelmann et al, 2008). Thus, the further clinical development of BIMI1 was terminated. RO5429083, also known as RG7356, is a humanised IgG1 mAb that is specific to CD44. In a phase I study, among 44 patients with refractory or relapsed acute myeloid leukemia, two patients treated with RG7356 had complete responses, and one had stable disease, and the side effects were generally mild or moderate (Vey et al, 2016). The study of RG7356 in patients with CD44+ advanced solid tumours, including GC, was sponsored by Roche (NCT01358903), and it is expected to reach a favourable outcome.
Epithelial cell adhesion molecule (EpCAM) is a transmembrane glycoprotein in normal epithelia and is upregulated in epithelial neoplasms. In addition, it is a marker widely expressed in CSCs and is involved in the activation of a stemness-related pathway (Gires et al, 2009; Munz et al, 2009). EpCAM has long been considered a potential target for antitumour therapy, and vaccination with EpCAM protein has been proposed (Mosolits et al, 2004). Furthermore, clinical trials of mAbs have ranged from testing the first murine IgG2a antibody, termed edrecolomab (Mosolits et al, 2004), to the EpCAM/CD3 bi-specific antibody MT110 (NCT00635596) and catumaxomab (Heiss et al, 2010; Goere et al, 2014; Bokemeyer et al, 2015), and ultimately to human engineered or fully human antibodies including ING-1 (Mosolits et al, 2004) and adecatumumab (MT201; Schmidt et al, 2012). Heiss et al (2010) found that catumaxomab provided a significant clinical benefit to GC patients with malignant ascites in a phase II/III trial. Another study has also demonstrated the safety and efficacy of catumaxomab in patients with resectable GC when serving as a part of a combined modality therapy (Bokemeyer et al, 2015). Moreover, a phase II trial of the intraperitoneal infusion of catumaxomab to treat postoperative GC patients with resectable primary lesions and peritoneal carcinomatosis is currently underway (Goere et al, 2014). In addition, a clinical study (NCT02725125) aiming to assess the safety and efficacy of EpCAM-targeted CAR-T for GC is currently recruiting patients.
CD133+ cells represent a subpopulation of CSCs in many solid tumours (Grosse-Gehling et al, 2013). Although several mAbs (Swaminathan et al, 2010; Huang et al, 2013) against CD133 have been developed, retrospective clinical trials are lacking. Recently, our research team successfully generated anti-CD133 CAR vector-transduced T cells, and a phase I clinical study (NCT02541370) on the safety and efficacy of our CART133 formulation for treating relapsed or chemotherapy-refractory advanced malignancies including GC is currently recruiting patients. In addition, the University of Minnesota has sponsored a clinical trial to test CD133KDEL toxin (an anti-CD133 ligand-directed, non-immunogenic pseudomonas toxin) in solid tumour patients (NCT02845414).
Programmed cell death-ligand 1 (PD-L1) is now considered a critical component of the immunosuppressive microenvironment in GAC (Thompson et al, 2016). Multiple inhibitors of immune checkpoints have been applied to clinical therapy, especially the anti-PD-1 mAb pembrolizumab, which has shown manageable side effects and a 22% (8/36) overall response rate in a phase I study of PD-L1+ AGC patients (Muro et al, 2016). However, another multicenter study on the anti-PD-L1 mAb nivolumab showed no benefit in 7 GC patients (Brahmer et al, 2012). Many promising studies of combination treatments of pembrolizumab or nivolumab with different therapies and new drugs including atezolizumab (a human engineered anti-PD-L1 mAb) and CA-170 (a PD-L1/2 and VISTA checkpoint inhibitor) are underway in GC patients.
Lgr5 and Sox2 are thought to be specific markers of GCSCs and normal stem cells (NSCs). A Sox2-targeted plasmid DNA vaccine has been developed and used to treat patients with advanced breast cancer (NCT02157051). In addition, an anti-Lgr5 humanised mAb, BNC101, is currently being tested in a phase I clinical study of patients with metastatic colorectal cancer (NCT02726334). The expected results may promote the application of these drugs in GC patients.
Targeting stemness-related signalling pathways
HH pathway inhibitors have been designed to target four major sites, specifically SMO, Hh ligand, Hh acyltransferase (HHAT) and GLI. To date, the drugs applied in clinical trials only include SMO inhibitors, such as vismodegib (GDC-0449), sonidegib (LDE225), saridegib (IPI-926), taladegib (LY2940680), glasdegib (PF-04449913), TAK-441, BMS-833923 (XL139) and LEQ506. Although some have exerted targeted efficacy against tumours (Justilien and Fields, 2015; Migden et al, 2015; Robinson et al, 2015), the clinical results have not shown clear improvements in terms of therapeutic effects in GC patients. A randomised, controlled, multicenter phase II clinical trial conducted by the NCI to evaluate the efficacy of chemotherapy (FOLFOX) with or without vismodegib in advanced gastric and gastroesophageal junction (GEJ) cancer patients has been completed, and it revealed no significant differences in the outcome measures, including progression-free survival (PFS), relative risk (RR) and OS (NCT00982592). Likewise, the GC patients in multiple phase I clinical trials rarely experienced a confirmed complete or partial response to other SMO inhibitors, such as the objective response being 0/1 for sonidegib by Rodon et al (2014) and 0/1 for TAK-441 by Goldman et al (2015). Nevertheless, some studies have been conducted, including a phase 1b/2 clinical trial of taladegib in combination with chemotherapy and radiotherapy to treat patients with oesophageal or GEJ adenocarcinoma (NCT02530437) and some GC-related clinical studies.
There are three major types of Notch pathway-targeted drugs: (1) Notch receptor inhibitors: brontictuzumab (OMP-52M51, anti-Notch1), tarextumab (OMP-59R5, anti-Notch2/3) and BMS906024 (pan-Notch inhibitor); (2) inhibitors of the Notch ligand DLL4: MED10639, demcizumab (OMP-21M18), OMP-305B83 and enoticumab (REGN421); and (3) γ-secretase inhibitors: MK0752, RO4929097, PF03084014, BMS986115, LY3039478 and LY900009. However, only a few solid tumour patients have experienced complete responses to selected drugs, including MK0752 (Krop et al, 2012; Piha-Paul et al, 2015) and PF03084014 (Messersmith et al, 2015). Moreover, many drugs induce a variety of severe adverse events such as intractable diarrhoea, vomiting, skin disorders and hypophosphatemia (Takebe et al, 2014), perhaps due to the wide distribution of the Notch pathway in normal tissues, especially NSCs. Hence, the antitumour activity of Notch pathway inhibitors must be confirmed based on the results of ongoing clinical trials.
Drugs targeting the Wnt pathway have been developed and have emerged for clinical use within the last 5 years. The mechanisms associated with targeting the Wnt pathway have been categorised as follows: (1) targeting the Wnt receptor: Foxy-5 (mimicking the effects of Wnt-5a), OTSA101 (anti-Fzd10), ipafricept (Fzd8-Fc, OMP-54F28); (2) β-catenin antagonists: PRI-724, CWP232291 (CWP291); (3) PORCN protein inhibitors: WNT974 (LGK974), ETC-1922159 (ETC-159), CGX1321; and (4) other inhibitors: DKN01 (LY2812176, anti-DKK1), LY2090314 (anti-GSK3β). A phase I clinical trial that assessed the combination treatment of LY2090314 with pemetrexed and carboplatin in patients with solid tumours reported an overall response rate of 5/41, but two GC cases had no objective response (Gray et al, 2015). Most of the clinical studies on targeting the Wnt pathway are in progress, although a few have been terminated or suspended due to severe adverse events.
The clinical trials of other agents aimed at targeting CSC-related pathways in GC patients are shown in Table 3. Although the majority of existing results have demonstrated negative or limited efficacy, we believe that targeted therapy for CSCs is a feasible and promising approach for developing cures for cancer in light of successful analogous research in multiple types of tumours.
Further perspectives
Currently, the primary problems with regard to the safety and efficacy of targeted therapies against solid tumours are off-target effects and drug resistance, a situation that largely reflects the complexity and dynamic variation of cancer cells. Technological advances in molecular biology have revolutionised our understanding of tumour development. In particular, sequencing techniques have confirmed multiple subclones with distinct genomes in tumours, and functional experiments have revealed rare subpopulations with stemness properties, indicating that tumour growth can be considered as an evolutionary tree, in which the most primitive CSCs, representing the trunk, continuously divide, self-renew and differentiate. Subsequently, new mutations emerge as branches, and the main subtypes are gradually formed due to selection by the TME. However, most of the mechanisms underlying the unified model remain unclear. Even within a single genotype, each individual cancer cell possesses different gene expression levels, phenotypes and functional characteristics, and this diversity is constantly regulated. Hence, many problems remain to be resolved, such as how heterogeneity is generated among different CSCs with the same genome, whether cancer could be cured by killing CSCs just as one may cut down the trunk of a tree, and so forth. Additional promising studies are currently focused on investigating the mechanisms of heterogeneity in CSCs at the single-cell level, which will likely aid in addressing the current challenge of developing clinical CSC-targeted treatments.
References
Aoi T, Yae K, Nakagawa M, Ichisaka T, Okita K, Takahashi K, Chiba T, Yamanaka S (2008) Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science 321: 699–702.
Arnold K, Sarkar A, Yram MA, Polo JM, Bronson R, Sengupta S, Seandel M, Geijsen N, Hochedlinger K (2011) Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 9: 317–329.
Baer C, Claus R, Plass C (2013) Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Res 73: 473–477.
Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, Sato T, Stange DE, Begthel H, van den Born M, Danenberg E, van den Brink S, Korving J, Abo A, Peters PJ, Wright N, Poulsom R, Clevers H (2010) Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 6: 25–36.
Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H (2007) Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449: 1003–1007.
Bessede E, Dubus P, Megraud F, Varon C (2015) Helicobacter pylori infection and stem cells at the origin of gastric cancer. Oncogene 34: 2547–2555.
Bessede E, Staedel C, Acuna Amador LA, Nguyen PH, Chambonnier L, Hatakeyama M, Belleannee G, Megraud F, Varon C (2014) Helicobacter pylori generates cells with cancer stem cell properties via epithelial-mesenchymal transition-like changes. Oncogene 33: 4123–4131.
Bintu L, Yong J, Antebi YE, McCue K, Kazuki Y, Uno N, Oshimura M, Elowitz MB (2016) Dynamics of epigenetic regulation at the single-cell level. Science 351: 720–724.
Bokemeyer C, Stein A, Ridwelski K, Atanackovic D, Arnold D, Woll E, Ulrich A, Fischer R, Kruger C, Schuhmacher C (2015) A phase II study of catumaxomab administered intra- and postoperatively as part of a multimodal approach in primarily resectable gastric cancer. Gastric Cancer 18: 833–842.
Bonnet D, Dick JE (1997) Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3: 730–737.
Borovski T, De Sousa EMF, Vermeulen L, Medema JP (2011) Cancer stem cell niche: the place to be. Cancer Res 71: 634–639.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366: 2455–2465.
Cancer Genome Atlas Research Network (2014) Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513: 202–209.
Cao L, Hu X, Zhang Y (2009) Omental milky spots—highly efficient ‘natural filter’ for screening gastric cancer stem cells. Med Hypotheses 73: 1017–1018.
Cao L, Hu X, Zhang Y, Sun XT (2011) Omental milky spots in screening gastric cancer stem cells. Neoplasma 58: 20–26.
Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D'Alessio AC, Young RA, Weinberg RA (2013) Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 154: 61–74.
Chen H, Xie GH, Wang WW, Yuan XL, Xing WM, Liu HJ, Chen J, Dou M, Shen LS (2015) Epigenetically downregulated Semaphorin 3E contributes to gastric cancer. Oncotarget 6: 20449–20465.
Chen T, Yang K, Yu J, Meng W, Yuan D, Bi F, Liu F, Liu J, Dai B, Chen X, Wang F, Zeng F, Xu H, Hu J, Mo X (2012) Identification and expansion of cancer stem cells in tumor tissues and peripheral blood derived from gastric adenocarcinoma patients. Cell Res 22: 248–258.
Chen W, Tang T, Eastham-Anderson J, Dunlap D, Alicke B, Nannini M, Gould S, Yauch R, Modrusan Z, DuPree KJ, Darbonne WC, Plowman G, de Sauvage FJ, Callahan CA (2011) Canonical hedgehog signaling augments tumor angiogenesis by induction of VEGF-A in stromal perivascular cells. Proc Natl Acad Sci USA 108: 9589–9594.
Choi YJ, Kim N, Chang H, Lee HS, Park SM, Park JH, Shin CM, Kim JM, Kim JS, Lee DH, Jung HC (2015) Helicobacter pylori-induced epithelial-mesenchymal transition, a potential role of gastric cancer initiation and an emergence of stem cells. Carcinogenesis 36: 553–563.
Chow KH, Shin DM, Jenkins MH, Miller EE, Shih DJ, Choi S, Low BE, Philip V, Rybinski B, Bronson RT, Taylor MD, Yun K (2014) Epigenetic states of cells of origin and tumor evolution drive tumor-initiating cell phenotype and tumor heterogeneity. Cancer Res 74: 4864–4874.
Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM (2006) Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 66: 9339–9344.
Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS, Liu J, Yue YG, Wang J, Yu K, Ye XS, Do IG, Liu S, Gong L, Fu J, Jin JG, Choi MG, Sohn TS, Lee JH, Bae JM, Kim ST, Park SH, Sohn I, Jung SH, Tan P, Chen R, Hardwick J, Kang WK, Ayers M, Hongyue D, Reinhard C, Loboda A, Kim S, Aggarwal A (2015) Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med 21: 449–456.
Cunningham D, Starling N, Rao S, Iveson T, Nicolson M, Coxon F, Middleton G, Daniel F, Oates J, Norman AR (2008) Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 358: 36–46.
Ehata S, Johansson E, Katayama R, Koike S, Watanabe A, Hoshino Y, Katsuno Y, Komuro A, Koinuma D, Kano MR, Yashiro M, Hirakawa K, Aburatani H, Fujita N, Miyazono K (2011) Transforming growth factor-beta decreases the cancer-initiating cell population within diffuse-type gastric carcinoma cells. Oncogene 30: 1693–1705.
Elowitz MB, Levine AJ, Siggia ED, Swain PS (2002) Stochastic gene expression in a single cell. Science 297: 1183–1186.
Endo H, Shiroki T, Nakagawa T, Yokoyama M, Tamai K, Yamanami H, Fujiya T, Sato I, Yamaguchi K, Tanaka N, Iijima K, Shimosegawa T, Sugamura K, Satoh K (2013) Enhanced expression of long non-coding RNA HOTAIR is associated with the development of gastric cancer. PLoS One 8: e77070.
Ernst M, Najdovska M, Grail D, Lundgren-May T, Buchert M, Tye H, Matthews VB, Armes J, Bhathal PS, Hughes NR, Marcusson EG, Karras JG, Na S, Sedgwick JD, Hertzog PJ, Jenkins BJ (2008) STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J Clin Invest 118: 1727–1738.
Fan Y, Wang YF, Su HF, Fang N, Zou C, Li WF, Fei ZH (2016) Decreased expression of the long noncoding RNA LINC00261 indicate poor prognosis in gastric cancer and suppress gastric cancer metastasis by affecting the epithelial-mesenchymal transition. J Hematol Oncol 9: 57.
Ferrand J, Lehours P, Schmid-Alliana A, Megraud F, Varon C (2011a) Helicobacter pylori infection of gastrointestinal epithelial cells in vitro induces mesenchymal stem cell migration through an NF-kappaB-dependent pathway. PLoS One 6: e29007.
Ferrand J, Noel D, Lehours P, Prochazkova-Carlotti M, Chambonnier L, Menard A, Megraud F, Varon C (2011b) Human bone marrow-derived stem cells acquire epithelial characteristics through fusion with gastrointestinal epithelial cells. PLoS One 6: e19569.
Fujii Y, Yoshihashi K, Suzuki H, Tsutsumi S, Mutoh H, Maeda S, Yamagata Y, Seto Y, Aburatani H, Hatakeyama M (2012) CDX1 confers intestinal phenotype on gastric epithelial cells via induction of stemness-associated reprogramming factors SALL4 and KLF5. Proc Natl Acad Sci USA 109: 20584–20589.
Fujita T, Chiwaki F, Takahashi RU, Aoyagi K, Yanagihara K, Nishimura T, Tamaoki M, Komatsu M, Komatsuzaki R, Matsusaki K, Ichikawa H, Sakamoto H, Yamada Y, Fukagawa T, Katai H, Konno H, Ochiya T, Yoshida T, Sasaki H (2015) Identification and characterization of CXCR4-positive gastric cancer stem cells. PLoS One 10: e0130808.
Fukamachi H, Seol HS, Shimada S, Funasaka C, Baba K, Kim JH, Park YS, Kim MJ, Kato K, Inokuchi M, Kawachi H, Yook JH, Eishi Y, Kojima K, Kim WH, Jang SJ, Yuasa Y (2013) CD49f(high) cells retain sphere-forming and tumor-initiating activities in human gastric tumors. PLoS One 8: e72438.
Fukamachi H, Shimada S, Ito K, Ito Y, Yuasa Y (2011) CD133 is a marker of gland-forming cells in gastric tumors and Sox17 is involved in its regulation. Cancer Sci 102: 1313–1321.
Fukuda K, Saikawa Y, Ohashi M, Kumagai K, Kitajima M, Okano H, Matsuzaki Y, Kitagawa Y (2009) Tumor initiating potential of side population cells in human gastric cancer. Int J Oncol 34: 1201–1207.
Gao P, Xing AY, Zhou GY, Zhang TG, Zhang JP, Gao C, Li H, Shi DB (2013) The molecular mechanism of microRNA-145 to suppress invasion-metastasis cascade in gastric cancer. Oncogene 32: 491–501.
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366: 883–892.
Gires O, Klein CA, Baeuerle PA (2009) On the abundance of EpCAM on cancer stem cells. Nat Rev Cancer 9: 143.
Goere D, Gras-Chaput N, Auperin A, Flament C, Mariette C, Glehen O, Zitvogel L, Elias D (2014) Treatment of gastric peritoneal carcinomatosis by combining complete surgical resection of lesions and intraperitoneal immunotherapy using catumaxomab. BMC Cancer 14: 148.
Goldman J, Eckhardt SG, Borad MJ, Curtis KK, Hidalgo M, Calvo E, Ryan DP, Wirth LJ, Parikh A, Partyka J, Faessel H, Gangolli E, Stewart S, Rosen LS, Bowles DW (2015) Phase I dose-escalation trial of the oral investigational Hedgehog signaling pathway inhibitor TAK-441 in patients with advanced solid tumors. Clin Cancer Res 21: 1002–1009.
Gray JE, Infante JR, Brail LH, Simon GR, Cooksey JF, Jones SF, Farrington DL, Yeo A, Jackson KA, Chow KH, Zamek-Gliszczynski MJ, Burris 3rd HA (2015) A first-in-human phase I dose-escalation, pharmacokinetic, and pharmacodynamic evaluation of intravenous LY2090314, a glycogen synthase kinase 3 inhibitor, administered in combination with pemetrexed and carboplatin. Invest New Drugs 33: 1187–1196.
Grosse-Gehling P, Fargeas CA, Dittfeld C, Garbe Y, Alison MR, Corbeil D, Kunz-Schughart LA (2013) CD133 as a biomarker for putative cancer stem cells in solid tumours: limitations, problems and challenges. J Pathol 229: 355–378.
Guo L, Bai H, Zou D, Hong T, Liu J, Huang J, He P, Zhou Q, He J (2014) The role of microRNA-133b and its target gene FSCN1 in gastric cancer. J Exp Clin Cancer Res 33: 99.
Guo LL, He ZC, Yang CQ, Qiao PT, Yin GL (2015) Epigenetic silencing of olfactomedin-4 enhances gastric cancer cell invasion via activation of focal adhesion kinase signaling. BMB Rep 48: 630–635.
Guo X, Yang Z, Zhi Q, Wang D, Guo L, Li G, Miao R, Shi Y, Kuang Y (2016) Long noncoding RNA OR3A4 promotes metastasis and tumorigenicity in gastric cancer. Oncotarget 7: 30276–30294.
Haam K, Kim HJ, Lee KT, Kim JH, Kim M, Kim SY, Noh SM, Song KS, Kim YS (2014) Epigenetic silencing of BTB and CNC homology 2 and concerted promoter CpG methylation in gastric cancer. Cancer Lett 351: 206–214.
Han ME, Jeon TY, Hwang SH, Lee YS, Kim HJ, Shim HE, Yoon S, Baek SY, Kim BS, Kang CD, Oh SO (2011) Cancer spheres from gastric cancer patients provide an ideal model system for cancer stem cell research. Cell Mol Life Sci 68: 3589–3605.
Han TS, Hur K, Xu G, Choi B, Okugawa Y, Toiyama Y, Oshima H, Oshima M, Lee HJ, Kim VN, Chang AN, Goel A, Yang HK (2015) MicroRNA-29c mediates initiation of gastric carcinogenesis by directly targeting ITGB1. Gut 64: 203–214.
Hasegawa T, Yashiro M, Nishii T, Matsuoka J, Fuyuhiro Y, Morisaki T, Fukuoka T, Shimizu K, Shimizu T, Miwa A, Hirakawa K (2014) Cancer-associated fibroblasts might sustain the stemness of scirrhous gastric cancer cells via transforming growth factor-beta signaling. Int J Cancer 134: 1785–1795.
Hashimoto Y, Akiyama Y, Otsubo T, Shimada S, Yuasa Y (2010) Involvement of epigenetically silenced microRNA-181c in gastric carcinogenesis. Carcinogenesis 31: 777–784.
Hayakawa Y, Ariyama H, Stancikova J, Sakitani K, Asfaha S, Renz BW, Dubeykovskaya ZA, Shibata W, Wang H, Westphalen CB, Chen X, Takemoto Y, Kim W, Khurana SS, Tailor Y, Nagar K, Tomita H, Hara A, Sepulveda AR, Setlik W, Gershon MD, Saha S, Ding L, Shen Z, Fox JG, Friedman RA, Konieczny SF, Worthley DL, Korinek V, Wang TC (2015) Mist1 expressing gastric stem cells maintain the normal and neoplastic gastric epithelium and are supported by a perivascular stem cell niche. Cancer Cell 28: 800–814.
Heiss MM, Murawa P, Koralewski P, Kutarska E, Kolesnik OO, Ivanchenko VV, Dudnichenko AS, Aleknaviciene B, Razbadauskas A, Gore M, Ganea-Motan E, Ciuleanu T, Wimberger P, Schmittel A, Schmalfeldt B, Burges A, Bokemeyer C, Lindhofer H, Lahr A, Parsons SL (2010) The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: results of a prospective randomized phase II/III trial. Int J Cancer 127: 2209–2221.
Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, Hoffmann EM, Lax S, Waldispuehl-Geigl J, Mauermann O, Lackner C, Hofler G, Eisner F, Sill H, Samonigg H, Pantel K, Riethdorf S, Bauernhofer T, Geigl JB, Speicher MR (2013) Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res 73: 2965–2975.
Hong KJ, Wu DC, Cheng KH, Chen LT, Hung WC (2014) RECK inhibits stemness gene expression and tumorigenicity of gastric cancer cells by suppressing ADAM-mediated Notch1 activation. J Cell Physiol 229: 191–201.
Hong X, Xu Y, Qiu X, Zhu Y, Feng X, Ding Z, Zhang S, Zhong L, Zhuang Y, Su C, Hong X, Cai J (2016) MiR-448 promotes glycolytic metabolism of gastric cancer by downregulating KDM2B. Oncotarget 7: 22092–22102.
Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC (2004) Gastric cancer originating from bone marrow-derived cells. Science 306: 1568–1571.
Hu N, Kadota M, Liu H, Abnet CC, Su H, Wu H, Freedman ND, Yang HH, Wang C, Yan C, Wang L, Gere S, Hutchinson A, Song G, Wang Y, Ding T, Qiao YL, Koshiol J, Dawsey SM, Giffen C, Goldstein AM, Taylor PR, Lee MP (2016) Genomic landscape of somatic alterations in esophageal squamous cell carcinoma and gastric cancer. Cancer Res 76: 1714–1723.
Hu Y, Wang J, Qian J, Kong X, Tang J, Wang Y, Chen H, Hong J, Zou W, Chen Y, Xu J, Fang JY (2014) Long noncoding RNA GAPLINC regulates CD44-dependent cell invasiveness and associates with poor prognosis of gastric cancer. Cancer Res 74: 6890–6902.
Huang J, Li C, Wang Y, Lv H, Guo Y, Dai H, Wicha MS, Chang AE, Li Q (2013) Cytokine-induced killer (CIK) cells bound with anti-CD3/anti-CD133 bispecific antibodies target CD133(high) cancer stem cells in vitro and in vivo. Clin Immunol 149: 156–168.
Huang TT, Ping YH, Wang AM, Ke CC, Fang WL, Huang KH, Lee HC, Chi CW, Yeh TS (2015) The reciprocal regulation loop of Notch2 pathway and miR-23b in controlling gastric carcinogenesis. Oncotarget 6: 18012–18026.
Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H, Masuko T, Shimizu T, Ishikawa T, Kai K, Takahashi E, Imamura Y, Baba Y, Ohmura M, Suematsu M, Baba H, Saya H (2011) CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(−) and thereby promotes tumor growth. Cancer Cell 19: 387–400.
Ishimoto T, Sugihara H, Watanabe M, Sawayama H, Iwatsuki M, Baba Y, Okabe H, Hidaka K, Yokoyama N, Miyake K, Yoshikawa M, Nagano O, Komohara Y, Takeya M, Saya H, Baba H (2014) Macrophage-derived reactive oxygen species suppress miR-328 targeting CD44 in cancer cells and promote redox adaptation. Carcinogenesis 35: 1003–1011.
Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, Chaudhuri S, Pujara K, Guillory J, Edgar KA, Janakiraman V, Scholz RP, Bowman KK, Lorenzo M, Li H, Wu J, Yuan W, Peters BA, Kan Z, Stinson J, Mak M, Modrusan Z, Eigenbrot C, Firestein R, Stern HM, Rajalingam K, Schaefer G, Merchant MA, Sliwkowski MX, de Sauvage FJ, Seshagiri S (2013) Oncogenic ERBB3 mutations in human cancers. Cancer Cell 23: 603–617.
Janowitz T, Thuss-Patience P, Marshall A, Kang JH, Connell C, Cook N, Dunn J, Park SH, Ford H (2016) Chemotherapy vs supportive care alone for relapsed gastric, gastroesophageal junction, and oesophageal adenocarcinoma: a meta-analysis of patient-level data. Br J Cancer 114: 381–387.
Jenkins BJ, Grail D, Nheu T, Najdovska M, Wang B, Waring P, Inglese M, McLoughlin RM, Jones SA, Topley N, Baumann H, Judd LM, Giraud AS, Boussioutas A, Zhu HJ, Ernst M (2005) Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-beta signaling. Nat Med 11: 845–852.
Ji Q, Hao X, Meng Y, Zhang M, Desano J, Fan D, Xu L (2008) Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer 8: 266.
Jia XH, Feng GW, Wang ZL, Du Y, Shen C, Hui H, Peng D, Li ZJ, Kong DL, Tian J (2016) Activation of mesenchymal stem cells by macrophages promotes tumor progression through immune suppressive effects. Oncotarget 7: 20934–20944.
Jiang J, Zhang Y, Chuai S, Wang Z, Zheng D, Xu F, Zhang Y, Li C, Liang Y, Chen Z (2012) Trastuzumab (herceptin) targets gastric cancer stem cells characterized by CD90 phenotype. Oncogene 31: 671–682.
Justilien V, Fields AP (2015) Molecular pathways: novel approaches for improved therapeutic targeting of Hedgehog signaling in cancer stem cells. Clin Cancer Res 21: 505–513.
Kaern M, Elston TC, Blake WJ, Collins JJ (2005) Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet 6: 451–464.
Kakiuchi M, Nishizawa T, Ueda H, Gotoh K, Tanaka A, Hayashi A, Yamamoto S, Tatsuno K, Katoh H, Watanabe Y, Ichimura T, Ushiku T, Funahashi S, Tateishi K, Wada I, Shimizu N, Nomura S, Koike K, Seto Y, Fukayama M, Aburatani H, Ishikawa S (2014) Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat Genet 46: 583–587.
Kang YK, Kang WK, Shin DB, Chen J, Xiong J, Wang J, Lichinitser M, Guan Z, Khasanov R, Zheng L, Philco-Salas M, Suarez T, Santamaria J, Forster G, McCloud PI (2009) Capecitabine/cisplatin versus 5-fluorouracil/cisplatin as first-line therapy in patients with advanced gastric cancer: a randomised phase III noninferiority trial. Ann Oncol 20: 666–673.
Kanzawa M, Semba S, Hara S, Itoh T, Yokozaki H (2013) WNT5A is a key regulator of the epithelial-mesenchymal transition and cancer stem cell properties in human gastric carcinoma cells. Pathobiology 80: 235–244.
Karam SM, Leblond CP (1993) Dynamics of epithelial cells in the corpus of the mouse stomach. I. Identification of proliferative cell types and pinpointing of the stem cell. Anat Rec 236: 259–279.
Katsuno Y, Ehata S, Yashiro M, Yanagihara K, Hirakawa K, Miyazono K (2012) Coordinated expression of REG4 and aldehyde dehydrogenase 1 regulating tumourigenic capacity of diffuse-type gastric carcinoma-initiating cells is inhibited by TGF-beta. J Pathol 228: 391–404.
Kiga K, Mimuro H, Suzuki M, Shinozaki-Ushiku A, Kobayashi T, Sanada T, Kim M, Ogawa M, Iwasaki YW, Kayo H, Fukuda-Yuzawa Y, Yashiro M, Fukayama M, Fukao T, Sasakawa C (2014) Epigenetic silencing of miR-210 increases the proliferation of gastric epithelium during chronic Helicobacter pylori infection. Nat Commun 5: 4497.
Kim TW, Lee SJ, Oh BM, Lee H, Uhm TG, Min JK, Park YJ, Yoon SR, Kim BY, Kim JW, Choe YK, Lee HG (2016) Epigenetic modification of TLR4 promotes activation of NF-kappaB by regulating methyl-CpG-binding domain protein 2 and Sp1 in gastric cancer. Oncotarget 7: 4195–4209.
Kirchner T, Muller S, Hattori T, Mukaisyo K, Papadopoulos T, Brabletz T, Jung A (2001) Metaplasia, intraepithelial neoplasia and early cancer of the stomach are related to dedifferentiated epithelial cells defined by cytokeratin-7 expression in gastritis. Virchows Arch 439: 512–522.
Klutstein M, Nejman D, Greenfield R, Cedar H (2016) DNA methylation in cancer and aging. Cancer Res 76: 3446–3450.
Kreso A, Dick JE (2014) Evolution of the cancer stem cell model. Cell Stem Cell 14: 275–291.
Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, Butowski N, Groves MD, Kesari S, Freedman SJ, Blackman S, Watters J, Loboda A, Podtelezhnikov A, Lunceford J, Chen C, Giannotti M, Hing J, Beckman R, Lorusso P (2012) Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol 30: 2307–2313.
Kurashige J, Mima K, Sawada G, Takahashi Y, Eguchi H, Sugimachi K, Mori M, Yanagihara K, Yashiro M, Hirakawa K, Baba H, Mimori K (2015) Epigenetic modulation and repression of miR-200b by cancer-associated fibroblasts contribute to cancer invasion and peritoneal dissemination in gastric cancer. Carcinogenesis 36: 133–141.
Lau WM, Teng E, Chong HS, Lopez KA, Tay AY, Salto-Tellez M, Shabbir A, So JB, Chan SL (2014) CD44v8-10 is a cancer-specific marker for gastric cancer stem cells. Cancer Res 74: 2630–2641.
Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang CY, Yaswen P, Goga A, Werb Z (2015) Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 526: 131–135.
Li J, Woods SL, Healey S, Beesley J, Chen X, Lee JS, Sivakumaran H, Wayte N, Nones K, Waterfall JJ, Pearson J, Patch AM, Senz J, Ferreira MA, Kaurah P, Mackenzie R, Heravi-Moussavi A, Hansford S, Lannagan TR, Spurdle AB, Simpson PT, da Silva L, Lakhani SR, Clouston AD, Bettington M, Grimpen F, Busuttil RA, Di Costanzo N, Boussioutas A, Jeanjean M, Chong G, Fabre A, Olschwang S, Faulkner GJ, Bellos E, Coin L, Rioux K, Bathe OF, Wen X, Martin HC, Neklason DW, Davis SR, Walker RL, Calzone KA, Avital I, Heller T, Koh C, Pineda M, Rudloff U, Quezado M, Pichurin PN, Hulick PJ, Weissman SM, Newlin A, Rubinstein WS, Sampson JE, Hamman K, Goldgar D, Poplawski N, Phillips K, Schofield L, Armstrong J, Kiraly-Borri C, Suthers GK, Huntsman DG, Foulkes WD, Carneiro F, Lindor NM, Edwards SL, French JD, Waddell N, Meltzer PS, Worthley DL, Schrader KA, Chenevix-Trench G (2016a) Point mutations in exon 1B of APC reveal gastric adenocarcinoma and proximal polyposis of the stomach as a familial adenomatous polyposis variant. Am J Hum Genet 98: 830–842.
Li P, Shan JX, Chen XH, Zhang D, Su LP, Huang XY, Yu BQ, Zhi QM, Li CL, Wang YQ, Tomei S, Cai Q, Ji J, Li JF, Chouchane L, Yu YY, Sun FZ, Xu ZH, Liu BY, Zhu ZG (2015) Epigenetic silencing of microRNA-149 in cancer-associated fibroblasts mediates prostaglandin E2/interleukin-6 signaling in the tumor microenvironment. Cell Res 25: 588–603.
Li Q, Jia Z, Wang L, Kong X, Li Q, Guo K, Tan D, Le X, Wei D, Huang S, Mishra L, Xie K (2012) Disruption of Klf4 in villin-positive gastric progenitor cells promotes formation and progression of tumors of the antrum in mice. Gastroenterology 142: 531–542.
Li X, Wu WK, Xing R, Wong SH, Liu Y, Fang X, Zhang Y, Wang M, Wang J, Li L, Zhou Y, Tang S, Peng S, Qiu K, Chen L, Chen K, Yang H, Zhang W, Chan MT, Lu Y, Sung JJ, Yu J (2016b) Distinct subtypes of gastric cancer defined by molecular characterization include novel mutational signatures with prognostic capability. Cancer Res 76: 1724–1732.
Liang Q, Yao X, Tang S, Zhang J, Yau TO, Li X, Tang CM, Kang W, Lung RW, Li JW, Chan TF, Xing R, Lu Y, Lo KW, Wong N, To KF, Yu C, Chan FK, Sung JJ, Yu J (2014) Integrative identification of Epstein-Barr virus-associated mutations and epigenetic alterations in gastric cancer. Gastroenterology 147: 1350–1362.e4.
Lim B, Kim C, Kim JH, Kwon WS, Lee WS, Kim JM, Park JY, Kim HS, Park KH, Kim TS, Park JL, Chung HC, Rha SY, Kim SY (2016a) Genetic alterations and their clinical implications in gastric cancer peritoneal carcinomatosis revealed by whole-exome sequencing of malignant ascites. Oncotarget 7: 8055–8066.
Lim CH, Cho YK, Kim SW, Choi MG, Rhee JK, Chung YJ, Lee SH, Kim TM (2016b) The chronological sequence of somatic mutations in early gastric carcinogenesis inferred from multiregion sequencing of gastric adenomas. Oncotarget 7: 39758–39767.
Lin M, Shi C, Lin X, Pan J, Shen S, Xu Z, Chen Q (2016) sMicroRNA-1290 inhibits cells proliferation and migration by targeting FOXA1 in gastric cancer cells. Gene 582: 137–142.
Liu H, Li F, Zhu Y, Li T, Huang H, Lin T, Hu Y, Qi X, Yu J, Li G (2016) Whole-exome sequencing to identify somatic mutations in peritoneal metastatic gastric adenocarcinoma: a preliminary study. Oncotarget 7: 43894–43906.
Liu J, Ma L, Xu J, Liu C, Zhang J, Liu J, Chen R, Zhou Y (2013a) Co-expression of CD44 and ABCG2 in spheroid body-forming cells of gastric cancer cell line MKN45. Hepatogastroenterology 60: 975–980.
Liu J, Ma L, Xu J, Liu C, Zhang J, Liu J, Chen R, Zhou Y (2013b) Spheroid body-forming cells in the human gastric cancer cell line MKN-45 possess cancer stem cell properties. Int J Oncol 42: 453–459.
Lu H, Samanta D, Xiang L, Zhang H, Hu H, Chen I, Bullen JW, Semenza GL (2015) Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc Natl Acad Sci USA 112: E4600–E4609.
Lu MH, Tang B, Zeng S, Hu CJ, Xie R, Wu YY, Wang SM, He FT, Yang SM (2016) Long noncoding RNA BC032469, a novel competing endogenous RNA, upregulates hTERT expression by sponging miR-1207-5p and promotes proliferation in gastric cancer. Oncogene 35: 3524–3534.
Magee JA, Piskounova E, Morrison SJ (2012) Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell 21: 283–296.
Mao J, Fan S, Ma W, Fan P, Wang B, Zhang J, Wang H, Tang B, Zhang Q, Yu X, Wang L, Song B, Li L (2014) Roles of Wnt/beta-catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis 5: e1039.
McDonald SA, Greaves LC, Gutierrez-Gonzalez L, Rodriguez-Justo M, Deheragoda M, Leedham SJ, Taylor RW, Lee CY, Preston SL, Lovell M, Hunt T, Elia G, Oukrif D, Harrison R, Novelli MR, Mitchell I, Stoker DL, Turnbull DM, Jankowski JA, Wright NA (2008) Mechanisms of field cancerization in the human stomach: the expansion and spread of mutated gastric stem cells. Gastroenterology 134: 500–510.
McMillan R, Matsui W (2012) Molecular pathways: the hedgehog signaling pathway in cancer. Clin Cancer Res 18: 4883–4888.
Meacham CE, Morrison SJ (2013) Tumour heterogeneity and cancer cell plasticity. Nature 501: 328–337.
Medema JP, Vermeulen L (2011) Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature 474: 318–326.
Messersmith WA, Shapiro GI, Cleary JM, Jimeno A, Dasari A, Huang B, Shaik MN, Cesari R, Zheng X, Reynolds JM, English PA, McLachlan KR, Kern KA, LoRusso PM (2015) A Phase I, dose-finding study in patients with advanced solid malignancies of the oral gamma-secretase inhibitor PF-03084014. Clin Cancer Res 21: 60–67.
Miao ZF, Wang ZN, Zhao TT, Xu YY, Gao J, Miao F, Xu HM (2014) Peritoneal milky spots serve as a hypoxic niche and favor gastric cancer stem/progenitor cell peritoneal dissemination through hypoxia-inducible factor 1alpha. Stem Cells 32: 3062–3074.
Migden MR, Guminski A, Gutzmer R, Dirix L, Lewis KD, Combemale P, Herd RM, Kudchadkar R, Trefzer U, Gogov S, Pallaud C, Yi T, Mone M, Kaatz M, Loquai C, Stratigos AJ, Schulze HJ, Plummer R, Chang AL, Cornelis F, Lear JT, Sellami D, Dummer R (2015) Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): a multicentre, randomised, double-blind phase 2 trial. Lancet Oncol 16: 716–728.
Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, Desai R, Fox DB, Brannigan BW, Trautwein J, Arora KS, Desai N, Dahl DM, Sequist LV, Smith MR, Kapur R, Wu CL, Shioda T, Ramaswamy S, Ting DT, Toner M, Maheswaran S, Haber DA (2015) RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 349: 1351–1356.
Mosolits S, Markovic K, Frodin JE, Virving L, Magnusson CG, Steinitz M, Fagerberg J, Mellstedt H (2004) Vaccination with Ep-CAM protein or anti-idiotypic antibody induces Th1-biased response against MHC class I- and II-restricted Ep-CAM epitopes in colorectal carcinoma patients. Clin Cancer Res 10: 5391–5402.
Munz M, Baeuerle PA, Gires O (2009) The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res 69: 5627–5629.
Muro K, Chung HC, Shankaran V, Geva R, Catenacci D, Gupta S, Eder JP, Golan T, Le DT, Burtness B, McRee AJ, Lin CC, Pathiraja K, Lunceford J, Emancipator K, Juco J, Koshiji M, Bang YJ (2016) Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol 17: 717–726.
Murray-Stewart T, Sierra JC, Piazuelo MB, Mera RM, Chaturvedi R, Bravo LE, Correa P, Schneider BG, Wilson KT, Casero RA (2016) Epigenetic silencing of miR-124 prevents spermine oxidase regulation: implications for Helicobacter pylori-induced gastric cancer. Oncogene 35: 5480–5488.
Nagarajan N, Bertrand D, Hillmer AM, Zang ZJ, Yao F, Jacques PE, Teo AS, Cutcutache I, Zhang Z, Lee WH, Sia YY, Gao S, Ariyaratne PN, Ho A, Woo XY, Veeravali L, Ong CK, Deng N, Desai KV, Khor CC, Hibberd ML, Shahab A, Rao J, Wu M, Teh M, Zhu F, Chin SY, Pang B, So JB, Bourque G, Soong R, Sung WK, Tean Teh B, Rozen S, Ruan X, Yeoh KG, Tan PB, Ruan Y (2012) Whole-genome reconstruction and mutational signatures in gastric cancer. Genome Biol 13: R115.
Nam KT, Lee HJ, Sousa JF, Weis VG, O'Neal RL, Finke PE, Romero-Gallo J, Shi G, Mills JC, Peek RM Jr, Konieczny SF, Goldenring JR (2010) Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 139: 2028–2037.e9.
Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M (2011) Tumour evolution inferred by single-cell sequencing. Nature 472: 90–94.
Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, Menzies A, Martin S, Leung K, Chen L, Leroy C, Ramakrishna M, Rance R, Lau KW, Mudie LJ, Varela I, McBride DJ, Bignell GR, Cooke SL, Shlien A, Gamble J, Whitmore I, Maddison M, Tarpey PS, Davies HR, Papaemmanuil E, Stephens PJ, McLaren S, Butler AP, Teague JW, Jonsson G, Garber JE, Silver D, Miron P, Fatima A, Boyault S, Langerod A, Tutt A, Martens JW, Aparicio SA, Borg A, Salomon AV, Thomas G, Borresen-Dale AL, Richardson AL, Neuberger MS, Futreal PA, Campbell PJ, Stratton MR (2012) Mutational processes molding the genomes of 21 breast cancers. Cell 149: 979–993.
Nishii T, Yashiro M, Shinto O, Sawada T, Ohira M, Hirakawa K (2009) Cancer stem cell-like SP cells have a high adhesion ability to the peritoneum in gastric carcinoma. Cancer Sci 100: 1397–1402.
Nishikawa S, Konno M, Hamabe A, Hasegawa S, Kano Y, Fukusumi T, Satoh T, Takiguchi S, Mori M, Doki Y, Ishii H (2015) Surgically resected human tumors reveal the biological significance of the gastric cancer stem cell markers CD44 and CD26. Oncol Lett 9: 2361–2367.
Nishikawa S, Konno M, Hamabe A, Hasegawa S, Kano Y, Ohta K, Fukusumi T, Sakai D, Kudo T, Haraguchi N, Satoh T, Takiguchi S, Mori M, Doki Y, Ishii H (2013) Aldehyde dehydrogenase high gastric cancer stem cells are resistant to chemotherapy. Int J Oncol 42: 1437–1442.
Notta F, Mullighan CG, Wang JC, Poeppl A, Doulatov S, Phillips LA, Ma J, Minden MD, Downing JR, Dick JE (2011) Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 469: 362–367.
Ohkuma M, Haraguchi N, Ishii H, Mimori K, Tanaka F, Kim HM, Shimomura M, Hirose H, Yanaga K, Mori M (2012) Absence of CD71 transferrin receptor characterizes human gastric adenosquamous carcinoma stem cells. Ann Surg Oncol 19: 1357–1364.
Ohtsu A, Ajani JA, Bai YX, Bang YJ, Chung HC, Pan HM, Sahmoud T, Shen L, Yeh KH, Chin K, Muro K, Kim YH, Ferry D, Tebbutt NC, Al-Batran SE, Smith H, Costantini C, Rizvi S, Lebwohl D, Van Cutsem E (2013) Everolimus for previously treated advanced gastric cancer: results of the randomized, double-blind, phase III GRANITE-1 study. J Clin Oncol 31: 3935–3943.
Okamoto R, Yajima T, Yamazaki M, Kanai T, Mukai M, Okamoto S, Ikeda Y, Hibi T, Inazawa J, Watanabe M (2002) Damaged epithelia regenerated by bone marrow-derived cells in the human gastrointestinal tract. Nat Med 8: 1011–1017.
Okugawa Y, Toiyama Y, Hur K, Toden S, Saigusa S, Tanaka K, Inoue Y, Mohri Y, Kusunoki M, Boland CR, Goel A (2014) Metastasis-associated long non-coding RNA drives gastric cancer development and promotes peritoneal metastasis. Carcinogenesis 35: 2731–2739.
Oshima H, Ishikawa T, Yoshida GJ, Naoi K, Maeda Y, Naka K, Ju X, Yamada Y, Minamoto T, Mukaida N, Saya H, Oshima M (2014) TNF-alpha/TNFR1 signaling promotes gastric tumorigenesis through induction of Noxo1 and Gna14 in tumor cells. Oncogene 33: 3820–3829.
Oue N, Mukai S, Imai T, Pham TT, Oshima T, Sentani K, Sakamoto N, Yoshida K, Yasui W (2016) Induction of KIFC1 expression in gastric cancer spheroids. Oncol Rep 36: 349–355.
Pant S, Patel MR, Kurkjian C, Hemphill MB, Flores MRC, Thompson DS, Bendell JC (2015) A phase II study of the c-Met inhibitor tivantinib (tiv) in combination with FOLFOX for the treatment of patients (pts) with previously untreated metastatic adenocarcinoma of the distal esophagus, gastroesophageal (GE) junction, or stomach. J Clin Oncol 33 (15 suppl): 4065.
Park JL, Kim HJ, Seo EH, Kwon OH, Lim B, Kim M, Kim SY, Song KS, Kang GH, Kim HJ, Choi BY, Kim YS (2015) Decrease of 5hmC in gastric cancers is associated with TET1 silencing due to with DNA methylation and bivalent histone marks at TET1 CpG island 3'-shore. Oncotarget 6: 37647–37662.
Park JW, Park JM, Park DM, Kim DY, Kim HK (2016) Stem cells antigen-1 enriches for a cancer stem cell-like subpopulation in mouse gastric cancer. Stem Cells 34: 1177–1187.
Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG (2005) Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res 65: 6207–6219.
Peterson AJ, Menheniott TR, O'Connor L, Walduck AK, Fox JG, Kawakami K, Minamoto T, Ong EK, Wang TC, Judd LM, Giraud AS (2010) Helicobacter pylori infection promotes methylation and silencing of trefoil factor 2, leading to gastric tumor development in mice and humans. Gastroenterology 139: 2005–2017.
Piha-Paul SA, Munster PN, Hollebecque A, Argiles G, Dajani O, Cheng JD, Wang R, Swift A, Tosolini A, Gupta S (2015) Results of a phase 1 trial combining ridaforolimus and MK-0752 in patients with advanced solid tumours. Eur J Cancer 51: 1865–1873.
Plaks V, Kong N, Werb Z (2015) The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 16: 225–238.
Qiao XT, Gumucio DL (2011) Current molecular markers for gastric progenitor cells and gastric cancer stem cells. J Gastroenterol 46: 855–865.
Qiao XT, Ziel JW, McKimpson W, Madison BB, Todisco A, Merchant JL, Samuelson LC, Gumucio DL (2007) Prospective identification of a multilineage progenitor in murine stomach epithelium. Gastroenterology 133: 1989–1998.
Quante M, Marrache F, Goldenring JR, Wang TC (2010) TFF2 mRNA transcript expression marks a gland progenitor cell of the gastric oxyntic mucosa. Gastroenterology 139: 2018–2027.e2.
Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, Friedman R, Varro A, Tycko B, Wang TC (2011) Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 19: 257–272.
Raggi C, Mousa HS, Correnti M, Sica A, Invernizzi P (2016) Cancer stem cells and tumor-associated macrophages: a roadmap for multitargeting strategies. Oncogene 35: 671–682.
Riechelmann H, Sauter A, Golze W, Hanft G, Schroen C, Hoermann K, Erhardt T, Gronau S (2008) Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma. Oral Oncol 44: 823–829.
Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I, Packer RJ, Goldman S, Prados MD, Desjardins A, Chintagumpala M, Takebe N, Kaste SC, Rusch M, Allen SJ, Onar-Thomas A, Stewart CF, Fouladi M, Boyett JM, Gilbertson RJ, Curran T, Ellison DW, Gajjar A (2015) Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase II Pediatric Brain Tumor Consortium studies PBTC-025B and PBTC-032. J Clin Oncol 33: 2646–2654.
Rodon J, Tawbi HA, Thomas AL, Stoller RG, Turtschi CP, Baselga J, Sarantopoulos J, Mahalingam D, Shou Y, Moles MA, Yang L, Granvil C, Hurh E, Rose KL, Amakye DD, Dummer R, Mita AC (2014) A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin Cancer Res 20: 1900–1909.
Rupp U, Schoendorf-Holland E, Eichbaum M, Schuetz F, Lauschner I, Schmidt P, Staab A, Hanft G, Huober J, Sinn HP, Sohn C, Schneeweiss A (2007) Safety and pharmacokinetics of bivatuzumab mertansine in patients with CD44v6-positive metastatic breast cancer: final results of a phase I study. Anticancer Drugs 18: 477–485.
Sauter A, Kloft C, Gronau S, Bogeschdorfer F, Erhardt T, Golze W, Schroen C, Staab A, Riechelmann H, Hoermann K (2007) Pharmacokinetics, immunogenicity and safety of bivatuzumab mertansine, a novel CD44v6-targeting immunoconjugate, in patients with squamous cell carcinoma of the head and neck. Int J Oncol 30: 927–935.
Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, Clevers H (2012) Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 337: 730–735.
Schmidt M, Ruttinger D, Sebastian M, Hanusch CA, Marschner N, Baeuerle PA, Wolf A, Goppel G, Oruzio D, Schlimok G, Steger GG, Wolf C, Eiermann W, Lang A, Schuler M (2012) Phase IB study of the EpCAM antibody adecatumumab combined with docetaxel in patients with EpCAM-positive relapsed or refractory advanced-stage breast cancer. Ann Oncol 23: 2306–2313.
Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, Rupec RA, Gerhard M, Schmid R, Barker N, Clevers H, Lang R, Neumann J, Kirchner T, Taketo MM, van den Brink GR, Sansom OJ, Arkan MC, Greten FR (2013) Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152: 25–38.
Shah MA, Bang YJ, Lordick F, Tabernero J, Chen M, Hack SP, Phan SC, Shames DS, Cunningham D (2015) METGastric: a phase III study of onartuzumab plus mFOLFOX6 in patients with metastatic HER2-negative (HER2−) and MET-positive (MET+) adenocarcinoma of the stomach or gastroesophageal junction (GEC). J Clin Oncol 33 (15 suppl): 4012.
Shah MA, Cho JY, Tan IB, Tebbutt NC, Yen CJ, Kang A, Shames DS, Bu L, Kang YK (2016) A randomized phase II study of FOLFOX with or without the MET inhibitor onartuzumab in advanced adenocarcinoma of the stomach and gastroesophageal junction. Oncologist 21: 1085–1090.
Shang Y, Zhang Z, Liu Z, Feng B, Ren G, Li K, Zhou L, Sun Y, Li M, Zhou J, An Y, Wu K, Nie Y, Fan D (2014) miR-508-5p regulates multidrug resistance of gastric cancer by targeting ABCB1 and ZNRD1. Oncogene 33: 3267–3276.
She JJ, Zhang PG, Wang X, Che XM, Wang ZM (2012) Side population cells isolated from KATO III human gastric cancer cell line have cancer stem cell-like characteristics. World J Gastroenterol 18: 4610–4617.
Shen J, Xiao Z, Wu WK, Wang MH, To KF, Chen Y, Yang W, Li MS, Shin VY, Tong JH, Kang W, Zhang L, Li M, Wang L, Lu L, Chan RL, Wong SH, Yu J, Chan MT, Chan FK, Sung JJ, Cheng AS, Cho CH (2015) Epigenetic silencing of miR-490-3p reactivates the chromatin remodeler SMARCD1 to promote Helicobacter pylori-induced gastric carcinogenesis. Cancer Res 75: 754–765.
Shibata W, Ariyama H, Westphalen CB, Worthley DL, Muthupalani S, Asfaha S, Dubeykovskaya Z, Quante M, Fox JG, Wang TC (2013) Stromal cell-derived factor-1 overexpression induces gastric dysplasia through expansion of stromal myofibroblasts and epithelial progenitors. Gut 62: 192–200.
Shimizu T, Marusawa H, Matsumoto Y, Inuzuka T, Ikeda A, Fujii Y, Minamiguchi S, Miyamoto S, Kou T, Sakai Y, Crabtree JE, Chiba T (2014) Accumulation of somatic mutations in TP53 in gastric epithelium with Helicobacter pylori infection. Gastroenterology 147: 407–17.e3.
Shitara K, Doi T, Nagano O, Imamura CK, Ozeki T, Ishii Y, Tsuchihashi K, Takahashi S, Nakajima TE, Hironaka S, Fukutani M, Hasegawa H, Nomura S, Sato A, Einaga Y, Kuwata T, Saya H, Ohtsu A (2016) Dose-escalation study for the targeting of CD44v+ cancer stem cells by sulfasalazine in patients with advanced gastric cancer (EPOC1205). Gastric Cancer 20: 341–349.
Sigal M, Rothenberg ME, Logan CY, Lee JY, Honaker RW, Cooper RL, Passarelli B, Camorlinga M, Bouley DM, Alvarez G, Nusse R, Torres J, Amieva MR (2015) Helicobacter pylori activates and expands Lgr5(+) stem cells through direct colonization of the gastric glands. Gastroenterology 148: 1392–404.e21.
Song Z, Yue W, Wei B, Wang N, Li T, Guan L, Shi S, Zeng Q, Pei X, Chen L (2011) Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PLoS One 6: e17687.
Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK (2009) Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459: 428–432.
Stange DE, Koo BK, Huch M, Sibbel G, Basak O, Lyubimova A, Kujala P, Bartfeld S, Koster J, Geahlen JH, Peters PJ, van Es JH, van de Wetering M, Mills JC, Clevers H (2013) Differentiated Troy+ chief cells act as reserve stem cells to generate all lineages of the stomach epithelium. Cell 155: 357–368.
Sun B, Zhang D, Zhao N, Zhao X (2016) Epithelial-to-endothelial transition and cancer stem cells: two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget e-pub ahead of print 29 March 2016; doi:10.18632/oncotarget.8461.
Sun W, Powell M, O'Dwyer PJ, Catalano P, Ansari RH, Benson 3rd AB (2010) Phase II study of sorafenib in combination with docetaxel and cisplatin in the treatment of metastatic or advanced gastric and gastroesophageal junction adenocarcinoma: ECOG 5203. J Clin Oncol 28: 2947–2951.
Swaminathan SK, Olin MR, Forster CL, Cruz KS, Panyam J, Ohlfest JR (2010) Identification of a novel monoclonal antibody recognizing CD133. J Immunol Methods 361: 110–115.
Takahashi-Yanaga F, Kahn M (2010) Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res 16: 3153–3162.
Takaishi S, Okumura T, Tu S, Wang SS, Shibata W, Vigneshwaran R, Gordon SA, Shimada Y, Wang TC (2009) Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 27: 1006–1020.
Takebe N, Nguyen D, Yang SX (2014) Targeting notch signaling pathway in cancer: clinical development advances and challenges. Pharmacol Ther 141: 140–149.
Thompson ED, Zahurak M, Murphy A, Cornish T, Cuka N, Abdelfatah E, Yang S, Duncan M, Ahuja N, Taube JM, Anders RA, Kelly RJ (2016) Patterns of PD-L1 expression and CD8 T cell infiltration in gastric adenocarcinomas and associated immune stroma. Gut e-pub ahead of print 22 January 2016; doi:10.1136/gutjnl-2015-310839.
Tijink BM, Buter J, de Bree R, Giaccone G, Lang MS, Staab A, Leemans CR, van Dongen GA (2006) A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin Cancer Res 12: 6064–6072.
Tomita H, Takaishi S, Menheniott TR, Yang X, Shibata W, Jin G, Betz KS, Kawakami K, Minamoto T, Tomasetto C, Rio MC, Lerkowit N, Varro A, Giraud AS, Wang TC (2011) Inhibition of gastric carcinogenesis by the hormone gastrin is mediated by suppression of TFF1 epigenetic silencing. Gastroenterology 140: 879–891.
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A (2015) Global cancer statistics, 2012. CA Cancer J Clin 65: 87–108.
Tseng YC, Tsai YH, Tseng MJ, Hsu KW, Yang MC, Huang KH, Li AF, Chi CW, Hsieh RH, Ku HH, Yeh TS (2012) Notch2-induced COX-2 expression enhancing gastric cancer progression. Mol Carcinog 51: 939–951.
Tsujimoto H, Hagiwara A, Shimotsuma M, Sakakura C, Osaki K, Sasaki S, Ohyama T, Ohgaki M, Imanishi T, Yamazaki J, Takahashi T (1996) Role of milky spots as selective implantation sites for malignant cells in peritoneal dissemination in mice. J Cancer Res Clin Oncol 122: 590–595.
Tye H, Kennedy CL, Najdovska M, McLeod L, McCormack W, Hughes N, Dev A, Sievert W, Ooi CH, Ishikawa TO, Oshima H, Bhathal PS, Parker AE, Oshima M, Tan P, Jenkins BJ (2012) STAT3-driven upregulation of TLR2 promotes gastric tumorigenesis independent of tumor inflammation. Cancer Cell 22: 466–478.
Ushijima T, Hattori N (2012) Molecular pathways: involvement of Helicobacter pylori-triggered inflammation in the formation of an epigenetic field defect, and its usefulness as cancer risk and exposure markers. Clin Cancer Res 18: 923–929.
Varon C, Dubus P, Mazurier F, Asencio C, Chambonnier L, Ferrand J, Giese A, Senant-Dugot N, Carlotti M, Megraud F (2012) Helicobacter pylori infection recruits bone marrow-derived cells that participate in gastric preneoplasia in mice. Gastroenterology 142: 281–291.
Vey N, Delaunay J, Martinelli G, Fiedler W, Raffoux E, Prebet T, Gomez-Roca C, Papayannidis C, Kebenko M, Paschka P, Christen R, Guarin E, Broske AM, Baehner M, Brewster M, Walz AC, Michielin F, Runza V, Meresse V, Recher C (2016) Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget 7: 32532–32542.
Voog J, Jones DL (2010) Stem cells and the niche: a dynamic duo. Cell Stem Cell 6: 103–115.
Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, Simeone DM, Zou W, Welling TH (2014) Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 147: 1393–1404.
Wang B, Liu J, Ma LN, Xiao HL, Wang YZ, Li Y, Wang Z, Fan L, Lan C, Yang M, Hu L, Wei Y, Bian XW, Chen D, Wang J (2013) Chimeric 5/35 adenovirus-mediated Dickkopf-1 overexpression suppressed tumorigenicity of CD44(+) gastric cancer cells via attenuating Wnt signaling. J Gastroenterol 48: 798–808.
Wang J, Guo LP, Chen LZ, Zeng YX, Lu SH (2007) Identification of cancer stem cell-like side population cells in human nasopharyngeal carcinoma cell line. Cancer Res 67: 3716–3724.
Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, Chan TL, Kan Z, Chan AS, Tsui WY, Lee SP, Ho SL, Chan AK, Cheng GH, Roberts PC, Rejto PA, Gibson NW, Pocalyko DJ, Mao M, Xu J, Leung SY (2011) Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet 43: 1219–1223.
Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, Siu HC, Deng S, Chu KM, Law S, Chan KH, Chan AS, Tsui WY, Ho SL, Chan AK, Man JL, Foglizzo V, Ng MK, Chan AS, Ching YP, Cheng GH, Xie T, Fernandez J, Li VS, Clevers H, Rejto PA, Mao M, Leung SY (2014) Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet 46: 573–582.
Wang Z, Liu C (2015) Lgr5-positive cells are cancer-stem-cell-like cells in gastric cancer. Cell Physiol Biochem 36: 2447–2455.
Wei B, Sun X, Geng Z, Shi M, Chen Z, Chen L, Wang Y, Fu X (2016) Isoproterenol regulates CD44 expression in gastric cancer cells through STAT3/MicroRNA373 cascade. Biomaterials 105: 89–101.
Wong SS, Kim KM, Ting JC, Yu K, Fu J, Liu S, Cristescu R, Nebozhyn M, Gong L, Yue YG, Wang J, Ronghua C, Loboda A, Hardwick J, Liu X, Dai H, Jin JG, Ye XS, Kang SY, Do IG, Park JO, Sohn TS, Reinhard C, Lee J, Kim S, Aggarwal A (2014) Genomic landscape and genetic heterogeneity in gastric adenocarcinoma revealed by whole-genome sequencing. Nat Commun 5: 5477.
Worthley DL, Ruszkiewicz A, Davies R, Moore S, Nivison-Smith I, Bik To L, Browett P, Western R, Durrant S, So J, Young GP, Mullighan CG, Bardy PG, Michael MZ (2009) Human gastrointestinal neoplasia-associated myofibroblasts can develop from bone marrow-derived cells following allogeneic stem cell transplantation. Stem Cells 27: 1463–1468.
Wu D, Mou YP, Chen K, Cai JQ, Zhou YC, Pan Y, Xu XW, Zhou W, Gao JQ, Chen DW, Zhang RC (2016a) Aldehyde dehydrogenase 3A1 is robustly upregulated in gastric cancer stem-like cells and associated with tumorigenesis. Int J Oncol 49: 611–622.
Wu K, Ma L, Zhu J (2016b) miR4835p promotes growth, invasion and selfrenewal of gastric cancer stem cells by Wnt/betacatenin signaling. Mol Med Rep 14: 3421–3428.
Wu Q, Yang Z, Wang F, Hu S, Yang L, Shi Y, Fan D (2013) MiR-19b/20a/92a regulates the self-renewal and proliferation of gastric cancer stem cells. J Cell Sci 126: 4220–4229.
Xu M, Gong A, Yang H, George SK, Jiao Z, Huang H, Jiang X, Zhang Y (2015a) Sonic hedgehog-glioma associated oncogene homolog 1 signaling enhances drug resistance in CD44(+)/Musashi-1(+) gastric cancer stem cells. Cancer Lett 369: 124–133.
Xu ZY, Tang JN, Xie HX, Du YA, Huang L, Yu PF, Cheng XD (2015b) 5-Fluorouracil chemotherapy of gastric cancer generates residual cells with properties of cancer stem cells. Int J Biol Sci 11: 284–294.
Xue Z, Yan H, Li J, Liang S, Cai X, Chen X, Wu Q, Gao L, Wu K, Nie Y, Fan D (2012) Identification of cancer stem cells in vincristine preconditioned SGC7901 gastric cancer cell line. J Cell Biochem 113: 302–312.
Yang C, Gu L, Deng D (2013) Bone marrow-derived cells may not be the original cells for carcinogen-induced mouse gastrointestinal carcinomas. PLoS One 8: e79615.
Yang W, Mok MT, Li MS, Kang W, Wang H, Chan AW, Chou JL, Chen J, Ng EK, To KF, Yu J, Chan MW, Chan FK, Sung JJ, Cheng AS (2016) Epigenetic silencing of GDF1 disrupts SMAD signaling to reinforce gastric cancer development. Oncogene 35: 2133–2144.
Yashiro M, Nishii T, Hasegawa T, Matsuzaki T, Morisaki T, Fukuoka T, Hirakawa K (2013) A c-Met inhibitor increases the chemosensitivity of cancer stem cells to the irinotecan in gastric carcinoma. Br J Cancer 109: 2619–2628.
Yeh TS, Wu CW, Hsu KW, Liao WJ, Yang MC, Li AF, Wang AM, Kuo ML, Chi CW (2009) The activated Notch1 signal pathway is associated with gastric cancer progression through cyclooxygenase-2. Cancer Res 69: 5039–5048.
Yong X, Tang B, Xiao YF, Xie R, Qin Y, Luo G, Hu CJ, Dong H, Yang SM (2016) Helicobacter pylori upregulates Nanog and Oct4 via Wnt/beta-catenin signaling pathway to promote cancer stem cell-like properties in human gastric cancer. Cancer Lett 374: 292–303.
Yoo YA, Kang MH, Kim JS, Oh SC (2008) Sonic hedgehog signaling promotes motility and invasiveness of gastric cancer cells through TGF-beta-mediated activation of the ALK5-Smad 3 pathway. Carcinogenesis 29: 480–490.
Yoon C, Park do J, Schmidt B, Thomas NJ, Lee HJ, Kim TS, Janjigian YY, Cohen DJ, Yoon SS (2014) CD44 expression denotes a subpopulation of gastric cancer cells in which Hedgehog signaling promotes chemotherapy resistance. Clin Cancer Res 20: 3974–3988.
Yoshida K, Tsujimoto H, Matsumura K, Kinoshita M, Takahata R, Matsumoto Y, Hiraki S, Ono S, Seki S, Yamamoto J, Hase K (2015) CD47 is an adverse prognostic factor and a therapeutic target in gastric cancer. Cancer Med 4: 1322–1333.
Yoshida S, Yamashita S, Niwa T, Mori A, Ito S, Ichinose M, Ushijima T (2017) Epigenetic inactivation of FAT4 contributes to gastric field cancerization. Gastric Cancer 20: 136–145.
Yu D, Shin HS, Lee YS, Lee YC (2014) miR-106b modulates cancer stem cell characteristics through TGF-beta/Smad signaling in CD44-positive gastric cancer cells. Lab Invest 94: 1370–1381.
Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, Ciciliano JC, Smas ME, Winokur D, Gilman AJ, Ulman MJ, Xega K, Contino G, Alagesan B, Brannigan BW, Milos PM, Ryan DP, Sequist LV, Bardeesy N, Ramaswamy S, Toner M, Maheswaran S, Haber DA (2012) RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature 487: 510–513.
Yun SM, Yoon K, Lee S, Kim E, Kong SH, Choe J, Kang JM, Han TS, Kim P, Choi Y, Jho S, Yoo H, Bhak J, Yang HK, Kim SJ (2014) PPP1R1B-STARD3 chimeric fusion transcript in human gastric cancer promotes tumorigenesis through activation of PI3K/AKT signaling. Oncogene 33: 5341–5347.
Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A, Lim KH, Ong CK, Huang D, Chin SY, Tan IB, Ng CC, Yu W, Wu Y, Lee M, Wu J, Poh D, Wan WK, Rha SY, So J, Salto-Tellez M, Yeoh KG, Wong WK, Zhu YJ, Futreal PA, Pang B, Ruan Y, Hillmer AM, Bertrand D, Nagarajan N, Rozen S, Teh BT, Tan P (2012) Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet 44: 570–574.
Zhan YY, He JP, Chen HZ, Wang WJ, Cai JC (2013) Orphan receptor TR3 is essential for the maintenance of stem-like properties in gastric cancer cells. Cancer Lett 329: 37–44.
Zhang C, Li C, He F, Cai Y, Yang H (2011) Identification of CD44+CD24+ gastric cancer stem cells. J Cancer Res Clin Oncol 137: 1679–1686.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X, Semenza GL (2016a) Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA 113: E2047–E2056.
Zhang H, Xi H, Cai A, Xia Q, Wang XX, Lu C, Zhang Y, Song Z, Wang H, Li Q, Chen L, Guo Z (2013) Not all side population cells contain cancer stem-like cells in human gastric cancer cell lines. Dig Dis Sci 58: 132–139.
Zhang L, Xu Z, Xu X, Zhang B, Wu H, Wang M, Zhang X, Yang T, Cai J, Yan Y, Mao F, Zhu W, Shao Q, Qian H, Xu W (2014) SALL4, a novel marker for human gastric carcinogenesis and metastasis. Oncogene 33: 5491–5500.
Zhang X, Hua R, Wang X, Huang M, Gan L, Wu Z, Zhang J, Wang H, Cheng Y, Li J, Guo W (2016b) Identification of stem-like cells and clinical significance of candidate stem cell markers in gastric cancer. Oncotarget 7: 9815–9831.
Zheng B, Liang L, Wang C, Huang S, Cao X, Zha R, Liu L, Jia D, Tian Q, Wu J, Ye Y, Wang Q, Long Z, Zhou Y, Du C, He X, Shi Y (2011) MicroRNA-148a suppresses tumor cell invasion and metastasis by downregulating ROCK1 in gastric cancer. Clin Cancer Res 17: 7574–7583.
Zhou J, Hayakawa Y, Wang TC, Bass AJ (2014) RhoA mutations identified in diffuse gastric cancer. Cancer Cell 26: 9–11.
Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, Rich JN, Bao S (2015) Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol 17: 170–182.
Zhou Y, Wang Y, Wen J, Zhao H, Dong X, Zhang Z, Wang S, Shen L (2016) Aquaporin 3 promotes the stem-like properties of gastric cancer cells via Wnt/GSK-3beta/beta-catenin pathway. Oncotarget 7: 16529–16541.
Zhu L, Cheng X, Shi J, Lin J, Chen G, Jin H, Liu AB, Pyo H, Ye J, Zhu Y, Wang H, Chen H, Fang J, Cai L, Wang TC, Yang CS, Tu SP (2016a) Crosstalk between bone marrow-derived myofibroblasts and gastric cancer cells regulates cancer stemness and promotes tumorigenesis. Oncogene 35: 5388–5399.
Zhu M, Wang M, Yang F, Tian Y, Cai J, Yang H, Fu H, Mao F, Zhu W, Qian H, Xu W (2016b) miR-155-5p inhibition promotes the transition of bone marrow mesenchymal stem cells to gastric cancer tissue derived MSC-like cells via NF-kappaB p65 activation. Oncotarget 7: 16567–16580.
Zuo QF, Zhang R, Li BS, Zhao YL, Zhuang Y, Yu T, Gong L, Li S, Xiao B, Zou QM (2015) MicroRNA-141 inhibits tumor growth and metastasis in gastric cancer by directly targeting transcriptional co-activator with PDZ-binding motif, TAZ. Cell Death Dis 6: e1623.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (Nos. 81272698, 81672319 and 81602507).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Song, Y., Wang, Y., Tong, C. et al. A unified model of the hierarchical and stochastic theories of gastric cancer. Br J Cancer 116, 973–989 (2017). https://doi.org/10.1038/bjc.2017.54
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.54
Keywords
This article is cited by
-
Celastrus orbiculatus extract suppresses gastric cancer stem cells through the TGF-β/Smad signaling pathway
Journal of Natural Medicines (2024)
-
PTBP1 drives c-Myc-dependent gastric cancer progression and stemness
British Journal of Cancer (2023)
-
Radioresistance or/and radiosensitivity of head and neck squamous cell carcinoma: biological angle
Oral and Maxillofacial Surgery (2023)
-
Stochastic model analysis of cancer oncolytic virus therapy: estimation of the extinction mean times and their probabilities
Nonlinear Dynamics (2022)
-
Translational models of 3-D organoids and cancer stem cells in gastric cancer research
Stem Cell Research & Therapy (2021)
