
Abstract
Background:
Photodynamic therapy (PDT) can lead to development of antigen-specific immune response and PDT-mediated immunity can be potentiated by T regulatory cell (Treg) depletion. We investigated whether the combination of PDT with cyclophosphamide (CY) could foster immunity against wild-type tumours expressing self-antigen (gp70).
Methods:
Mice with CT26 tumours were treated with PDT alone or in combination with low-dose CY. T regulatory cell numbers and transforming growth factor-β (TGF-β) levels were measured at several time points after treatment. Mice cured by PDT+CY were rechallenged with CT26 and monitored for long-term survival.
Results:
Photodynamic therapy+CY led to complete tumour regression and long-term survival in 90% of treated mice while the absolute numbers of Treg decreased after PDT+CY and the TGF-β levels were reduced to a level comparable to naïve mice. Sixty-five percent of the mice treated with PDT+CY that survived over 90 days tumour free rejected the rechallenge with the same tumour when a second dose of CY was administered before rechallenge but not without.
Conclusion:
Administration of CY before PDT led to depletion of Treg and potentiated PDT-mediated immunity, leading to long-term survival and development of memory immunity that was only uncovered by second Treg depletion.
Similar content being viewed by others


Main
Photodynamic therapy (PDT) is an effective anti-cancer treatment that involves the administration of a photosensitiser dye (PS), followed by visible light irradiation of the tumour (Dougherty et al, 1998; Dolmans et al, 2003; Agostinis et al, 2011). The light activation of the PS triggers a photochemical reaction that culminates in the production of highly reactive single oxygen (1O2) and/or reactive oxygen species that cause immediate cell damage (Henderson and Dougherty, 1992). Additionally, PDT leads to destruction of tumour vessels and the induction of acute inflammatory responses (Jalili et al, 2004; Castano et al, 2006). Photodynamic therapy has been approved as a treatment modality by US Food and Drug Administration for the use in oesophageal and bronchial cancer and also for pre-invasive and invasive malignant conditions of oral cavity, stomach, bladder, breast and skin (actinic keratosis) (Pass, 1993; Dougherty, 2002; Huang, 2005; Agostinis et al, 2011). One interesting aspect of PDT is its ability to stimulate a systemic immune response against a locally treated tumour (Castano et al, 2006). In fact, PDT has been shown to effectively stimulate both the innate and the adaptive immune systems of the host (Korbelik, 1996; Dougherty et al, 1998; Korbelik et al, 2005) by triggering the release of various pro-inflammatory and acute-phase response mediators (Cecic and Korbelik, 2002; Korbelik et al, 2008; Garg et al, 2011) that lead to infiltration of the treated site by a large number of neutrophils, dendritic cells and other inflammatory cells (Krosl et al, 1995; Cecic et al, 2006; Gollnick et al, 2006).
It has been suggested that tumour development and progression may be strongly dependent on the immune system ability to recognise and destroy malignant cells. However, several lines of evidence suggest that tumours can escape immune surveillance by decreasing or losing the expression of tumour antigens or MHC molecules or by inducing highly immunosuppressive tumour microenvironment by producing high levels of immunosuppressive cytokines like transforming growth factor-β (TGF-β) or IL-10 that in turn promote the development and proliferation of CD4+CD25+FoxP3+ T regulatory cells (Tregs) and immature dendritic cells (Curiel et al, 2004). In healthy individuals, Treg has an important role in maintaining immune homeostasis and tolerance to self-antigens, and preventing autoimmune diseases (Woo et al, 2001; Golgher et al, 2002; Sakaguchi et al, 2008); while in cancer patients the fine balance between Treg and effector T cells is usually distorted and the unrestrained expansion of Treg may foster cancer progression (Valzasina et al, 2006). This immunosuppressive tumour environment presents therefore a therapeutic challenge, but several reports have indicated that the depletion of Treg in vivo facilitates tumour eradication and enhances anti-tumour immunity (Shimizu et al, 1999; Golgher et al, 2002; Tanaka et al, 2002). More recently, the anti-Treg antibody, ipilimumab that targets CTLA-4 has been approved by the FDA and the European Medicine Agency (EMEA) for the treatment of metastatic malignant melanoma (Hodi et al, 2010).
In this report, we further investigate a potential relationship between PDT and Treg. There are limited reports that suggest that PDT on its own may have some immunosuppressive effects (Mroz and Hamblin, 2011), but its effects on Treg have not been thoroughly elucidated. We have previously shown that if Treg is depleted by low-dose cyclophosphamide (CY, 50 mg kg−1) (a traditional cytotoxic cancer drug that at low doses selectively depletes Tregs in mice; Lutsiak et al, 2005), the PDT-induced anti-tumour immune responses are potentiated and a memory immunity is generated against metastatic J774 tumours (Castano et al, 2008). This effect was not observed when PDT was combined with high-dose CY (150 mg kg−1). We also showed (Mroz et al, 2010) that PDT of CT26 tumours expressing a model tumour antigen (Chen et al, 1996) led to long-term survival and spontaneous regression of remote, untreated antigen-positive tumours. However, PDT of CT26 wild-type tumours led only to local regression followed by recurrence, despite the fact that several reports showed that CT26 tumours also express tumour antigens, in particular a single peptide epitope known as AH-1, a non-mutated nonamer derived from the envelope protein (gp70) of an endogeneous ecotropic murine leukaemia provirus (Huang et al, 1996). Recently, a paper by McWilliams et al (2008) described the expression of gp70 mRNA in several tissues of BALB/c mice resulting in immunologic tolerance that affects anti-tumour immunity. In view of these reports, it is possible that gp70 antigen in CT26 tumours behaves like a self-antigen and can serve as a model of cancer shared/auto-antigen.
In this report, we hypothesised that combination of 50 mg kg−1 CY leading to previously shown depletion of Treg with PDT may result in uncovering a PDT-mediated immune response to tumours expressing a model shared tumour antigen gp70.
We also investigated whether the potentiation of PDT by CY is related to the levels of Treg in the spleen and lymph nodes (LNs) and/or to the secretion of the immunosuppressive cytokine, TGF-β, in the serum.
Materials and methods
Cell lines
CT26 wild-type cell line (ATCC, Manassas, VA, USA) were cultured in RPMI medium with L-glutamine and NaHCO3 supplemented with 10% heat inactivated fetal bovine serum, penicillin (100 U ml−1) and streptomycin (100 μg ml−1) (all from Sigma, St Louis, MO, USA) at 37 °C in 5% CO2 in 75 cm2 flasks (Falcon; Invitrogen, Carlsbad, CA, USA).
Mouse tumour model
BALB/c mice (6–8 weeks old) were purchased from Charles River Laboratories (Boston, MA, USA). All experiments were carried out according to a protocol approved by the Subcommittee on Research Animal Care (IACUC) at MGH and were in accord with NIH guidelines. Mice were inoculated with 350 000 cells subcutaneously into the depilated right thigh. Two orthogonal dimensions (a and b) of the tumour were measured 2–3 times a week with vernier calipers. Tumour volumes were calculated as follows, volume=4π/3 × [(a+b)/4]3. When tumours reached a diameter of 5–7 mm (10 days after inoculation) PDT was performed.
Photosensitiser and light source
Liposomal benzoporphyrin derivative mono acid ring A (BPD) was a generous gift from QLT Inc. (Vancouver, BC, Canada) and was prepared by diluting the powder to a concentration of 0.3 mg ml−1 in sterile 5% dextrose. A 1 W 690-nm diode laser (B&W Tek Inc., Newark, DE, USA) was coupled into a 0.8-mm diameter fibre.
PDT and CY treatment
Tumour-bearing mice were anaesthetised with intraperitoneal (i.p.) injection of 87.5 mg kg−1 of ketamine and 12.5 mg kg−1 of xylazine and BPD (1 mg kg−1 in 5% dextrose solution) was administered i.v. via the supraocular plexus. Control mice received 5% dextrose only. Fifteen minutes after BPD injection, illumination was performed. We measured the light power with a power meter (THORLABS GmbH, Dachau, Germany; PM 100D) and calculated the correct distance between the optic fibre and the tumour, to use 100 mW cm−2 fluence rate in the exposed area. A total fluence of 120 J cm−2 was delivered at a fluence rate of 100 mW cm−2. The mice were killed when any of the tumour diameters exceeded 1.5 cm or tumour volume reached 1500 mm3. In certain groups of mice, 50 mg of CY was administered via i.p. injection 2 days before PDT.
Surgical operation
When tumours reached 5–7 mm in diameter tumour was excised under general anaesthesia with i.p. injection of 87.5 mg kg−1 of ketamine and 12.5 mg kg−1 of xylazine. After the surgical operation, mouse was killed at five different time points (0, 1, 4, 10 and 14 days).
Rechallenge
Mice surviving 90 days after PDT were subsequently rechallenged with 350 000 cells of CT26 in the left thigh and monitored for another 60 days. Naïve control mice were inoculated with the same sample of cells as a control for tumorigenicity. A group of mice received 50 mg kg−1 of CY via i.p. injection 2 days before rechallenge
Flow cytometry analysis of Treg
There were 18 mice per group killed in groups of 3 mice per time point. Spleen and inguinal LNs were harvested at six different time points (−2 days, 0 h, 1, 4, 10 and 14 days). Both organs were homogenised separately by gently pressing them through the 70-μm cell strainer, using a syringe plunger to make single-cell suspensions. Cells were counted by trypan blue exclusion on a haemocytometer after red blood cell lysis. In all, 1 × 106 of either splenocytes or lymphocytes from LNs was washed and resuspended in staining buffer (eBioscience, San Diego, CA, USA). The cells were incubated with rat anti-mouse CD16/CD32 (FcRγIII/II) mAb (eBioscience) to minimise the binding to the Fc receptor by the staining antibodies.
This was followed by labelling with fluorescent dye conjugated primary antibodies against the markers CD4 (RM4-5, FITC conjugated; eBioscience) and CD25 (PC61.5, phycoerythrin conjugated; eBioscience). After washing and fixation/permeabilisation, the cells were incubated with primary conjugated anti-Foxp3 antibody (FJK-16s). An FACSAria flow cytometer was used for data acquisition (Becton Dickinson, San Antonio, TX, USA). In all, 50 000 events were counted in the case of splenocytes and 20 000 for the lymphocytes. All data were analysed with either FACSAria or FlowJo (TreeStar, Ashland, OR, UAS) software. Isotype controls were used to set proper regional gates.
ELISA for TGF-β
Blood samples were drawn from the aorta and were centrifuged at 6000 r.p.m. for 20 min to extract serum. All samples of sera were stored in the −80 °C degree freezer until ELISA was performed. ELISA for TGF-β (R&D Systems, Minneapolis, MN, USA; DuoSet) was performed according to the manufacturer’s instructions. Briefly, 20 μl of serum was incubated with 20 μl of 2.5 N acetic acid/10 M urea for 10 min to activate TGF-β. After 10 min of incubation, the samples were neutralised by adding 20 μl of 2.7 N NaOH/1 M HEPES (at this stage the serum is diluted 1 : 3). After 10 min of incubation, 30 μl of diluted samples was 15-fold further diluted with Reagent diluents (1.4% bovine serum albumin, 0.05% Tween-20 in PBS) and used for the assay.
RT–PCR
Total RNA was extracted from cultured cancer cells using the RNeasy Plus Mini kit (Qiagen, Gaithersburg, MD, USA) and reversely transcribed by using the Seniscript RT kit (Qiagen). Primers for specific murine TGF-β (sense, TGCTTCAGCTCCACAGAGAA: antisense, TGGTTGTAGAGGGCAAGGAC) (Sieber et al, 2011) and murine gp70 (sense, AAGGTCCAGCGTTCTCAAAAC; antisense, AGGTGGCGTTAGCTGTTTGT) (Abe et al, 2012) were used with 2 μg of sample cDNA and amplified with Taq polymerase (Qiagen) using Thermal Cycler (GeneAmp PCR system 9700; Applied Biosystems, Woburn, MA, USA).
Statistics
Comparisons of Treg and TGF-β values were done by one-way ANOVA followed by Tukey post hoc test. Survival analysis was carried out by plotting in the ordinate the percentage of surviving mice out of the total number of mice per treatment group. According to the protocol, when the tumours reached 1500 mm3, the mice were euthanised and therefore counted as dead. In the statistical analysis, we used the log-rank test and in all cases the significance level was set at P<0.05.
Results
The combination of 50 mg kg−1 CY with PDT leads to tumour eradication and survival advantage
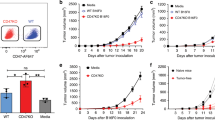
We employed a vascular BPD-PDT regimen delivered alone, or in combination with CY (50 mg kg−1). Cyclophosphamide was administered on day 8 after CT26 tumour inoculation and PDT performed at day 10 (Figure 1). Photodynamic therapy produced a good local response in CT26 tumours as manifested by development of oedema followed by a circumscribed black eschar and a marked reduction in tumour size lasting until day 18 (Figure 2A). However, local tumour regrowth occurred in all treated mice and the median survival time of the PDT-treated mice was 29 days vs 25 days for control untreated CT26 (Figure 2B).

(A) Experimental design scheme of in vivo PDT, CY administration and tumour rechallenge. (B) RT–PCR expression of gp70 in CT26 cells.

PDT and CY treatment (alone or combined) of CT26 tumours. (A) Plots of mean tumour volumes in mice bearing CT26 tumours. Points are means from 10 to 15 mice and bars are s.d. (B) Kaplan–Meier survival curves of the % of mice surviving after no treatment, PDT alone, CY alone or PDT plus CY. The median survival time of PDT-treated mice was 29 days vs 25 days for control untreated CT26 (P=0.0304).
When mice were treated with the combination of BPD-PDT and CY, the outcome was completely different from that seen in mice treated with PDT alone. Figure 2A shows that PDT and CY together led to greater local oedema compared with PDT alone followed by tumour regression in 9 out of 10 mice with all 9 mice surviving tumour free for >90 days (Figure 2B). Mice treated with CY alone showed some tumour growth delay (Figure 2A), but the survival of this group of mice was not significantly different from control untreated mice, or treated with PDT alone (Figure 2B). These results taken together suggest that PDT combined with 50 mg kg−1 of CY led to a dramatic improvement in long-term survival when compared with either treatment alone.
CY combined with PDT abrogates the increase in Treg induced by PDT alone
To elucidate the immunostimulatory/immunosuppressive effects of PDT alone, and combined with 50 mg kg−1 of CY, we examined the absolute number of CD4+CD25+Foxp3+ Tregs in both spleen and LNs at different time points after PDT (0, 1, 4, 10 and 14 days). We included five groups of mice in the study: (A) tumour-free mice, that received PDT (PDT no cancer); (B) mice inoculated with cancer and treated with PDT (PDT cancer); (C) tumour-bearing mice that received injection of 5% dextrose and laser therapy alone (sham PDT cancer); (D) tumour-bearing mice that were treated with PDT and CY combined (PDT+CY cancer) and (E) tumour-bearing mice injected with CY alone (CY cancer). We also measured the numbers of Tregs in naïve mice (i.e., mice without cancer that had not received any kind of treatment) and the values obtained were 0.152±0.011 × 106 (n=3) in the spleen and 0.199±0.020 × 106 (n=3) in the LNs.
The absolute numbers of Treg measured from spleen and LNs in the five groups are depicted in Figure 3A and B, respectively, and the P-values obtained from statistical analyses are displayed in Table 1 for spleen and Table 2 for LNs.

Quantification of Tregs in the spleens and lymph nodes. Absolute numbers of CD4+CD25+Foxp3+ Tregs from spleens (A) and lymph nodes (B) extracted from the mouse groups: PDT no cancer, PDT cancer, sham PDT cancer, PDT+CY cancer and CY cancer. Bars are s.e.m. Statistical comparisons are shown in Table 1 (spleen) and Table 2 (lymph nodes). Significance was determined by one-way ANOVA and Tukey post hoc correction.
We found that PDT alone led to a significant increase in CD4+CD25+Foxp3+ Treg between day 0 and day 4 after treatment in the spleen and also in the LNs. At later time points, the numbers of Treg in both organs dropped back to values comparable to the naive mice. The ‘PDT no cancer’ group showed that Treg was highly, but not significantly increased in the spleen at day 4, and significantly increased in the LNs at day 4.
The ‘sham PDT cancer’ group displayed a gradual and significant raise of Treg in both the spleen (Figure 3A) and the LNs (Figure 3B), to levels much higher than in PDT-treated mice (at 14 days, P<0.001 in both spleen and LNs) coinciding for the progression and increase in size of tumour. In the ‘CY alone’ group, the absolute numbers of Treg decreased between −2 days and 1 day after PDT in both spleen (Figure 3A) and LNs (Figure 3B), confirming that CY is effectively depleting Tregs. Then, similarly to the sham PDT cancer group, Tregs increased steadily between 1 day and 14 days, in both organs, to levels higher than in naïve mice (at 14 days P<0.001 in both spleen and LNs).
When CY was combined with PDT, Tregs decreased in both spleen and LNs reaching a minimum at 0 day and then remained within the ‘naïve’ range during PDT and all the time points after the treatment
These results indicate that tumour alone as well as PDT alone may lead to an increase in Treg population, and that this unfavourable effect can be abrogated by administration of CY before PDT.
PDT combined with CY, but not PDT alone, decreases TGF-β to a baseline level
Since TGF-β is the major cytokine involved in Treg-mediated immunosuppression as well as it can lead to an increase in numbers of Treg, we measured the level of TGF-β in the serum of mice treated with PDT, PDT+CY or mice with tumour that was surgically removed. We also measured the level of TGF-β in naïve mice and the value was 1412.41±93.31 pg ml−1 (n=3). Our results demonstrate that PDT alone led to moderate elevation of TGF-β levels in serum as compared with naïve mice while the combination of PDT+CY led to a significant decrease in TGF-β levels at day 1 (Figure 4B; Table 3) and the TGF-β levels in this group remained low throughout the entire period of the experiment.

Detection of TGF- β. (A) RT–PCR analysis of TGF-β expression in CT26 cells. (B) Mean levels of TGF-β cytokine measured in the serum from mice at different time points after PDT, PDT+CY, or surgically operated, as well as in control naïve mice. Statistical comparisons are shown in Table 3 and significance was determined by one-way ANOVA and Tukey post hoc correction.
To determine to which extent PDT is responsible for TGF-β secretion and not the tumour itself, we analysed the serum samples from mice whose tumours were surgically removed (Surgery cancer). Upon tumour resection, TGF-β decreased gradually and significantly (at 4 days P<0.01 and 14 days P<0.001 compared with 0 day), becoming comparable to the levels from naïve mice at 14 days after treatment.
Treg suppresses the reactivation of the long-term anti-tumour memory immunity generated by PDT plus CY
To investigate whether the combination of PDT and CY induces the formation of memory immunity, mice that were treated by PDT+CY and remained tumour free for 90 days were subsequently rechallenged with the same tumour cells (CT26). Naïve mice were used as a control. As shown in Figure 5, the tumours were not rejected in the PDT+CY cured mice (CT26 WT CY cured rechall) and the median survival time of PDT+CY treated rechallenged mice was 18 days vs 25 days for naõve mice. When the rechallenge was preceded by administration of 50 mg kg−1 of CY, 65% of mice rejected the rechallenge (CT26 WT CY cured CY rechall) and remained tumour free for another 60 days of observation. When CY was given in naïve mice before tumour inoculation, all tumours progressed and all the mice were killed due to the tumour burden (median survival time=22 days). These data show that PDT+CY leads to development of memory immunity T cells, that is however suppressed by the Treg. The administration of CY to deplete Treg again at the time of rechallenge abrogated the immunosuppressive effects of Treg and uncovered the activity of memory T cell leading to rechallenge rejection in 65% of cases,

Tumour rechallenge of cured mice. Kaplan–Meier survival curves of the % of mice surviving after rechallenge with CT26 in the groups: naïve; naïve+CY; CT26 cured; CT26 cured+CY. The median survival time of PDT+CY rechallenged mice was 18 days vs 25 days for naïve mice (P=0.0009).
Discussion
In a previous work, we showed that PDT combined with low-dose CY produced a dramatic improvement in survival and formation of anti-tumour immunity manifested by rejection of tumour rechallenge in mice bearing the highly metastatic J774 tumour (Castano et al, 2008). In the present study, we investigated whether this potentiating effect of CY on PDT-induced immunity could also be extended to other mouse tumour models. Therefore, we employed CT26 colon adenocarcinoma and found that the combination of PDT with low-dose CY produced a dramatic improvement in long-term survival, whereas treatment with PDT or 50 mg kg−1 of CY alone did not lead to a significant survival advantage.
The CY administration has been suggested to have a specific effect in depleting the Treg (Awwad and North, 1988; Awwad and North, 1989; Ghiringhelli et al, 2004) and has been used in several studies to induce tumour regression and increase the immunological responses against cancer, that could otherwise be suppressed by Treg.
To our knowledge, the effect of PDT on Treg has not been described so far. Here, we show for the first time that PDT led to a significant increase of Treg in the first few days post treatment in both spleen and LNs, reaching a peak at 4 days after PDT. In the following days, Treg numbers reduced back to levels comparable to those before the treatment.
Interestingly, this transient PDT effect on Treg levels was not related to the presence of cancer cells as PDT had the same effect in tumour-free mice. These results corroborate the hypothesis that PDT does not act only as a local therapy, capable of destroying tumours at the site of light exposure, but PDT may also have profound systemic immunological effects.
Several pre-clinical and clinical studies have shown that PDT is capable to influence the adaptive arm of the immune system in many different ways; some regimens leads to potentiation of adaptive immunity, while others produce immunosuppressive effects (Agostinis et al, 2011; Mroz and Hamblin, 2011). The underlying mechanism responsible for potentiation or suppression is not yet known; however, it has been suggested that they may be multifactorial and dependent on PDT treatment regimen, the area treated and the photosensitiser used. In our case, we saw that Treg reach a peak value between 0 and 4 days just in two animal groups: PDT no cancer and PDT cancer. The same two groups reach the peak of Treg at the same time points also in the LNs. We hypothesise that this early immunosuppressive effect of PDT may result from a strong inflammatory response induced by PDT at site of the treatment. The host’s immune system might produce more Tregs to limit the local acute inflammation. Obviously, after day 4 the withdrawal of the inflammatory response and the tumour healing restored the normal immunological conditions. This may explain why Treg was increased in the first days after PDT, even in the absence of tumour and after 4 days decreased again to the ‘naïve’ level.
The administration of 50 mg kg−1 of CY led to a considerable depletion of Treg in both spleen and LNs and nadir was reached at 2 days after CY administration (the time we selected to deliver PDT). The reduction in the absolute number of Treg was more remarkable in LNs than in spleen; the reason could be that while LNs are more pure pools of T cells, the spleen is acting mainly as a blood filter and contains a wider variety of different subsets of cells. The literature suggest three possible mechanisms of selective depletion of Treg exerted by low-dose CY: (i) inhibition of proliferation of the more rapid proliferating Tregs as compared with other classes of lymphocytes (Ercolini et al, 2005, ii) induction of apoptosis and functional inhibition of Tregs (Lutsiak et al, 2005) and (iii) inhibition of inducible nitric oxide synthase that controls the number of Tregs (Loeffler et al, 2005).
It remains to be fully elucidated how long the effect of low-dose CY on Treg lasts for. Peng et al (2013) have recently found that a single dose administration of CY leads to a reduction in the number of Tregs in the tumour microenvironment in a transient manner. Eight days after CY injection they observed comparable levels of Tregs in mice treated with CY or untreated, suggesting that a repopulation of Treg occurs some days after CY administration. Here, we saw that after the initial depletion of Treg, from day 3 onwards following CY administration the number of Treg has steadily increased over time; however, this increase coincided with progression of tumour and increase in tumour volume. It was only when PDT was combined with 50 mg kg−1 of CY, that the increase in Treg was not seen, either in spleen or in LNs, at any time point after the therapy. This observation correlated with the PDT-mediated regression of the tumour and decrease in tumour volume.
To further investigate the relationship of Treg and tumour microenvironment in the context of PDT and CY therapy, we measured the levels of TGF-β in the serum of the mice at different time points after PDT. The TGF-β signalling has a complex role in carcinogenesis, having both tumour suppressor and oncogenic activities. It has been shown that TGF-β exerts tumour suppressor effects in normal epithelial cells and in early stage of tumour progression by inducing cell-cycle arrest, senescence and apoptosis. Conversely, during the progression of cancer the anti-proliferative effects of TGF-β are selectively lost and TGF-β induces many activities leading to growth, invasion and metastasis of cancer cells (Nagaraj and Datta, 2010). Cancer cells often escape from the growth inhibitory function of TGF-β by mutational inactivation or dysregulated expression of components in its signalling. It also known that increased levels of TGF-β lead to adverse effects on anti-tumour-immunity in multiple ways: inhibiting the proliferation and differentiation of T lymphocytes, natural killer cells, neutrophils, macrophages and B cells (Letterio and Roberts, 1998) and promoting the function of Tregs (Wan and Flavell, 2008). Moreover, Tregs mediate their suppressive activity by producing TGF-β themselves. We expected that any effect induced by PDT and/or CY on the function and level of Tregs could be reflected also in the level of TGF-β in the serum. However, it is necessary to consider that also other cell types are capable of secreting TGF-β, such as cancer cells (Elliott and Blobe, 2005), macrophages (Wahl et al, 1990) and B lymphocytes (Weitzmann et al, 2000); therefore, the kinetics of TGF-β may not exactly reflect the kinetics of Treg numbers.
We have confirmed that indeed the CT26 cancer cells do express TGF-β (Figure 4A), a characteristic that may strongly contribute to the development of immunosuppressive microenvironment of this tumour. The fact that CT26 tumours are a significant source of TGF-β in tumour-bearing mice may explain why the TGF-β and Treg levels continued to increase unabated with tumour progression and returned to ‘naïve’ levels when the tumour was removed by surgery or PDT. Photodynamic therapy and PDT+CY had similar impacts on TGF-β; however, the two effects were not identical. Photodynamic therapy+CY caused a more pronounced reduction in TGF-β compared with PDT alone, decreasing the level to a range comparable to naïve mice. When PDT was applied alone, we observed a reduction in TFG-β, however, it remained significantly higher than in naïve mice.
In CT26 tumour model, the combined PDT+CY treatment led to long-term tumour regression, however, none of the mice treated by combination therapy rejected the subsequent tumour rechallenge. It was only when we administered a second dose of CY before rechallenge that 65% of mice in long-term remission after PDT+CY treatment successfully rejected the rechallenge. We attribute this effect to the fact that CT26 cells express a tumour rejection antigen gp70 (Figure 1B) that appears to behave like a shared auto-antigen. It appears that Treg can suppress the development of anti-gp70 immunity and/or reactivation of anti-gp 70 memory immunity; however, this effect can be uncovered when Treg is depleted before PDT and again before rechallenge. This observation may have far-reaching clinical implication as it suggests that in patients treated with PDT there may be developing anti-tumour immunity that could be uncovered at the time of relapse by appropriately designed anti-Treg therapy. If PDT was ever combined with low-dose CY in patients, then it may be possible to use a metronomic CY regimen (Ge et al, 2012) rather than a bolus administration to extend the length of time that Tregs were controlled until the possibility of tumour recurrence was no longer a real threat. In conclusion, the presented results further enrich our understanding of the effects of the role of Treg in PDT-mediated anti-tumour immune response. Further studies, however, are certainly necessary to fully understand the complicated interaction between PDT, Treg, immunosuppressive microenvironment of the tumour and the role of different types of tumour antigens to better design and exploit PDT-based immunotherapy to improve patient outcome.
Change history
15 October 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Abe S, Kurata M, Suzuki S, Yamamoto K, Aisaki K, Kanno J, Kitagawa M (2012) Minichromosome maintenance 2 bound with retroviral Gp70 is localized to cytoplasm and enhances DNA-damage-induced apoptosis. PLoS One 7 (6): e40129.
Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson BC, Golab J (2011) Photodynamic therapy of cancer: an update. CA Cancer J Clin 61 (4): 250–281.
Awwad M, North RJ (1988) Cyclophosphamide (Cy)-facilitated adoptive immunotherapy of a Cy-resistant tumour. Evidence that Cy permits the expression of adoptive T-cell mediated immunity by removing suppressor T cells rather than by reducing tumour burden. Immunology 65 (1): 87–92.
Awwad M, North RJ (1989) Cyclophosphamide-induced immunologically mediated regression of a cyclophosphamide-resistant murine tumor: a consequence of eliminating precursor L3T4+ suppressor T-cells. Cancer Res 49 (7): 1649–1654.
Castano AP, Mroz P, Hamblin MR (2006) Photodynamic therapy and anti-tumour immunity. Nat Rev Cancer 6 (7): 535–545.
Castano AP, Mroz P, Wu MX, Hamblin MR (2008) Photodynamic therapy plus low-dose cyclophosphamide generates antitumor immunity in a mouse model. Proc Natl Acad Sci USA 105 (14): 5495–5500.
Cecic I, Korbelik M (2002) Mediators of peripheral blood neutrophilia induced by photodynamic therapy of solid tumors. Cancer Lett 183 (1): 43–51.
Cecic I, Stott B, Korbelik M (2006) Acute phase response-associated systemic neutrophil mobilization in mice bearing tumors treated by photodynamic therapy. Int Immunopharmacol 6 (8): 1259–1266.
Chen PW, Wang M, Bronte V, Zhai Y, Rosenberg SA, Restifo NP (1996) Therapeutic antitumor response after immunization with a recombinant adenovirus encoding a model tumor-associated antigen. J Immunol 156 (1): 224–231.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W (2004) Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10 (9): 942–949.
Dolmans DE, Fukumura D, Jain RK (2003) Photodynamic therapy for cancer. Nat Rev Cancer 3 (5): 380–387.
Dougherty TJ (2002) An update on photodynamic therapy applications. J Clin Laser Med Surg 20 (1): 3–7.
Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J, Peng Q (1998) Photodynamic therapy. JNCI 90 (12): 889–905.
Elliott RL, Blobe GC (2005) Role of transforming growth factor Beta in human cancer. J Clin Oncol 23 (9): 2078–2093.
Ercolini AM, Ladle BH, Manning EA, Pfannenstiel LW, Armstrong TD, Machiels JP, Bieler JG, Emens LA, Reilly RT, Jaffee EM (2005) Recruitment of latent pools of high-avidity CD8(+) T cells to the antitumor immune response. J Exp Med 201 (10): 1591–1602.
Garg AD, Krysko DV, Vandenabeele P, Agostinis P (2011) DAMPs and PDT-mediated photo-oxidative stress: exploring the unknown. Photochem Photobiol Sci 10 (5): 670–680.
Ge Y, Domschke C, Stoiber N, Schott S, Heil J, Rom J, Blumenstein M, Thum J, Sohn C, Schneeweiss A, Beckhove P, Schuetz F (2012) Metronomic cyclophosphamide treatment in metastasized breast cancer patients: immunological effects and clinical outcome. Cancer Immunol Immunother 61 (3): 353–362.
Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F (2004) CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol 34 (2): 336–344.
Golgher D, Jones E, Powrie F, Elliott T, Gallimore A (2002) Depletion of CD25+ regulatory cells uncovers immune responses to shared murine tumor rejection antigens. Eur J Immunol 32 (11): 3267–3275.
Gollnick SO, Owczarczak B, Maier P (2006) Photodynamic therapy and anti-tumor immunity. Lasers Surg Med 38 (5): 509–515.
Henderson BW, Dougherty TJ (1992) How does photodynamic therapy work? Photochem Photobiol 55 (1): 145–157.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363 (8): 711–723.
Huang AY, Gulden PH, Woods AS, Thomas MC, Tong CD, Wang W, Engelhard VH, Pasternack G, Cotter R, Hunt D, Pardoll DM, Jaffee EM (1996) The immunodominant major histocompatibility complex class I-restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proc Natl Acad Sci USA 93 (18): 9730–9735.
Huang Z (2005) A review of progress in clinical photodynamic therapy. Tech Canc Res Treat 4 (3): 283–293.
Jalili A, Makowski M, Switaj T, Nowis D, Wilczynski GM, Wilczek E, Chorazy-Massalska M, Radzikowska A, Maslinski W, Bialy L, Sienko J, Sieron A, Adamek M, Basak G, Mroz P, Krasnodebski IW, Jakobisiak M, Golab J (2004) Effective photoimmunotherapy of murine colon carcinoma induced by the combination of photodynamic therapy and dendritic cells. Clin Cancer Res 10 (13): 4498–4508.
Korbelik M (1996) Induction of tumor immunity by photodynamic therapy. J Clin Laser Med Surg 14 (5): 329–334.
Korbelik M, Cecic I, Merchant S, Sun J (2008) Acute phase response induction by cancer treatment with photodynamic therapy. Int J Cancer 122 (6): 1411–1417.
Korbelik M, Sun J, Cecic I (2005) Photodynamic therapy-induced cell surface expression and release of heat shock proteins: relevance for tumor response. Cancer Res 65 (3): 1018–1026.
Krosl G, Korbelik M, Dougherty GJ (1995) Induction of immune cell infiltration into murine SCCVII tumour by photofrin-based photodynamic therapy. Br J Cancer 71 (3): 549–555.
Letterio JJ, Roberts AB (1998) Regulation of immune responses by TGF-beta. Annu Rev Immunol 16: 137–161.
Loeffler M, Kruger JA, Reisfeld RA (2005) Immunostimulatory effects of low-dose cyclophosphamide are controlled by inducible nitric oxide synthase. Cancer Res 65 (12): 5027–5030.
Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H (2005) Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 105 (7): 2862–2868.
McWilliams JA, Sullivan RT, Jordan KR, McMahan RH, Kemmler CB, McDuffie M, Slansky JE (2008) Age-dependent tolerance to an endogenous tumor-associated antigen. Vaccine 26 (15): 1863–1873.
Mroz P, Hamblin MR (2011) The immunosuppressive side of PDT. Photochem Photobiol Sci 10 (5): 751–758.
Mroz P, Szokalska A, Wu MX, Hamblin MR (2010) Photodynamic therapy of tumors can lead to development of systemic antigen-specific immune response. PLoS One 5 (12): e15194.
Nagaraj NS, Datta PK (2010) Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin Investig Drugs 19 (1): 77–91.
Pass HI (1993) Photodynamic therapy in oncology: mechanisms and clinical use. J Natl Cancer Inst 85 (6): 443–456.
Peng S, Lyford-Pike S, Akpeng B, Wu A, Hung CF, Hannaman D, Saunders JR, Wu TC, Pai SI (2013) Low-dose cyclophosphamide administered as daily or single dose enhances the antitumor effects of a therapeutic HPV vaccine. Cancer Immunol Immunother 62 (1): 171–182.
Sakaguchi S, Yamaguchi T, Nomura T, Ono M (2008) Regulatory T cells and immune tolerance. Cell 133 (5): 775–787.
Shimizu J, Yamazaki S, Sakaguchi S (1999) Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol 163 (10): 5211–5218.
Sieber MW, Claus RA, Witte OW, Frahm C (2011) Attenuated inflammatory response in aged mice brains following stroke. PLoS One 6 (10): e26288.
Tanaka H, Tanaka J, Kjaergaard J, Shu S (2002) Depletion of CD4+ CD25+ regulatory cells augments the generation of specific immune T cells in tumor-draining lymph nodes. J Immunother 25 (3): 207–217.
Valzasina B, Piconese S, Guiducci C, Colombo MP (2006) Tumor-induced expansion of regulatory T cells by conversion of CD4+CD25- lymphocytes is thymus and proliferation independent. Cancer Res 66 (8): 4488–4495.
Wahl SM, McCartney-Francis N, Allen JB, Dougherty EB, Dougherty SF (1990) Macrophage production of TGF-beta and regulation by TGF-beta. Ann NY Acad Sci 593: 188–196.
Wan YY, Flavell RA (2008) TGF-beta and regulatory T cell in immunity and autoimmunity. J Clin Immunol 28 (6): 647–659.
Weitzmann MN, Cenci S, Haug J, Brown C, DiPersio J, Pacifici R (2000) B lymphocytes inhibit human osteoclastogenesis by secretion of TGFbeta. J Cell Biochem 78 (2): 318–324.
Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, Rubin SC, Kaiser LR, June CH (2001) Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res 61 (12): 4766–4772.
Acknowledgements
Research in the Hamblin laboratory is supported by US NIH grant AI050875. ER was supported by the PhD program of the Medical University of Graz, Graz, Austria.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Reginato, E., Mroz, P., Chung, H. et al. Photodynamic therapy plus regulatory T-cell depletion produces immunity against a mouse tumour that expresses a self-antigen. Br J Cancer 109, 2167–2174 (2013). https://doi.org/10.1038/bjc.2013.580
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.580
Keywords
This article is cited by
-
Inhibition of IDO leads to IL-6-dependent systemic inflammation in mice when combined with photodynamic therapy
Cancer Immunology, Immunotherapy (2020)
-
Anti-tumor immunity of BAM-SiPc-mediated vascular photodynamic therapy in a BALB/c mouse model
Cellular & Molecular Immunology (2017)
-
Inhibition of mouse breast adenocarcinoma growth by ablation with intratumoral alpha-irradiation combined with inhibitors of immunosuppression and CpG
Cancer Immunology, Immunotherapy (2016)
